
Abstract
Peloruside A (PLA) and Laulimalide (LAU) are novel microtubule-stabilizing agents with promising properties against different cancer types. These ligands share a non-taxoid binding site at the outer surface of β-tubulin and promote microtubule stabilization by bridging two adjacent αβ-tubulin dimers from parallel protofilaments. Recent site-directed mutagenesis experiments confirmed the existence of a unique β-tubulin site mutation (Gln293Met) that specifically increased the activity of PLA and caused resistance to LAU, without affecting the stability of microtubules in the absence of the ligands. In this work, fully atomistic molecular dynamics simulations were carried out to examine the PLA and LAU association with native and mutated αβ-tubulin in the search for structural and energetic evidence to explain the role of Gln293Met mutation on determining the activity of these ligands. Our results revealed that Gln293Met mutation induced the loss of relevant LAU–tubulin contacts but exerted negligible changes in the interaction networks responsible for PLA–tubulin association. Binding free energy calculations (MM/GBSA and MM/PBSA), and weak interaction analysis (aNCI) predicted an increased affinity for PLA, and a weakened association for LAU after mutation, thus suggesting that Gln293Met mutation exerts its action by a modulation of drug–tubulin interactions. These results are valuable to increase understanding about PLA and LAU activity and to assist the future design of novel agents targeting the PLA/LAU binding pocket.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Microtubules are tube-shaped protein polymers that play a relevant role in a number of cell processes such as cellular transport, cell structure maintenance, signaling and mitosis. Microtubules are formed by the polymerization of αβ-tubulin heterodimers organized in a polar head-to-tail fashion, and assembled into linear protofilaments that interact laterally to form a cylindrical structure [1]. Because of their key role on cell division, microtubules are well-known molecular targets for antimitotic agents, which disrupt microtubule dynamics by either stabilizing the microtubule structure (e.g. paclitaxel, epothilones) or inhibiting the microtubule polymerization (e.g. colchicine, vinblastine) [1,2,3].
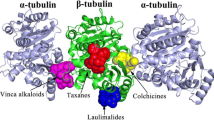
Peloruside A (PLA) and Laulimalide (LAU) are novel lead non-taxoid-site microtubule-stabilizing agents with promising antitumor properties against a wide range of cancer cell lines (Fig. 1) [4,5,6]. In the past years, PLA and LAU have been the subject of extensive research due to their advantageous features compared to taxol-like compounds, such as higher aqueous solubility, greater activity against taxol-resistant cancer cells, and better tolerability, among others [7, 8]. As microtubule-stabilizing agents, PLA and LAU exert their action by binding to polymerized microtubules at a unique binding site located on the outer surface of β-tubulin, which is different and does not overlap the well-known taxol pocket at the luminal side of the β-subunit [9, 10]. Recent high-resolution crystallographic structures of PLA and LAU complexes with αβ-tubulin, the stathmine-like protein RB3 and tubulin tyrosine ligase (T2T-TTL)—with PDB codes 4O4J (2.2 Å) and 4O4H (2.1 Å), respectively—confirmed the location of the PLA/LAU binding site and the identity of the amino acid residues involved in their association with β-tubulin. These models revealed that both ligands overlap within the binding pocket, and locate part of their macrocycles outside the protein cavity enabling the interaction between adjacent αβ-tubulin dimers from neighboring protofilaments, which is the molecular mechanism through which PLA and LAU binding triggers microtubule stabilization. Additionally, crystallographic data showed that PLA and LAU induce the allosteric stabilization of the M-loop region in β-tubulin allowing favorable lateral contacts across protofilaments [9].

Structure of Laulimalide (LAU) and Peloruside A (PLA)
In the past years, diverse reports addressed the role of specific binding site residues on determining the activity of PLA and LAU by generation of resistant cell lines and site-directed mutagenesis experiments [11,12,13]. In 2015, Kanakkanthara et al. studied the activity of PLA, LAU, and other tubulin-binding agents in four β-tubulin mutants (Gln293Met, Glu297Ile, Val335Trp and Asn339Leu) using site-directed mutagenesis, and transfection of each mutant into HEK and HeLa cells [13]. Their results revealed that Val335Trp and Asn339Leu mutations affected the stability of microtubules in the absence of the ligands, which resulted in increased resistance to PLA and LAU. On the other hand, the Glu297Ile mutation did not interfere with microtubule stability in a ligand-free state, but specifically conferred resistance to PLA and LAU, as expected. Surprisingly, the Gln293Met mutation had no effect on microtubule dynamics in the absence of the ligands but specifically increased the activity of PLA and exerted resistance to LAU. This unique result constituted the first example of a single site mutation that specifically increased the activity of a microtubule-stabilizing agent, and was attributed to a modulation of drug–tubulin interactions by the Gln293Met mutation. Nevertheless, further information is required to support this hypothesis and to gain insight into the molecular mechanism through which this site mutation exerts its action.
In this work, fully atomistic molecular dynamics (MD) simulations, combined with binding free energy calculations, and weak interaction analysis were carried out to examine the role of the Gln293Met mutation on the binding properties of PLA and LAU complexes with native and mutated αβ-tubulin, aimed at providing molecular-level information to explain the enhanced activity of PLA in the mutant system. Despite MD simulations have been widely used as complementary tools to examine the binding properties of diverse microtubule-stabilizing/destabilizing agents [14,15,16,17,18,19], studies on PLA and LAU association are scarce and the molecular details for their efficacious interaction with αβ-tubulin remain unclear [20, 21]. In the present report, 300 ns MD simulations were performed using the high-resolution 4O4J (2.2 Å) and 4O4H (2.1 Å) crystallographic models as reference structures to build the initial coordinates for PLA and LAU complexes with native αβ-tubulin, respectively. Trajectory analysis was conducted during the last 100 ns of MD trajectories revealing that both ligands retained their global binding poses and bound conformations after Gln293Met mutation, despite some relevant drug–tubulin intermolecular contacts were lost in the LAU complex with the mutant. Binding free energy calculations (MM/GBSA and MM/PBSA) and weak interaction analysis (aNCI) suggested that Gln293Met mutation increased the affinity towards PLA but weakened the interaction with LAU, thus confirming that this mutation mediates PLA and LAU activity by altering drug–tubulin interactions. To the best of our knowledge, this work constitutes the first molecular modeling approach dealing with the binding properties of PLA and the specific role of the Gln293Met site mutation on modulating its activity as microtubule-stabilizing agent. Furthermore, the results herein discussed are valuable to enlarge understanding about the molecular mechanism of action of PLA and LAU, and to assist the design novel more potent ligands targeting the PLA/LAU binding pocket.
Methods and materials
Simulated systems
Starting structures for the αβ-tubulin complexes with PLA and LAU were retrieved from the crystallographic models of αβ-tubulin–T2T-TTL complexes with the corresponding ligands (PDB codes 4O4J and 4O4H, respectively). Coordinates for missing residues and Gln293Met mutant were modeled using the SWISS-MODEL server [22, 23]. The initial coordinates of PLA and LAU complexes with Gln293Met mutant were obtained from protein–ligand docking studies using the Autodock Vina software [24]. A cubic box of 20 × 20 × 20 Å centered on the binding site was defined, Gasteiger charges were assigned to all atoms, and rotatable bonds were identified using AutoDockTools. The highest-ranked structure for each complex was selected to conduct further simulation protocols. To assess the validity of the docking protocol, the structures of LAU and PLA complexes with native αβ-tubulin were simulated and compared to the corresponding crystallographic models.
Molecular dynamics simulations
MD simulations were carried out to examine the PLA and LAU association with native and mutated tubulin using AMBER16 software. Protonation states of ionisable residues corresponding to pH 6.5 were determined by H++13–15 web interface [25, 26]. Protein structure parameters were described using ff14SB force field. Parameters compatible with the GAFF force field were employed for LAU and PLA residues. Assignation of atom types and calculation of partial charges for each ligand were carried out using the Antechamber implementation of Ambertools15 using the standard AM1 semiempirical method. Protein–ligand systems were solvated in a cubic box of 10 Å length using TIP3P water model. To ensure overall the charge neutrality, Na+ counterions were added. Non-bonded terms were calculated with a 10 Å cutoff, and long-range electrostatics were treated using the simulations were carried out using the Particle-Mesh Ewald. SHAKE algorithm was enabled to constrain all bonds involving hydrogen during simulations. MD protocol consisted of: (a) 1000 steepest descent minimization steps followed by 1000 conjugate gradient minimization steps, (b) 600 ps of progressive NVT heating from 0 to 300 K (e) 300 ns of NPT production dynamics at 300 K and 1 bar from which production data were collected.
Trajectory analysis
MD trajectories were analyzed using VMD software implementations. RMSD calculations were carried out considering protein backbone atoms and ligands heavy atoms. The Timeline plugin of VMD was employed to identify hydrogen bond and salt bridge events during the last 100 ns of MD trajectories in the systems under study considering cutoff distances of 3.0 and 3.2 Å, respectively, and a hydrogen bond angle cutoff of 20°. Hydrogen bond lengths are reported as the average distance between heavy atoms. The bound conformations of the ligands in equilibrated systems were examined through the analysis of the backbone dihedrals of PLA and LAU macrocycles along MD trajectories. Polar histograms corresponding to each dihedral were built using MATLAB R2008b software.
Protein–ligand binding free energy calculations were estimated using MM/GBSA and MM/PBSA approaches following a single trajectory protocol. Binding free energies were estimated from 2000 snapshots retrieved from the last 100 ns of each MD production using the mm_pbsa.pl module in Amber16. GB calculations were carried out using the modified GB model (igb = 5) with α, β, and γ values of 1.0, 0.8, and 4.85, respectively. Dielectric constants for the solvent and the protein were set to 80 and 5, respectively. These values considered appropriate to describe the interaction between PLA and LAU with a highly solvent-exposed binding site, as previously discussed in the literature. Preliminary work was done aimed at evaluating the performance of different GB models (igb = 1, 2) showing that igb = 5 was the best choice for the systems under study.
The Multiwfn software was used to examine the non-covalent interactions in PLA and LAU complexes with native and mutated αβ-tubulin using the last 500 frames of the corresponding MD trajectories. To reduce the computational cost the average promolecular densities from atoms of protein residues within a 5 Å cutoff radius of the ligands were used to compute the non-covalent interaction indices under the approach of fluctuating systems (aNCI). The cube grid was set to 250 × 250 × 250 Å, with a 0.1 Å step size along x, y, and z coordinates. The visual representations of averaged NCI were obtained using VMD software.
Results and discussion
MD simulations on PLA and LAU complexes with native and mutated αβ-tubulin were carried out to provide molecular-level insight into the role of the Gln293Met mutation on determining the activity of these ligands as microtubule-stabilizing agents. Initial coordinates for MD simulations were retrieved from the high-resolution crystallographic models 4O4J and 4O4H for PLA– and LAU–tubulin complexes, respectively. The use of high-resolution crystallographic data is relevant to obtain reliable information from MD simulations, considering that these models detail the specific features of PLA and LAU association and the unique allosteric effects observed on the protein upon binding. Additionally, the 4O4J and 4O4H models are representative for the interaction of PLA and LAU with assembled microtubules, which activates microtubule stabilization, since it has been demonstrated that the overall conformation of the protein and the PLA/LAU binding pocket are not affected by the curved-to-straight transition that occurs when αβ-tubulin is organized in microtubules [9]. Thus, our MD simulations are expected to provide valuable structural, dynamical and energetic information to explain the activity trends observed for PLA and LAU in native and Gln293Met mutated systems.
Systems equilibration along 300 ns simulation runs was monitored by checking the protein, ligand, M-loop, and binding site root mean square deviation (RMSD). Looking at Fig. S1 it seems this happens after 150 ns of NPT simulation. Consequently, the structure and energetic properties of PLA and LAU complexes with wild type and mutated αβ-tubulin were analyzed during last 100 ns of production trajectories.
To assess the quality of our simulations results, the final MD structures of PLA and LAU complexes with wild type αβ-tubulin were compared to the corresponding crystallographic models 4O4J and 4O4H, showing a high level of structural agreement (Fig. 2). MD simulations reproduced the most relevant drug–tubulin interactions found in the crystals and evidenced that PLA/LAU complexation occurs through the penetration of the side chains and parts of the macrocycles in the binding pocket. Details about the intermolecular interactions observed in PLA and LAU complexes with native αβ-tubulin are displayed in Fig. S2. In the case of the PLA-tubulin association, MD simulations revealed the formation of two hydrogen bonds involving the PLA side chain hydroxyl group, the side chain of Tyr312, and the C=O of Phe296, with average lengths of 2.7 Å and occupancies of 100%. Other hydrogen bonds involving the macrocycle of PLA were observed during MD runs, namely the interaction between: (a) the C1 carbonyl and the amide NH group of Ser298 (2.8 Å, 90% of occupancy); (b) the C2 hydroxyl group and Asp297 side chain (2.6 Å, 99% of occupancy); and (c) the C3 methoxyl group and Arg308 side chain (2.6 Å, 90% occupancy). Finally, a low occupancy (20%) hydrogen bond was observed between the C9 hydroxyl group of PLA and the side chain of Gln293 residue with an average length of 2.9 Å. Regarding van der Waals contacts, the ethyl group of PLA side chain was found to be immersed in a hydrophobic pocket formed by Phe296, Tyr312, Tyr342, and Phe343 residues. In the case of the LAU–tubulin complex, MD simulations reproduced the hydrogen bonds observed in the crystal between the side chain hydroxyl group of LAU and the side chains of residues Ser298 (3.0 Å, 64% of occupancy) and Asp297 (2.8 Å, 60% of occupancy). The water-mediated hydrogen bonds formed between the oxygen atom of the side chain’s dihydropyranyl ring and Phe296 and Tyr312 residues were observed as well with occupancies of 98 and 90%, respectively. Regarding the macrocycle of LAU, MD simulations reproduced the hydrogen bond found in the crystal between the dihydropyranyl ring of the macrocycle and the side chain of Asn339, with an average length of 2.9 Å and 90% of occupancy. Finally, a van der Waals contact was observed between the dihydropyranosyl ring of the macrocycle with the side chain of Gln293 and a second hydrophobic contact was observed between the side chain’s ring of LAU and Phe343 residue.

Structure of LAU and PLA complexes with native αβ-tubulin obtained from the last 50 ns of MD trajectories compared to the corresponding 4O4J and 4O4H crystallographic models
Besides drug–tubulin interactions, MD simulations revealed the formation of an intra-protein salt bridge in the binding site region of PLA and LAU complexes involving the side chains of residues Asp306–Arg308 (2.8 Å). In the case of PLA complex, a second salt bridge was observed in the binding region between residues Asp297–Lys299 (2.7 Å) (Fig. S3). A relevant feature of LAU and PLA association with αβ-tubulin is the structuring of the M-loop region, which is a critical molecular process to establish effective lateral contacts across protofilaments in microtubules. M-loop structuring is triggered by the stabilization of the Gln294–Phe296 segment in helical conformation in the ligand–bound state of β-tubulin, which is referred in the literature as the “polymerization-competent” state [9]. According to MD simulations, the Gln294–Phe296 region retained its helical conformation in 100% of the analyzed trajectories and the M-loop remained stable in a “polymerization-competent” conformation in both LAU– and PLA–tubulin complexes. This stabilization was assisted by two electrostatic intra-protein interactions involving the side chains of residues Asp226–Arg278, and Glu290–Arg284 with average distances of 2.8 Å in both LAU and PLA systems (Fig. 2).
Altogether, the aforementioned results show high consistency with crystallographic information, and support the validity of our current computational approach to proceed further with the study of PLA and LAU association with Gln293Met mutated αβ-tubulin, as detailed next.
Effect of Gln293Met mutation on the structure of PLA and LAU–tubulin complexes
To examine the role of Gln293Met mutation on the structure of LAU and PLA complexes with αβ-tubulin, the last 100 ns of MD runs were analyzed in the search for alterations in the drug–tubulin interactions found in the wild-type complexes. Figure 3 shows a comparison between PLA and LAU complexes with native αβ-tubulin and its Gln293Met mutant. According to MD results, both ligands remained associated in the same binding region without significant displacements o reorientations after Gln293Met mutation. Additionally, PLA and LAU retained their bound conformations almost unaltered in the mutant systems, as revealed from the analysis of PLA and LAU backbone dihedrals during the last 100 ns of MD trajectories (Fig. S4).

Structure of PLA and LAU complexes with the Gln293Met αβ-tubulin mutant obtained from the last 50 ns of MD trajectories. The bound structures of PLA (green) and LAU (orange) in wild-type complexes are included as reference. The reorientation of the Met295 residue in the LAU–tubulin complex after Gln293Met mutation is highlighted
Nevertheless, Gln293Met mutation exerted different effects in the drug–tubulin interaction networks observed in each system. In the case of the PLA–tubulin complex, the side chain of the Met293 residue gained flexibility compared to the side chain of Gln293 in the native system and was reoriented towards engaging in van der Waals contacts with the C8–C11 region of PLA (Fig. S5). After Gln293Met mutation the hydrogen bond formed between the side chain of Gln293 and the C9-hydroxyl group of the ligand in the native system was lost. Other PLA–tubulin interactions remain unaltered after Gln293Met mutation, namely (a) the two hydrogen bonds involving the PLA side chain hydroxyl group, the side chain of Tyr312, and the C=O of Phe296; (b) the hydrogen bond between the C1 carbonyl of PLA and the amide NH group of Ser298; (c) the hydrogen bond between the C2 hydroxyl group of the ligand and Asp297 side chain; and (d) the C3 methoxyl group of the ligand and Arg308 side chain (Fig. S6). In the case of the LAU–tubulin system, the Met293 side chain in mutant remained located in the same orientation as the Gln293 side chain in the native complex, thus engaging in in van der Waals contacts with C11-methylene group of LAU (Fig. S5). Compared to the native complex, most LAU–tubulin intermolecular interactions were retained in the mutant, namely (a) the hydrogen bond between the side chain hydroxyl group of LAU and Ser298; (b) the water-mediated hydrogen bonds formed between LAU, Phe296 and Tyr312; and (c) the hydrogen bond between the dihydropyranyl ring of the macrocycle and the side chain of Asn339. On the contrary, the hydrogen bond formed between the side chain hydroxyl group of LAU and the side chain of Asp297 was lost in 70% of the trajectory after Gln293Met mutation (Fig. S6). An additional change was observed in the LAU–tubulin complex after Gln293Met mutation, which is the reorientation of the side chain of the Met295 residue that is located near the mutation site. In the Gln293Met mutant, the side chain of Met295 was reoriented in such a way that the intermolecular contact with the ligand was lost in 77% of the trajectory (Fig. 3). The Met295 residue is part of the Gln294–Phe296 helix that induces structuring of the M-loop to its “polymerization-competent” state, so the loss of the LAU–Met295 interaction might induce a conformational perturbation in M-loop region that could explain the decreased activity of LAU after Gln293Met mutation.
Regarding intra-protein interactions, Gln293Met mutation induced the loss of the salt bridge in the binding site region between Asp306–Arg308 residues in 32% of the trajectory frames retrieved from the LAU–tubulin complex, whereas this interaction was retained in 97% of the structures sampled from the PLA–tubulin complex trajectory (Fig. 3). In the case of the PLA–tubulin complex, Gln293Met mutation induced the partial loss of the salt bridge between Asp297–Lys299 residues in 74% of the MD trajectory of the mutant complex but this loss was compensated by the formation of an additional salt bridge between Asp211–Lys299 residues in the binding site region. An additional effect of Gln293Met mutation was observed in the structure of the M-loop in both PLA and LAU tubulin complexes, in which the salt bridge between Glu290–Arg284 residues was lost in almost 100 and 51% of the corresponding MD trajectories, respectively. On the other hand, the Asp226–Arg278 electrostatic interaction in the M-loop region remained unaltered in both PLA and LAU complexes after Gln293Met mutation. In the case of the PLA–tubulin complex, the loss of the Glu290–Arg284 interaction increased the flexibility of the M-loop region, which might favor the accommodation of this region to establish effective contacts between protofilaments in stable microtubules.
To provide additional information about the role of Gln293Met mutation on the αβ-tubulin structure, supplementary MD simulations were conducted on the ligand-free state of this protein showing that negligible conformational changes were observed in the structure of αβ-tubulin after mutation in the absence of the ligands (Fig. S7). Thus, MD results suggest that Gln293Met exerts its action by altering the interactions that govern the PLA/LAU–tubulin association, inducing noticeable changes in the case of LAU and a less pronounced effect in the case of PLA.
Binding free energy calculations and weak interaction analysis
MM/GBSA and MM/PBSA binding free energy calculations were employed to assess the energetic aspects of PLA and LAU association with native and Gln293Met mutated αβ-tubulin. These methods have been widely employed in the past to deal with ligand–tubulin interactions and have proven to provide reliable information to account for the effect of tubulin mutations on the binding affinity towards diverse tubulin binding agents [14, 27,28,29,30] Under this approach binding free energy values were from Eq. (1)
The corresponding free energy \(G\) for each molecule (i.e. complex, receptor and ligand) was decomposed as follows:
where the E MM is the sum of molecular mechanical gas-phase energies, ΔG solv is the solvation free energy and −TS is the entropy contribution to the free energy. ΔG solv is the sum of polar and non-polar contributions. Polar contributions were estimated using the Poisson–Boltzmann (PB) or Generalized-Born (GB) implicit solvation models, whereas non-polar terms included components due to cavity formation and solute–solvent van der Waals interactions. Regarding to MM/GBSA and MM/PBSA calculations, our work is aimed at calculating relative binding free energy values (\(\Delta \Delta {G_{bind}}\)) for PLA–tubulin and LAU–tubulin complexes by comparing native and mutated systems as shown next:
Relative binding free energy are relevant to examine the specific role of Gln293Met mutation on the association of PLA and LAU to the tubulin receptor instead of absolute binding free energies values. Under this approach, the calculation entropic contributions can be ignored considering that entropy terms corresponding to native and mutated complexes with the same ligand will probably be similar and cancel out in the process of estimating the effect of Gln293Met mutation on PLA– and LAU–tubulin association. Additionally, given that Gln293Met mutation did not induce significant changes in the binding modes and bound conformations of the ligands, it is expected that entropy terms have a negligible contribution in the process of estimating \(\Delta \Delta {G_{bind}}\) values [27, 31].
Results of MM/GBSA and MM/PBSA estimates for the absolute \((\Delta {G_{bind~}})~\) and relative (\(\Delta \Delta {G_{bind}}\)) binding free energies for PLA– and LAU–tubulin complexes with the native and Gln293Met systems are reported in Table 1. According to MM/GBSA and MM/PBSA estimates, a slight increase in affinity was predicted for the PLA–tubulin system after Gln293Met mutation, whereas a weakened interaction was suggested for the LAU complex in the mutant system. These results support the formerly presented hypothesis regarding the modulation of drug–tubulin interactions as the origin of the enhanced activity of PLA and decreased activity of LAU in the Gln293Met mutant.
With the aim of providing an insightful description of the relative contribution of each binding site residue to the association of PLA and LAU to native and mutated tubulin, MM/GBSA and MM/PBSA estimates were decomposed per residue, as detailed in Table S1. The largest \(\Delta \Delta {G_{bind}}\) values per residue are displayed in Fig. 4. In the case of the PLA–tubulin system (orange bars), the Gln293Met mutation increased the affinity of the ligand towards Thr292, Met293, Asp297, Val335, Lys338 and Asn339 residues. On the other hand, weaker interactions are predicted for Ser298, Lys299, Arg308 and Tyr342 residues. In the case of the LAU–tubulin system (blue bars), the Gln293Met mutation was predicted to induce an increased affinity towards Met293, Asp297, Ser298, Arg308 and Tyr342, whereas a reduced interaction was estimated for Thr292, Lys299, Pro307, Val335, Lys338, and Ans339 residues. These results suggest that the Gln293Met site mutation induced specific changes in the interaction networks of both PLA– and LAU–tubulin complexes that are related to the overall stabilization and destabilization of these complexes after mutation.

ΔΔG bind (kcal mol−1) values decomposed per residue for PLA– and LAU–tubulin complexes. ΔΔG bind values were obtained as the difference between binding free energy values in the Gln293Met mutant system and in the wild-type complex
To further examine the contribution of weak intermolecular interactions (i.e. hydrogen bonds, van der Waals interactions, and steric clashes) in the stabilization of PLA/LAU–tubulin complexes, non-covalent interaction (NCI) analysis was carried out during the last 25 ns of MD trajectories. NCI is a valuable tool to study the favorable and unfavorable intermolecular interactions within biological systems with low computational cost [32,33,34]. This method is based on plotting the reduced density gradient (RDG or s(r)) with respect to the electron density ρ(r) for a supramolecular system as follows:
To obtain a more accurate description of weak interactions along the MD trajectories, the average NCI analysis (aNCI) is recommended, based on averaged density gradients \(\overline {\nabla \rho ({\varvec{r}})}\), and averaged densities \(\overline {\rho ({\varvec{r}})}\) [33, 34] retrieved from multiple frames of MD simulations. To visualize aNCI results, strongly attractive hydrogen bonding interactions are customarily displayed in blue, weakly attractive van der Waals interactions are displayed in green, and strongly repulsive steric clashes are colored in red, as shown in Fig. 5. According to aNCI results, van der Waals and hydrogen bonding interactions play a relevant role in the stabilization of both PLA and LAU complexes with native αβ-tubulin. After Gln293Met mutation, the PLA–tubulin complex experienced a decrease in repulsive steric clashes and an enhancement of van der Waals attractive interactions with Met293 and Arg308 residues, as consequence of the replacement of a polar glutamine residue by a non-polar methionine counterpart. On the other hand, the LAU–tubulin complex underwent a pronounced increase in repulsive steric clashes with Met293 and Arg308 residues, which accounted for the weakened ligand–protein interaction predicted from MM/GBSA and MM/PBSA calculations. Thus, the combined results of binding free energy calculations and weak interaction analysis indicated that Gln293Met mutation modulates PLA and LAU activity by altering drug–tubulin interactions and inducing an enhanced affinity towards PLA association compared to the native protein.

aNCI plots for the PLA and LAU association with native and mutated αβ-tubulin. The isosurfaces are colored on a blue–red–green scale according to the values of λH, ρ ranging from −0.05 to +0.05 a.u. Blue indicates strong attractive interactions (hydrogen bonds), green indicates weak interactions (van der Waals), and red indicates strong repulsive interactions (steric clashes)
Conclusions
MD simulations were carried out to examine the association of PLA and LAU with native and mutated αβ-tubulin in the search for structural and energetic evidence to explain the role of the Gln293Met mutation on increasing the activity of PLA and exerting resistance to LAU. MD results revealed that Gln293Met mutation did not induce noticeable changes in the binding modes of PLA and LAU but exerted the loss of significant drug–tubulin interactions in the LAU–tubulin complex. Binding free energy calculations and weak interaction analysis confirmed that Gln293Met mutation reduced the binding strength of the LAU–tubulin complex but induced an increased binding affinity for the PLA–tubulin system. These results suggest that Gln293Met mutation modulates PLA and LAU activity by altering drug–tubulin interactions in consistency with literature information reported from site-directed mutagenesis experiments. Additionally, our results provide valuable atomic-level information to increase understanding about the mechanism of action of PLA and LAU and to assist the design of novel more potent analogues that take advantage of the favorable interactions that were predicted for the systems under study.
References
Dumontet C, Sikic BI (1999) Mechanisms of action of and resistance to antitubulin agents: microtubule dynamics, drug transport, and cell death. J Clin Oncol 17:1061–1070
Dumontet C, Jordan MA (2010) Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 9:790–803
Jordan A, Hadfield JA, Lawrence NJ, McGown AT (1998) Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. Med Res Rev 18:259–296
Hood KA, West LM, Rouwe B, Northcote PT, Berridge MV, Wakefield SJ, Miller JH (2002) Peloruside A, a novel antimitotic agent with paclitaxel-like microtubule-stabilizing activity. Cancer Res 62(12):3356–3360
Mooberry SL, Tien G, Hernandez AH, Plubrukarn A, Davidson BS (1999) Laulimalide and isolaulimalide, new paclitaxel-like microtubule stabilizing agents. Cancer Res 59(3):653–660
Zhao Y, Mu X, Du GH (2016) Microtubule-stabilizing agents: new drug discovery and cancer therapy. Pharmacol Therapeut 162:134–143
Gaitanos TN, Buey RM, Diaz JF, Northcote PT, Teesdale-Spittle P, Andreu JM, Miller JH (2004) Peloruside A does not bind to the taxoid site on beta-tubulin and retains its activity in multidrug-resistant cell lines. Cancer Res 64(15):5063–5067
Pryor DE, O’Brate A, Bilcer G, Diaz JF, Wang YF, Wang Y, Kabaki M, Jung MK, Andreu JM, Ghosh AK, Giannakakou P, Hamel E (2002) The microtubule stabilizing agent Laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochem US 41:9109–9115
Prota AE, Bargsten K, Northcote PT, Marsh M, Altmann KH, Miller JH, Diaz JF, Steinmetz MO (2014) Structural basis of microtubule stabilization by Laulimalide and Peloruside A. Angew Chem Int Edit 53:1621–1625
Bennett MJ, Barakat K, Huzil JT, Tuszynski J, Schriemer DC (2010) Discovery and characterization of the Laulimalide-microtubule binding mode by mass shift perturbation mapping. Chem Biol 17:725–734
Kanakkanthara A, Wilmes A, O’Brate A, Escuin D, Chan A, Gjyrezi A, Crawford J, Rawson P, Kivell B, Northcote PT, Hamel E, Giannakakou P, Miller JH (2011) Peloruside- and Laulimalide-resistant human ovarian carcinoma cells have βI-tubulin mutations and altered expression of βII- and βIII-tubulin isotypes. Mol Cancer Ther 10:1419–1429
Begaye A, Trostel S, Zhao Z, Taylor RE, Schriemer DC, Sackett DL (2011) Mutations in the β-tubulin binding site for Peloruside A confer resistance by targeting a cleft significant in side chain binding. Cell Cycle 10:3387–3396
Kanakkanthara A, Rowe MR, Field JJ, Northcote PT, Teesdale-Spittle PH, Miller JH (2015) Beta I-tubulin mutations in the Laulimalide/Peloruside binding site mediate drug sensitivity by altering drug-tubulin interactions and microtubule stability. Cancer Lett 365:251–260
Hassanzadeh M, Bagherzadeh K, Amanlou M (2016) A comparative study based on docking and molecular dynamics simulations over HDAC-tubulin dual inhibitors. J Mol Graph Model 70:170–180
Kumbhar BV, Borogaon A, Panda D, Kunwar A (2016) Exploring the origin of differential binding affinities of human tubulin isotypes alpha beta II, alpha beta III and alpha beta IV for DAMA-colchicine using homology modelling, molecular docking and molecular dynamics simulations. PLoS ONE. doi:10.1371/journal.pone.0156048
Costa KM, Alves CN, Silva JRA, Lameira J (2016) A Computational analysis of indomethacin derivative as tubulin inhibitor: insights into development of chemotherapeutic agents. Comb Chem High Throughput Screen J 19:431–436
Churchill CDM, Klobukowski M, Tuszynski JA (2015) Elucidating the mechanism of action of the clinically approved taxanes: a comprehensive comparison of local and allosteric effects. Chem Biol Drug Des 86:1253–1266
Li DD, Qin YJ, Zhang X, Yin Y, Zhu HL, Zhao LG (2015) Combined molecular docking, 3D-QSAR, and pharmacophore model: design of novel tubulin polymerization inhibitors by binding to colchicine-binding site. Chem Biol Drug Des 86:731–745
Navarrete KR, Alderete JB, Jimenez VA (2015) Structural basis for drug resistance conferred by beta-tubulin mutations: a molecular modeling study on native and mutated tubulin complexes with epothilone B. J Biomol Struct Dyn 33:2530–2540
Churchill CDM, Klobukowski M, Tuszynski JA (2015) The unique binding mode of Laulimalide to two tubulin protofilaments. Chem Biol Drug Des 86:190–199
Churchill CDM, Klobukowski M, Tuszynski JA (2016) Analysis of the binding mode of Laulimalide to microtubules: establishing a Laulimalide-tubulin pharmacophore. J Biomol Struct Dyn 34:1455–1469
Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258
Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T (2009) The SWISS-MODEL repository and associated resources. Nucleic Acids Res 37:D387–D392
Trott O, Olson AJ (2010) Software news and update autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461
Anandakrishnan R, Aguilar B, Onufriev AV (2012) H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res 40:W537–W541
Gordon JC, Myers JB, Folta T, Shoja V, Heath LS, Onufriev A (2005) H++: a server for estimating pK(a)s and adding missing hydrogens to macromolecules. Nucleic Acids Res 33:W368–W371
Gilson MK, Zhou HX (2007). Calculation of protein-ligand binding affinities. Annu Rev Biophys Biomol 36:21–42
Kumbhar BV, Borogaon A, Panda D, Kunwar A (2016) Exploring the origin of differential binding affinities of human tubulin isotypes alpha beta II, alpha beta III and alpha beta IV for DAMA-colchicine using homology modelling, molecular docking and molecular dynamics simulations. PLoS ONE 11(5):22
Tripathi S, Kumar A, Kumar BS, Negi AS, Sharma A (2016) Structural investigations into the binding mode of novel neolignans Cmp10 and Cmp19 microtubule stabilizers by in silico molecular docking, molecular dynamics, and binding free energy calculations. J Biomol Struct Dyn 34(6):1232–1240
Tripathi S, Srivastava G, Sharma A (2016) Molecular dynamics simulation and free energy landscape methods in probing L215H, L217R and L225M βI-tubulin mutations causing paclitaxel resistance in cancer cells. Biochem Biophys Res Commun 476(4):273–279
Mobley DL, Dill KA (2009) Binding of small-molecule ligands to proteins: “what you see” is not always “what you get”. Structure 17(4):489–498
Bai Q, Yao X (2016) Investigation of allosteric modulation mechanism of metabotropic glutamate receptor 1 by molecular dynamics simulations, free energy and weak interaction analysis. Sci. Rep. UK 6:21763
Bian C, Wang SJ, Liu YH, Se KH, Jing XL (2016) Role of nonbond interactions in the glass transition of novolac-type phenolic resin: a molecular dynamics study. Ind Eng Chem Res 55:9440–9451
Wu P, Chaudret R, Hu XQ, Yang WT (2013) Noncovalent interaction analysis in fluctuating environments. J Chem Theory Comput 9:2226–2234
Acknowledgements
The authors thank FONDECYT Grant No. 1160060.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zúñiga, M.A., Alderete, J.B., Jaña, G.A. et al. Structural insight into the role of Gln293Met mutation on the Peloruside A/Laulimalide association with αβ-tubulin from molecular dynamics simulations, binding free energy calculations and weak interactions analysis. J Comput Aided Mol Des 31, 643–652 (2017). https://doi.org/10.1007/s10822-017-0029-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-017-0029-2