
Abstract
Purpose
The goal of this study is to determine whether any balanced translocation (BT) had been missed by previous karyotyping in patients with unexplained recurrent pregnancy loss (uRPL).
Methods
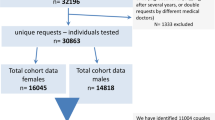
This case series included 48 uRPL-affected couples with normal karyotypes. The embryos from these couples have all undergone preimplantation testing for aneuploidies (PGT-A). Based on the PGT-A’s results, 48 couples could be categorized into two groups: 17 couples whose multiple embryos were detected with similar structural variations (SVs, segmental/complete) and 31 couples without such findings but who did not develop any euploid embryo despite at least three high-quality blastocysts being tested. The peripheral blood sample of each partner was then collected for mate-pair sequencing (MPseq) to determine whether any of them were BT carriers.
Results
MPseq analyses identified 13 BTs in the 17 couples whose multiple embryos had similar SVs detected (13/17, 76.47%) and three BTs in the 31 couples without euploid embryo obtained (3/31, 9.7%). Among the 16 MPseq-identified BTs, six were missed due to the limited resolution of G-banding karyotyping analysis, and the rest were mostly owing to the similar banding patterns and/or comparable sizes shared by the two segments exchanged.
Conclusion
A normal karyotype does not eliminate the possibility of carrying BT for couples with uRPL. The use of PGT-A allows us to perceive the “carrier couples” missed by karyotyping analysis, providing an increased risk of finding cryptic BTs if similar SVs are always detected on two chromosomes among multiple embryos. Nonetheless, certain carriers with translocated segments of sub-resolution may still go unnoticed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Balanced translocation (BT) is defined as the exchange of two segments originating from two or more non-homologous chromosomes [1]. The prevalence is about 1.6 to 2 per 1000 newborns [2] but significantly increased to 3 ~ 7% in couples with recurrent pregnancy loss (RPL) [3]. BT carriers are prone to experience adverse pregnancies and spontaneous miscarriages due to the formation of quadrivalents and the resultant segregation modes that may produce unbalanced gametes. It has been reported that the proportion of normal haploid sperms and oocytes produced by BT carriers is 45% [4] and 30% [5], respectively, suggesting that more than half of the gametes are abnormal. Therefore, knowing the carrier status thus enables the otherwise unwary couple to be informed of such risk and of the various reproductive options available to them.
Parental karyotyping analysis remains the first-tier clinical strategy for screening BT carriers [3]. However, its maximum resolution can only reach about 4 Mb (under ideal conditions) and it still has limited capability in detecting submicroscopic structural variations (SVs) [1]. As for the alternatives, fluorescence in situ hybridization (FISH) requires prior knowledge of target regions of chromosomes for probe design, whereas chromosomal microarray analysis (CMA) does not possess the ability to detect the balanced changes. Next-generation sequencing (NGS) has been recently used for the detection of balanced translocations and inversions; nevertheless, the mainstream of current NGS relies on the construction of small-insert DNA libraries (200 ~ 500 bp), which limits the detection capability of BTs, especially in those cases mediated by repetitive elements [6]. Despite the longer read-length (even up to 250 kb [7]) being a natural advantage for BT detection, third-generation sequencing technologies are not particularly well-suited for the purpose of carrier screening due to the cost factors. In comparison, mate-pair sequencing (MPseq), an optimized protocol of NGS, provides improved sensitivity in detecting balanced SVs by utilizing longer DNA fragments (from ~ 1 to 10 kb). Multiple laboratories have demonstrated its capability in detecting BTs with favorable results reported [8, 9]. Based on our previous work [10], MPseq provided a higher detection yield of balanced translocations and inversions (up to 11.7%) than the routine G-banded karyotyping analysis. However, whether it could be served as the first-tier test for RPL-affected populations (Table 1; [11,12,13,14,15,16,17]), remains inconclusive.
Cryptic balanced translocations [1] refer to those that are easily missed or not readily identified by conventional cytogenetic analysis, usually because of (i) (sub)telomere translocations, the segment(s) translocated is/are with a small size, near to or below the resolution limit (e.g., only a G-light band is visible in the translocated segment(s), thus rendering it difficult to recognize or being mistakenly reported as chromosomal polymorphism); (ii) two segments exchanged are characterized by comparable sizes and similar banding patterns; and (iii) complex chromosomal rearrangements, which involve three or more chromosomes or having more than two breakpoints. Once cryptic BT carriers with fertility intentions were missed, their RPLs are typically considered “unexplained” (uRPL). When the individuals in this subpopulation seek medical assistance, a potential strategy to prevent miscarriages might involve the use of in vitro fertilization (IVF) combined with preimplantation genetic testing (PGT) to rule out the embryos with aneuploidies (PGT-A). Their embryos, like those from carriers with apparently BTs, are expected to present with “unbalanced translocation-like” characteristics (similar SVs are always detected on two chromosomes among multiple embryos) from PGT-A analysis [18, 19]. In this regard, the “unbalanced translocation-like” embryos might be an indicator for further follow-up for these individuals. Additionally, the confirmation of carrier status in these patients might also be helpful to subject the embryos to PGT for structural rearrangements (PGT-SR) in their future IVF cycles. While it is noteworthy that the segments of cryptic BTs are often with small size and different clinics/laboratories adopt a wide range of resolutions for PGT-A (ranging from 1 to 20 Mb), as well as the fact that there must be an adequate number of embryos to be aware of such characteristic, carriers of cryptic BT may still exist in couples in whom such embryonic characters were not observed.
In this study, 48 couples with normal karyotypes who underwent PGT-A for uRPL were included for MPseq.
Patient profile
This case series consisted of 48 couples who were diagnosed with RPL (defined as the occurrence of three or more clinical pregnancy losses) (Table 2). They were gradually found and noticed by us during the clinical consultation process from November 2017 to December 2020 owing to their shared dilemma of facing the tough decision-making process of selecting the most suitable embryo for transfer with PGT-A results available.
Previous RPL workup
As recommended by the American Society for Reproductive Medicine (ASRM) [3], we firstly administered an RPL workup for them.
G-banded karyotyping analysis
Cell culture, smear preparation, and Giemsa staining were performed according to standard procedures [20]. At least five cells from two separate cultures were analyzed, and at least two images from each karyotype were saved. The initial reviews were completed independently by two qualified cytogeneticists and confirmed by the laboratory chief. Karyotypes were described following the ISCN guideline (2016) [21]. Results were reported at a resolution of 350 ~ 550 bands [22].
Other tests in RPL workup
The other tests contained in the RPL workup included the assessments of female sex hormones, prolactin, and thyroid-related hormones (via chemiluminescence assay, Roche-Diagnostics, Basel, Switzerland), alongside HbA1c (via liquid chromatography, Bio-Rad, Hercules, CA, USA), thrombophilia markers, and antiphospholipid antibodies (via coagulation tests, chromogenic assays, and turbidimetry, Diagnostica Stago, Asnières sur Seine, France). The endometrial conditions (e.g., endometritis) were determined based on hysteroscopy and immunohistochemical analyses of specimens obtained from endometrial biopsy and curettage.
After the completion of the above examinations, the couples had the following features in common: (i) maternal age was under 38 years old; (ii) received negative findings from the G-banded karyotype analysis; (iii) unexplained causes of RPL despite a comprehensive workup as proposed by the ASRM. In light of the uRPL being a common indication for PGT, the PGT-A procedures were subsequently added into their IVF cycles.
NGS-based PGT-A and clinical findings prior to the enrollment
NGS-based PGT-A
The protocols of controlled ovarian hyperstimulation (COH) and oocyte retrieval had been described in our previous study [23]. After being fertilized via intracytoplasmic sperm injection (ICSI), zygotes were cultured in vitro to the blastocyst stage (5 ~ 6 days). Several cells of trophectoderm (TE) were harvested from the blastocysts and the rest of the embryonic components were cryopreserved until a decision was made based on PGT-A results. All couples in this study had at least three high-quality (> 4BC, Gardner’s standard [24]) blastocysts being tested. Their TE samples were washed three times in polyvidone and transferred to the EP tubes containing 2.5 µL phosphate buffer solution for subsequent DNA extraction and single-cell whole genome amplification (WGA, SurePlex DNA Amplification System, Illumina, San Diego, CA, USA). Sequencing proceeded on Illumina Miseq platform (VeriSeq PGS Kit, MiSeq Reagent Kit, Illumina, San Diego, CA, USA). For all full or partial chromosomal regions detected, the difference value (DV) parameter was generated by calculating “control normalized mean coverage for one copy” multiplied by “expected ploidy,” plus “sample normalized mean coverage.” The embryos with DVs below 20% and above 80% were defined as euploidy and aneuploidy, and those with DVs in-between were classified as mosaic embryos. Deletions and duplications of chromosomal segments larger than 4 Mb were reported.
Clinical findings
Upon receiving PGT-A results, the embryos of a total of 17 couples were observed with abovementioned “unbalanced translocation-like” characteristics (hereafter referred to as “category-1”). The results for all embryos from the 17 couples are listed in Supplementary Table 1. As cryptic BTs often involve small translocated segments (< 5 Mb), we proposed MPseq prior to their embryo transfer to determine whether they were BT carriers and to avoid the risks of cryptic imbalances in their presumely balanced or euploid embryos at the current PGT-A’s resolution of 4 Mb.
Despite the embryos of the remaining 31 couples (hereafter referred to as “category-2”) did not exhibit apparent “unbalanced translocation-like” characteristics, none of them developed into euploid embryos. As the female partners in these couples were with young age and had obtained at least three high-quality blastocysts being tested, we found it is difficult to give them a medically grounded explanation. Due to the fact that these couples can only select mosaic embryos for transfer in addition to initiating a new COH cycle, we also recommended MPseq for them for seeking any additional information to potentially guide their next steps.
Diagnostic interventions
The study was approved by the Ethics Committee of the Reproductive Medicine Center of Shandong University ([2017] IRB No. 121), and all couples provided written informed consent before the peripheral blood collection.
Mate-pair sequencing
Genomic DNA (gDNA) from each partner was extracted with the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) and quantified with the Qubit dsDNA HS Assay kit (Invitrogen, Carlsbad, CA, USA), and the DNA integrity was evaluated by agarose gel electrophoresis. After QC, 1 µg DNA (measured by Qubit) from each sample was sheared with an E220e Focused-Ultrasonicator with miniTUBE Red (Covaris, Woburn, MA, USA) into 3 ~ 8 kb (median size of 5 kb) fragments and subjected to mate-pair library construction according to our previous study [25]. At least 60 million read-pairs were generated for each sample, equivalent to an average of 4 × read-depth on the MGISEQ-2000 platform (MGI Tech Co., Ltd., Shenzhen, China). Thereafter, copy number variations (CNVs at 50-kb), SVs (10-kb), and regions with absence of heterozygosity (AOHs, 5-Mb) were identified through our reported bioinformatics pipelines [26], and they were further annotated and interpreted based on the guidelines of the American College of Medical Genetics and Genomics (ACMG).
Verification of the translocations
Breakpoint junction-specific PCR with Sanger sequencing was used to verify the translocations detected by MPseq (all primer sequences designed for this study are listed in Supplementary Table 2), while FISH was employed if there were commercial probes (the Vysis locus-specific identifier DNA probe, Abbott Molecular Inc., Des Plaines, IL, USA) available.
Characterization of translocations detected in two categories
In total, MPseq identified 16 BTs in 16 couples, all of which were validated by breakpoint-junction specific PCR with Sanger sequencing and/or FISH. Thirteen BTs were revealed in 13 couples from category-1 (13/17, 76.47%), and three BTs in three couples from category-2 (3/31, 9.68%) (Table 3).
In category-2, the translocations identified in 20SD0026 and 20SD0082 both involved a cryptic segment that was smaller than 4Mb the PGT-A’s resolution, while both segments of the translocation identified in 20SD0002 were smaller than 4 Mb. It might explain why the previous PGT-A did not indicate any apparent “unbalanced translocation-like” characteristics from the embryos (Fig. 1). Previous PGT-A results for the three couples were provided in Supplementary Table 3.

Two cryptic BTs identified by MPseq. A to C show the detection result in sample 20SD0080 (from category-1), while D to F indicate the result in sample 20SD0002 (from category-2). A, D Previous G-banded chromosomes; B, E ideograms of normal and derivative chromosomes; C, F translocated segments indicated in UCSC browser. Red line indicates the breakpoint junction, while 2-bp microhomology is highlighted in yellow. In figure F, white arrows indicate the fluorescent signals in normal and derivative chromosomes. BT, balanced translocation; MPseq, mate-pair sequencing; FISH, fluorescence in situ hybridization
In category-1, the translocations identified in samples 20SD0016, 20SD0040, and 20SD0093 were missed mainly because one or both segments were with a size near to or smaller than the detection limit of karyotyping analysis (6 ~ 9 Mb). The translocations found in samples 20SD0001, 20SD0004, 20SD0007, 20SD0030, and 20SD0080 were missed possibly because the two segments shared a similar banding pattern and/or comparable size. In the case of samples 20SD0023, 20SD0024, 20SD0029, and 20SD0091, only a barely visible G-dark band could be observed in one of the translocated segments because of unsatisfactory terminal staining, whereas the other segment contained only a G-light band, which may elucidate why the previous karyotyping analysts failed to report them. The last translocation in sample 20SD0076 was likely missed due to the limited banding level and poor staining quality in the chromosome preparation. No complex chromosomal rearrangement was found in this study. Of note, the involvement of bands 4p16, 8q24.2, and 13q33 were reported twice in different samples, and the segments involving these bands were with a size almost near to the limit of detection, or located adjacent to the telomeric regions (Fig. 1).
Recognition of chromosome regions with AOHs
Based on our newly developed bioinformatic algorithm [26], MPseq also revealed the regions with AOHs that involved known imprinted genes and exceeded 5 Mb in size (Supplementary Table 4). There were multiple AOH regions found in four samples, while the overall sizes observed in three samples (20SD0001, 20SD0029, and 20SD0077) did not meet the reported cut-off for parental consanguinity, typically around ~ 89 Mb. In comparison, a total of eight AOH regions were identified in sample 20SD0042 with a combined size of 154.4 Mb, suggesting that the parents of this male patient probably married consanguineously. Given that the presence of AOHs potentially contributes to miscarriages, further mechanism studies are warranted.
Follow-up of pregnancy outcomes
We also conducted follow-up studies on the 16 couples with cryptic BTs identified (Cat.1 = 13; Cat.2 = 3).
As indicated by MPseq, the existing PGT-A platform’s resolution was feasible for the segments carried by the 13 couples in category-1, whose earlier PGT-A results could be adopted directly. Ten of them were with euploid embryos transferred. As current resolution (4 Mb) was insufficient, three couples in category-2 chose to initiate their second cycle but pursued PGT-SR. Based on the exact locations and flanking sequence contexts of knowing the breakpoint junctions from MPseq, their embryos with aneuploidies, balanced karyotypes, and normal karyotypes were distinguished as previously described [27]. Results showed that only the couple 20SD0026&27 had euploid embryo (balanced) obtained and transferred.
Among the 11 couples who underwent embryo transfers, a total of 11 clinical pregnancies (11/11, 100%) were achieved with an outcome of two miscarriages (2/11, 18.18%) and nine live births (9/11, 81.82%). The live birth rate (LBR) of “carrier couples” in this study was higher (81.82%) than the average LBR reported (59.28% [28]) for euploidy transfers in younger women.
Timeline
A timeline for the entire diagnostic and treatment process of these couples is depicted in Supplementary Fig. 1.
Review of literature on (sub)telomeric/cryptic rearrangements associated with RPL
Based on the results retrieved, the publications regarding cases with (sub)telomeric/cryptic rearrangements were more commonly found in diseases like intellectual disabilities (ID) [29] or congenital anomalies [30, 31], etc., while the studies focusing solely on RPL were relatively limited. Most of these studies are the reports that consist of a few to several dozen cases, which were predominantly published around the early 2000s, a time when FISH technique was clinically mature and widely promoted. This might be due to the fact that the individuals studied in the former category were typically genetically unbalanced and phenotypically noticeable, leading to a well-defined molecular diagnosis. In comparison, couples affected by RPL tend to be genetically balanced, with no evident phenotypic manifestations other than fertility problems, thus resulting in the difficulty of diagnosis (Table 4; Supplementary Fig. 2).
In these studies, parental carriers of cryptic BT were identified by tracing their abnormal fetuses or offspring with birth defects. In a study led by Shaffer et al. [33], a couple with a history of RPL and loss of an infant because multiple abnormalities underwent FISH testing; thus, the father was identified as a carrier of cryptic BT involving chromosomes 7 and 11. Brackley et al. [35] reported a case where the father was detected to carry a cryptic BT between the long arms of chromosomes 2 and 7 in relation to multiple fetal anomalies observed in his wife’s pregnancies. Similarly, five cryptic BT carriers were found in five families with a history of RPL and/or offspring with unbalanced karyotypes by Wakui et al. [34]. Bacino et al. [36] reported on a 4 ½-year-old girl with severe mental retardation and minor anomalies, who inherited the unbalanced product of a cryptic translocation involving chromosomes 2 and 17 from her father. According to a study from Kilby et al. [37], the prenatal FISH test of the chorionic villous sampling cultures showed the presence of an unbalanced karyotype, 46, XX.ish der(7) t(2;7)(q37;q36)pat derived from the balanced subtelomeric translocation in the father. Joyce et al. [41] described two families, whose offspring were affected by Miller-Dieker syndrome (MDS). The molecular analysis of two mothers revealed cryptic BTs between the subtelomeres of 11p and 17p. Bruyere et al. [43] reported a young woman who had experienced three spontaneous miscarriages and two neonatal deaths. The woman was demonstrated as the carrier of cryptic BT between the long arms of chromosomes 2 and 17. In a study by Alkuray et al. [45], a 36-year-old Greek woman with a history of three early miscarriages gave birth to a male infant with multiple anomalies in her last pregnancy. She was diagnosed with a cryptic BT between 10q26.3 and 17q25. In another study by Hajlaoui et al. [49], FISH was employed for 21 clinically normal couples exhibiting a “normal” karyotype with at least two abortions and detected one cryptic rearrangement between chromosomes 3q and 4p in the female partner of a couple.
There are several other studies that investigated couples only affected by RPL. Yakut et al. [40] performed FISH analysis on five families who had suffered at least five prior miscarriages. One couple was found to carry a cryptic BT between chromosomes 3 and 10, while in another couple, a signal of chromosome 20 was observed in the D-group chromosomes. Cockwell et al. [42] conducted a re-evaluation (FISH) of 50 couples with normal karyotypes who experienced three or more miscarriages. One female patient was identified to carry a subtelomeric BT, and five patients (one female and four males) were found to have cryptic abnormalities involving the pericentromeric regions of acrocentric chromosome. In a study by Monfort et al. [46], 18 couples with normal karyotypes who had suffered four or more spontaneous miscarriages were subjected to FISH analysis. A cryptic BT between 2 and 3p was detected in a woman with a history of seven miscarriages. Cryptic BTs were not reported in these studies [32, 38, 39, 44, 47, 48].
With the introduction of second- [10] and third-generation sequencing [50] since the 2010s, an increasing number of studies are taking advantage of these new technologies for the validation of cryptic BTs.
Discussion
Our findings demonstrated the significance of PGT-A for the subpopulation of couples with undetermined causes of RPL due to the cryptic BT in one of the partners. Once “carrier couples” were missed in the initial screening round of parental karyotyping, the use of PGT-A can be taken as an opportunity to re-identify them.
BT is presently one of the few well-established etiologies that is strongly associated with RPL; however, some of them could not be readily diagnosed by the gold-standard test (karyotyping analysis) [51]. PGT-SR has been demonstrated to be a management option for effectively reducing miscarriage rates in carrier populations [52]. Given the limitations of chromosome banding techniques, observation of embryonic patterns may provide reassurance against the possibility of missing cryptic BT carriers if the couples decide to pursue pregnancy through IVF and PGT. According to a previous study, Sundheimer et al. [18] identified BTs in eight couples with exactly the same mutations as indicated by their “unbalanced translocation-like” embryos, while most of the subjects they included were not subjected to parental karyotyping prior to IVF, which limits our knowledge of the spectrum of the cryptic BTs missed by G-banded chromosome analysis.
Despite the embryonic patterns of PGT-A could be regarded as an indicator of parental carrier status, there were still three cryptic BT carriers identified in category-2. Two were with one translocated segment cryptic to the PGT-A’s resolution, while the other one was with both segments that were smaller than the cutoff. The inability to obtain transferable euploid embryo despite multiple high-quality blastocysts being tested may also present a potential indication for finding cryptic BTs. In a recent study [50], a total of 11 cryptic BTs were uncovered, with seven out of the 22 translocated segments being smaller than the routine resolution of PGT-A (5 Mb). Although a small size of the unbalanced segment may less likely to result in a lethal consequence (embryonic demise), adverse birth outcomes are often reported. A segmental aneuploidy of larger than 2% of haploid autosomal length (HAL) is highly lethal and often leads to intrauterine death, while an unbalanced segment less than 1% of the HAL may not be lethal but could increase the risks of birth defects [53]. A substantial amount of evidence has been demonstrated that cryptic imbalances/rearrangements are responsible for multiple congenital anomalies, such as mental retardation [31, 54], leukemia [55], autism spectrum disorder [56], and fetal ultrasound abnormalities [57]. In these studies, karyotyping analysis had been performed on a large proportion of subjects in advance, with most of them having “normal” karyotypes. The resolution of PGT-A in our study was enhanced to 4 Mb, which is higher than that adopted in other studies (e.g., 5 ~ 10 Mb [58]), while there is still a fraction of translocated segments measured between 1 and 5 Mb [59]. If one (out of four) segment exchanged is with a size below the detection range or there were limited embryos obtainable for testing, the embryonic patterns unique to BT carriers may also not be indicated by PGT-A, thus resulting in the adverse outcomes listed above.
Before the advent of NGS and third-generation sequencing technologies, the method most used for BT detection is FISH. We selected MPseq to identify cryptic BTs in this study since there was no prior knowledge of target chromosomes for probe selection in category-2 (PGT-A’s results did not point to the derivative chromosomes). Furthermore, as a test that can only pinpoint a few loci, FISH confers a risk of missing BT again if PGT-A-indicated chromosomes do not match the chromosomes that translocation truly involved. Additionally, as cryptic BTs often involve small translocated segments (< 5 Mb, almost 1/3 in our study), the genomic contexts and structure of BT obtained from MPseq could also be served as the basis for subsequent PGT-SR to discern embryos with small-segment rearrangements. Third-generation sequencing technologies [60, 61] can accomplish the same goal, but the related testing costs are considerably higher.
Limitations remain. In this study, a relatively higher detection rate of cryptic BTs (13/17 in category-1 and 3/31 in category-2) was observed in uRPL patients. However, it should be noted that this observation was based on a small sample of susceptible individuals and may not accurately represent the true prevalence of cryptic BTs in the whole population with uRPL. Given the fact that nearly all current research on cryptic rearrangements are in the form of the case report. We expect our study to contribute to future systematic reviews/meta-analyses in this field, thus allowing for an accumulated number of cryptic BT cases. In fairness, MPseq analysis is not perfect either. Specifically, it has limited capability of detecting Robertsonian translocations and the recurrent t(11;22)(q23;q11.2) [62] due to its unsatisfactory performance in the extensive length of repetitive sequences, which has been known to be the limitation of currently available molecular technologies [63]. In some cases (e.g., with similar banding patterns), the identification of cryptic BT could also be achieved by the use of C-banding, silver nitrate (AgNOR) staining, or by improving the resolution of the initial method. In clinical settings, the product of conception (POC) of some couples with uRPL could be preserved sometimes. “Unbalanced translocation-like” POCs might be another indicator of the existence of cryptic BT. Nevertheless, to recognize the “unbalanced translocation-like” characteristics, a minimum accumulation of two to approximately three POCs is required, which would result in a longer turnaround time (more than one PGT cycle). In addition, the patients need to be educated and well-informed to make them aware of the importance of preserving POCs; the potential impact of maternal cell contamination should also be considered. In contrast, the use of PGT-A allows us to view the genetic results of multiple embryos in a shorter time, enabling it easier to realize this characteristics. For those “carrier couples” missed by karyotyping analysis, the stress and anxiety of failing to determine the cause of RPL, as well as the additional expenses for consultations, examinations, and transportation, merit attention too. Regrettably, we neglected to collect questionnaires from them, which is another limit of our study.
Conclusion
To conclude, clinicians taking care of patients with uRPL should be aware that a normal karyotype does not eliminate the possibility of a BT. In the setting of the presence of embryos with “unbalanced translocation-like” characteristics following PGT-A, there is a potential for MPseq to provide valuable information with a significant impact on the reproductive choices of couples seeking medical attention for RPL.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Morin SJ, Eccles J, Iturriaga A, Zimmerman RS. Translocations, inversions and other chromosome rearrangements. Fertil Steril. 2017;107:19–26.
Gupta N, Dalvi R, Koppaka N, Mandava S. Balanced reciprocal translocation: multiple chromosome rearrangements in an infertile female. J Hum Reprod Sci. 2019;12:72–4.
Practice Committee of the American Society for Reproductive Medicine. Evaluation and treatment of recurrent pregnancy loss: a committee opinion. Fertil Steril. 2012;98:1103–11.
Benet J, Oliver-Bonet M, Cifuentes P, Templado C, Navarro J. Segregation of chromosomes in sperm of reciprocal translocation carriers: a review. Cytogenet Genome Res. 2005;111:281–90.
Escudero T, Lee M, Sandalinas M, Munné S. Female gamete segregation in two carriers of translocations involving 2q and 14q. Prenat Diagn. 2000;20:235–7.
Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012;13:36–46.
Lee H, Gurtowski J, Yoo S, Nattestad M, Marcus S, Goodwin S, et al. Third-generation sequencing and the future of genomics. bioRxiv. 2016 [cited 2022 Jul 23]. p. 048603. Available from: https://www.biorxiv.org/content/10.1101/048603v1.
Smadbeck J, Peterson JF, Pearce KE, Pitel BA, Figueroa AL, Timm M, et al. Mate pair sequencing outperforms fluorescence in situ hybridization in the genomic characterization of multiple myeloma. Blood Cancer J. 2019;9:1–18.
Aypar U, Smoley SA, Pitel BA, Pearce KE, Zenka RM, Vasmatzis G, et al. Mate pair sequencing improves detection of genomic abnormalities in acute myeloid leukemia. Eur J Haematol. 2019;102:87–96.
Dong Z, Yan J, Xu F, Yuan J, Jiang H, Wang H, et al. Genome sequencing explores complexity of chromosomal abnormalities in recurrent miscarriage. Am J Hum Genet. 2019;105:1102–11.
Caspersson T, Zech L, Johansson C, Modest EJ. Identification of human chromosomes by DNA-binding fluorescent agents. Chromosoma. 1970;30:215–27.
Drets ME, Shaw MW. Specific banding patterns of human chromosomes. Proc Natl Acad Sci U S A. 1971;68:2073–7.
Pardue ML, Gall JG. Molecular hybridization of radioactive DNA to the DNA of cytological preparations. Proc Natl Acad Sci U S A. 1969;64:600–4.
Kawashima E, Farinelli L, Mayer P. Method of nucleic acid amplification. 1998 [cited 2022 Jun 4]. Available from: https://patents.google.com/patent/WO1998044151A1/en.
Kris A. W. DNA sequencing costs: data from the NHGRI Genome Sequencing Program (GSP). DNA Sequencing Costs: Data. [cited 2022 Jun 4]. Available from: https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data.
Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7:461–5.
Schwartz DC, Li X, Hernandez LI, Ramnarain SP, Huff EJ, Wang YK. Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science. 1993;262:110–4.
Sundheimer LW, Liu L, Buyalos RP, Hubert G, Al-Safi Z, Shamonki M. Diagnosis of parental balanced reciprocal translocations by trophectoderm biopsy and comprehensive chromosomal screening. J Assist Reprod Genet. 2018;35:165–9.
Snider AC, Darvin T, Spor L, Akinwole A, Cinnioglu C, Kayali R. Criteria to evaluate patterns of segmental and complete aneuploidies in preimplantation genetic testing for aneuploidy results suggestive of an inherited balanced translocation or inversion. F S Rep. 2021;2:72–9.
Arsham MS, Barch MJ, Lawce HJ. The AGT cytogenetics laboratory manual. John Wiley & Sons; 2017. Available from: https://onlinelibrary.wiley.com/doi/book/10.1002/9781119061199 .
McGowan-Jordan J, Simons A, Schmid M, editors. ISCN 2016: an international system for human cytogenomic nomenclature. S. Karger AG; 2016 [cited 2022 Jun 22]. Available from: https://www.karger.com/Book/Home/271658.
Geiersbach KB, Gardiner AE, Wilson A, Shetty S, Bruyère H, Zabawski J, et al. Subjectivity in chromosome band-level estimation: a multicenter study. Genet Med. 2014;16:170–5.
Yan J, Qin Y, Zhao H, Sun Y, Gong F, Li R, et al. Live birth with or without preimplantation genetic testing for aneuploidy. N Engl J Med. 2021;385:2047–58.
Gardner DK, Lane M, Stevens J, Schlenker T, Schoolcraft WB. Reprint of: Blastocyst score affects implantation and pregnancy outcome: towards a single blastocyst transfer. Fertil Steril. 2019;112:e81–4.
Dong Z, Zhao X, Li Q, Yang Z, Xi Y, Alexeev A, et al. Development of coupling controlled polymerizations by adapter-ligation in mate-pair sequencing for detection of various genomic variants in one single assay. DNA Res. 2019;26:313–25.
Dong Z, Chau MHK, Zhang Y, Yang Z, Shi M, Wah YM, et al. Low-pass genome sequencing-based detection of absence of heterozygosity: validation in clinical cytogenetics. Genet Med. 2021;23:1225–33.
Wang L, Shen J, Cram DS, Ma M, Wang H, Zhang W, et al. Preferential selection and transfer of euploid noncarrier embryos in preimplantation genetic diagnosis cycles for reciprocal translocations. Fertil Steril. 2017;108:620-627.e4.
Reig A, Franasiak J, Scott RT, Seli E. The impact of age beyond ploidy: outcome data from 8175 euploid single embryo transfers. J Assist Reprod Genet. 2020;37:595–602.
Shao L, Shaw CA, Lu X-Y, Sahoo T, Bacino CA, Lalani SR, et al. Identification of chromosome abnormalities in subtelomeric regions by microarray analysis: a study of 5,380 cases. Am J Med Genet A. 2008;146A:2242–51.
Moog U, Arens YHJM, van Lent-Albrechts JCM, Huijts PEA, Smeets EEJ, Schrander-Stumpel CTRM, et al. Subtelomeric chromosome aberrations: still a lot to learn. Clin Genet. 2005;68:397–407.
Menten B, Maas N, Thienpont B, Buysse K, Vandesompele J, Melotte C, et al. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: a new series of 140 patients and review of published reports. J Med Genet. 2006;43:625–33.
Rivera H, Sitch FL, Crolla JA. Telomeric translocations are uncommon. Genet Couns. 1995;6:343–7.
Shaffer LG, Spikes AS, Macha M, Dunn R. Identification of a subtle chromosomal translocation in a family with recurrent miscarriages and a child with multiple congenital anomalies. A case report. J Reprod Med. 1996;41:367–71.
Wakui K, Tanemura M, Suzumori K, Hidaka E, Ishikawa M, Kubota T, et al. Clinical applications of two-color telomeric fluorescence in situ hybridization for prenatal diagnosis: identification of chromosomal translocation in five families with recurrent miscarriages or a child with multiple congenital anomalies. J Hum Genet. 1999;44:85–90.
Brackley KJ, Kilby MD, Morton J, Whittle MJ, Knight SJ, Flint J. A case of recurrent congenital fetal anomalies associated with a familial subtelomeric translocation. Prenat Diagn. 1999;19:570–4.
Bacino CA, Kashork CD, Davino NA, Shaffer LG. Detection of a cryptic translocation in a family with mental retardation using FISH and telomere region-specific probes. Am J Med Genet. 2000;92:250–5.
Kilby MD, Brackley KJ, Walters JJ, Morton J, Roberts E, Davison EV. First-trimester prenatal diagnosis of a familial subtelomeric translocation. Ultrasound Obstet Gynecol. 2001;17:531–3.
Benzacken B, Carbillon L, Dupont C, Siffroi JP, Monier-Gavelle F, Bucourt M, et al. Lack of submicroscopic rearrangements involving telomeres in reproductive failures. Hum Reprod. 2002;17:1154–7.
Fan Y-S, Zhang Y. Subtelomeric translocations are not a frequent cause of recurrent miscarriages. Am J Med Genet. 2002;109:154.
Yakut S, Berker-Karauzum S, Simsek M, Zorlu G, Trak B, Luleci G. Telomere-specific fluorescence in situ hybridization analysis of couples with five or more recurrent miscarriages. Clin Genet. 2002;61:26–31.
Joyce CA, Dennis NR, Howard F, Davis LM, Thomas NS. An 11p;17p telomeric translocation in two families associated with recurrent miscarriages and Miller-Dieker syndrome. Eur J Hum Genet. 2002;10:707–14.
Cockwell AE, Jacobs PA, Beal SJ, Crolla JA. A study of cryptic terminal chromosome rearrangements in recurrent miscarriage couples detects unsuspected acrocentric pericentromeric abnormalities. Hum Genet. 2003;112:298–302.
Bruyere H, Rajcan-Separovic E, Doyle J, Pantzar T, Langlois S. Familial cryptic translocation (2;17) ascertained through recurrent spontaneous abortions. Am J Med Genet A. 2003;123A:285–9.
Jalal SM, Harwood AR, Sekhon GS, Pham Lorentz C, Ketterling RP, Babovic-Vuksanovic D, et al. Utility of subtelomeric fluorescent DNA probes for detection of chromosome anomalies in 425 patients. Genet Med. 2003;5:28–34.
Alkuraya FS, Martin CL, Kimonis VE. Recurrent miscarriage in a carrier of a balanced cytogenetically undetectable subtelomeric rearrangement: how many are we missing? Prenat Diagn. 2006;26:291–3.
Monfort S, Martínez F, Roselló M, Badia L, Prieto F, Orellana C. A subtelomeric translocation apparently implied in multiple abortions. J Assist Reprod Genet. 2006;23:97–101.
Wise JL, Crout RJ, McNeil DW, Weyant RJ, Marazita ML, Wenger SL. Cryptic subtelomeric rearrangements and X chromosome mosaicism: a study of 565 apparently normal individuals with fluorescent in situ hybridization. PLoS ONE. 2009;4:e5855.
Durmaz B, Karaca E, Durmaz A, Atik T, Akin H, Cogulu O, et al. Subtelomeric rearrangements in patients with idiopathic intellectual disabilitiy/multiple congenital anomalies and recurrent miscarriages: seven years’ experience. Genet Couns. 2013;24:167–77.
Hajlaoui A, Slimani W, Kammoun M, Sallem A, El Amri F, Chaieb A, et al. Subtelomeric rearrangements in patients with recurrent miscarriage. Int J Fertil Steril. 2018;12:218–22.
Zhang S, Pei Z, Lei C, Zhu S, Deng K, Zhou J, et al. Detection of cryptic balanced chromosomal rearrangements using high-resolution optical genome mapping. J Med Genet. 2022;jmedgenet-2022-108553.
Coonen E, Rubio C, Christopikou D, Dimitriadou E, Gontar J, Goossens V, et al. ESHRE PGT Consortium good practice recommendations for the detection of structural and numerical chromosomal aberrations†. Hum Reprod Open. 2020;2020:hoaa017.
Huang C, Jiang W, Zhu Y, Li H, Lu J, Yan J, et al. Pregnancy outcomes of reciprocal translocation carriers with two or more unfavorable pregnancy histories: before and after preimplantation genetic testing. J Assist Reprod Genet. 2019;36:2325–31.
Gardner RM, Sutherland GR, Shaffer LG. Chromosome abnormalities and genetic counseling. Oxford University Press; 2011. Available from: https://academic.oup.com/book/25214 .
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64.
Gross M, Mkrtchyan H, Glaser M, Fricke HJ, Höffken K, Heller A, et al. Delineation of yet unknown cryptic subtelomere aberrations in 50% of acute myeloid leukemia with normal GTG-banding karyotype. Int J Oncol. 2009;34:417–23.
Jacquemont M-L, Sanlaville D, Redon R, Raoul O, Cormier-Daire V, Lyonnet S, et al. Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J Med Genet. 2006;43:843–9.
Faas BHW, Nillesen W, Vermeer S, Weghuis DO, de Leeuw N, Smits APT, et al. Detection of cryptic subtelomeric imbalances in fetuses with ultrasound abnormalities. Eur J Med Genet. 2008;51:511–9.
Scott RT. Introduction: subchromosomal abnormalities in preimplantation embryonic aneuploidy screening. Fertil Steril. 2017;107:4–5.
Xie P, Liu P, Zhang S, Cheng D, Chen D, Tan Y-Q, et al. Segmental aneuploidies with 1 Mb resolution in human preimplantation blastocysts. Genet Med. 2022;S1098-3600(22)00899-1
Chow JFC, Cheng HHY, Lau EYL, Yeung WSB, Ng EHY. Distinguishing between carrier and noncarrier embryos with the use of long-read sequencing in preimplantation genetic testing for reciprocal translocations. Genomics. 2020;112:494–500.
Yanfei Cheng MM, Yu Q, Ma M, Wang H, Tian S, Zhang W, et al. Variant haplophasing by long-read sequencing: a new approach to preimplantation genetic testing workups. Fertil Steril. 2021;116:774–83.
Kurahashi H, Inagaki H, Ohye T, Kogo H, Tsutsumi M, Kato T, et al. The constitutional t(11;22): implications for a novel mechanism responsible for gross chromosomal rearrangements. Clin Genet. 2010;78:299–309.
Johnson SH, Smadbeck JB, Smoley SA, Gaitatzes A, Murphy SJ, Harris FR, et al. SVAtools for junction detection of genome-wide chromosomal rearrangements by mate-pair sequencing (MPseq). Cancer Genet. 2018;221:1–18.
Acknowledgements
We appreciate Miss Zihan Chen for her invaluable help in sample preparation and all participants involved in this study.
Funding
This study was supported by the National Key R&D Program of China (2021YFC2700604), the Collaborative Research Fund (C4062-21GF), and the National Natural Science Foundation of China (82171648 and 32270678).
Author information
Authors and Affiliations
Contributions
S. L., H. L., Z.-J. C., K. W. C., Z. D., and J. Y. designed the whole study. J. Y. recruited patients. S. L. collected samples and variant verification and data curation. H. Li contributed to FISH validation. Z. D. and K. W. C. completed MPseq, data analysis, and variant interpretation. Y. G., Y. Z., and X. Y. collected and analyzed the PGT results. S. L., H. L., K. W. C., Z. D., and J. Y. wrote the manuscript. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, S., Li, H., Gao, Y. et al. Identification of cryptic balanced translocations in couples with unexplained recurrent pregnancy loss based upon embryonic PGT-A results. J Assist Reprod Genet 41, 171–184 (2024). https://doi.org/10.1007/s10815-023-02999-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-023-02999-2