
Abstract
Purpose
Gap junctions and transzonal projections play a crucial role in intercellular communication between different follicular components and are necessary for follicle development. We aimed to demonstrate gap junction protein connexin 43 (Cx43) and transzonal projections (TZPs) in viable, category 1, isolated bovine pre-antral follicles (PAFs) during short-term culture and after vitrification and warming.
Methods
This study involved four experimental groups: fresh control, 2-day culture, 4-day culture, and vitrified secondary PAFs. Isolated PAFs were vitrified using a simple and efficient cryopreservation method by means of mini cell strainers.
Results
Cx43 and TZPs were detected in pre-antral follicles of all stages, as well as in every experimental group. The group fresh follicles showed a higher percentage of follicles that were positive for Cx43 (91.7%) than the follicles that were vitrified (77.4%). All follicles that were cultured for 2 days were Cx43-positive (100%). Follicles cultured for 4 days (65.8%) (P = 0.002) showed the lowest percentage of follicles that were Cx43-positive. The percentages of the presence or (partial) absence of the TZP network were shown to be very heterogeneous between follicles in different treatment groups.
Conclusions
These results suggest the maintenance of communication between the oocyte and the somatic companion cells after vitrification and warming. The varying percentages of the expression of the TZP network within groups suggests that it will be of interest to investigate whether this is truly due to variability in TZP integrity and follicle quality or due to methodological limitations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
It has been estimated that the number of cancer survivors diagnosed during childhood and adolescence in Europe is between 300 and 500,000, with approximately one in every 640 young adults being a survivor of childhood cancer [1]. Although surviving remains the main objective, quality of life in the increasing population of young female survivors has become an equally important consideration. To varying degrees, these former patients experience a wide range of long-term adverse health effects [2]. Chemo- and radiotherapy often damage ovarian tissue [3, 4] which might render patients infertile. A Childhood Cancer Survivor Study showed that, compared to their siblings, the relative risk for survivors of ever being pregnant following radiotherapy with 5–10 Gy is 0.56, with a decrease to 0.18 when more than 10 Gy is used to a radiation field including the ovaries [5]. As a consequence, the interest in fertility preservation (FP) strategies in women has been sparked to the extent that it has now become a key medical subdiscipline [6]. Prior to cancer therapy, current fertility-preserving options for women include oocyte and embryo cryopreservation. However, for prepubertal girls and women that cannot delay the start of chemotherapy, ovarian tissue cryopreservation (OTC) is offered as an experimental option [7] (while the experimental status of OTC is still under debate [8,9,10,11]). While more than 130 births have already been reported worldwide [12, 13], this technique is not advisable for patients with certain types of cancer with medium-to-high risk of ovarian metastasis, such as leukemia, as there is a considerable risk of reintroducing malignant cells contained in the cryopreserved tissue following autotransplantation [14, 15]. For these patients, a safer alternative to allow fertility restoration could be the isolation, cryopreservation, and reintroduction of pre-antral follicles or pre-antral follicles (PAFs) in remaining ovarian tissue “in situ” following chemo- or radiotherapy. In addition, it is expected that effective means of avoiding reseeding of malignant cells with ovarian grafts, such as an artificial ovary and an in vitro culture system for primordial follicles, will become available in the near future [16,17,18,19,20,21]. Thus far, individual follicle cryopreservation techniques are labor-intensive and time-consuming, and a substantial proportion of isolated follicles is lost during handling and after warming [22]. A vitrification protocol, successfully used for non-embedded isolated PAFs, resulted in higher efficiency, but lower viability when PAFs were vitrified encapsulated in alginate beads [23]. A recent meta-analysis comparing vitrification and slow freezing suggests that vitrification is the best strategy for oocyte cryopreservation in terms of clinical outcomes [24]. However, cell vitrification bares the risk of cell damage, while cell-cell connections are crucial for subsequent follicle survival and developmental capacity [25,26,27]. Therefore, the possible effects of follicle vitrification on cell-cell contacts need to be thoroughly investigated. Firstly, cell-cell interactions need to be morphologically visualized before and after vitrification [28]. The presence of transzonal projections (TZPs) and gap junctions are predicted to be good biomarkers of follicle health, because they are necessary for bidirectional communication between the oocyte and granulosa cells. TZPs extend through the glycoprotein-rich zona pellucida and connect via gap junctions on the oocyte membrane [29,30,31,32]. Starting in the primordial follicle stage, TZPs form initial, simple cell-cell gap junctions that are continuously remodeled and elongated during the formation of the zona pellucida and de course of oocyte growth [29]. In humans [33] and bovines [34, 35], the zona pellucida starts to develop from the secondary follicle stage onwards. TZPs are essential for the bidirectional transport of molecules such as sugars, amino acids, and nucleotides [36,37,38], which are essential for oocyte growth. Gap junctions are intercellular membrane channels directly connecting the cytoplasm of adjacent cells, thus allowing the exchange of ions, second messengers, and small metabolites [39]. They play a crucial role in intercellular communication between different follicular components [40]. A single gap junction channel is composed of two hemichannels (connexins), each of which is composed of six protein subunits called connexins. Among gap junction proteins identified in ovarian follicular cells, two connexins (Cx, Cx37 and Cx43) seem to be critical at each step of normal folliculogenesis [41]. Investigating Cx expression in the bovine ovary, Nuttinck et al. [42] found that Cx43 expression was restricted to granulosa cells, while Cx37 staining was observed in both the oocyte and granulosa cell compartments. Vitrification of follicles can lead to cryoinjuries, resulting in the loss of membrane proteins, such as Cx37 and Cx43 [43].
TZPs are composed of F-actin and microtubules [32, 38, 44], which are very sensitive to changes in temperature and sheer stress and, as a consequence, vulnerable during cryopreservation [45]. To date, the literature on the visualization of cell-cell connections such as Cxs and TZPs in isolated PAFs is very scarce. In addition, to the best of our knowledge, the influence of vitrification on the preservation of cell-cell connections in isolated PAFs has never been investigated. Recently, there has been an increasing interest for bovine in vitro models in human studies on assisted reproductive techniques (ARTs) and FP, as reviewed by Langbeen et al. [46]. It is well established that a number of similarities exist between bovine and human species with regard to ovarian morphology and function, and that the bovine species could serve as an effective model for human follicular dynamics [47, 48]. Moreover, the easy access to slaughterhouse ovaries guarantees unrestricted availability of study specimens such as PAFs [46]. Because of the importance of the presence of intact cell-cell contacts for further development of PAFs after warming and the possible advantages of vitrification of PAFs, the current study aimed to (1) visualize Cx43 and TZPs using immunohistochemical methods in follicles upon isolation and in follicles that were cultured in vitro for 2 and 4 days, respectively; and (2) assess the presence of Cx43 and TZPs in isolated PAFs following vitrification and warming, as a prerequisite for future follicle developmental capacity.
Materials and methods
Collection of ovaries, ovarian follicle isolation, and culture
As described earlier [49], adult bovine ovaries were collected upon slaughter and transported in warm physiologic saline (0.9% NaCl, Braun) to the laboratory within 3 h at 25 °C. The procedure to isolate the follicles from the ovaries was carried out 36 times. For each isolation procedure, follicles were derived from 10 ovaries that originated from 10 different cows. Follicle isolation was carried out on all 10 ovaries together. Two isolation procedures were performed subsequently on different ovaries, from the same slaughterhouse batch. No separate follicle isolations were performed for each separate ovary. As a consequence, there is no specific information on which follicle originated from which ovary. Following removal of the adnexa, the ovaries were washed in a warm (38.5 °C) physiological solution supplemented with kanamycin (0.25%) and rinsed in alcohol (70%). Active ovaries were selected, free of abundant antral follicles, corpora lutea, or scar tissues. The ovarian cortex was cut into pieces of approximately 1 mm3, using a scalpel. The pieces of ovarian cortex were transferred to isolation medium: M199; supplemented with HEPES (0.04 M), gentamycin (50 μL/mL), bovine serum albumin (10 mg/mL), and polyvinylpyrrolidone (4 mg/mL); and filtered through a 0.2-μm filter. Ovarian cortex tissue was mixed and dispersed using an Ultra Turrax T18 Basic device (IKA®, VWR, Leuven, Belgium) with a medium-sized plastic dispersing tool (IKA®, S18D-14G-KS) for 2 min and with a smaller size (IKA®, S18D-10G-KS) for 1 min. The resulting follicle suspension was subsequently filtered through a 100-μm and a 70-μm filter (BD Falcon®, Corning, NY, USA) and a 20-μm nylon filter (Millipore®, Cork, Ireland). Early PAFs were recovered from the 20-μm mesh filter by rinsing with isolation medium. Follicles were visualized using standard inverted light microscopy (Olympus, Aartselaar, Belgium). Primary PAFs with an oocyte surrounded by one layer of cuboidal granulosa cells and intact basal membrane were selected on day 0 [50]. They were individually transferred and cultured in 70 μL culture medium in 96-well plates (Greiner Bio-One, Germany) at 38.5 °C and 5% CO2. The culture medium consisted of equal parts DMEM and Ham’s F12 nutrient supplemented with penicillin G (240 U/mL) and streptomycin (240 μg/mL), fungizone (5 μg/mL), fetal calf serum (2.3(v/v)%) and newborn calf serum (2.3(v/v)%), bovine serum albumin (0.75(w/v)%), insulin (0.01 mg/mL), transferrin (0.55 μg/mL), and selenium (6.7 ng/mL). Two plates were used per isolation. Per plate, 40 wells were used for culture. The two plates were filled alternately per row to shorten follicle light exposure time and preserve the equilibration of the culture medium. Follicles were cultured for 2 days to recover from the isolation procedure and grow to secondary stage follicles. To evaluate follicle morphology from day 2 onwards, follicles were microscopically evaluated and assigned to three categories based on the morphological assessment of the connection between the oocyte and the surrounding granulosa cells and the integrity of the basal membrane as reported by Jorssen et al. [49]. From day 2 onwards, only secondary category 1 follicles were selected for further use, having the right size range (40–80 μm). Category 2 and 3 follicles were not further used.
Study design
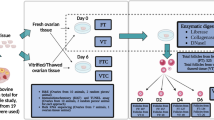
The experimental design is described and summarized in a flow chart (Fig. 1). Follicles were fixed in 4 different groups: immediately upon isolation (fresh control); following 2 days of culture (2-day culture); following vitrification, warming, and a 24-h recovery period on day 3 (vitrified); and following 4 days of culture (4-day culture). On days 2 and 4 and day 3 for vitrified follicles, follicles were assigned to three categories based on follicle morphology only when the basement membrane or connection between the oocyte and granulosa cells could be completely assessed. In addition, the follicle diameter was determined on days 2 and 4 and day 3 for vitrified follicles. Two perpendicular measures were recorded for each follicle, and the average of the two values was reported as follicular diameter (μm). When the diameter of a follicle could not be measured (when a follicle was located to the side of a well and the basal membrane could not be fully defined), it was reevaluated on the next evaluation day. The fresh follicles fixed on day 0 were primary PAFs with an oocyte surrounded by one layer of cuboidal granulosa cells and intact basal membrane; they were also analyzed for diameter immediately upon isolation. To minimize exposure to light and low temperatures and enhance follicle survival, all other follicles were cultured as soon as possible to equilibrate in the incubator. Follicles were also evaluated for follicle viability by Neutral Red staining before fixation [51]. Cx43 and TZP fluorescent staining were carried out to analyze the presence of cell-cell connections. Hoechst staining was carried out to visualize cell nuclei. For practical reasons, time constraints, and to minimize exposure to light and low temperatures, follicles were not followed up individually but as a group.

Experimental design. Follicles were fixed immediately following isolation (fresh control); following 2 days of culture (2-day culture); following vitrification, warming, and a 24-h recovery period on day 3 (vitrified); or following 4 days of culture (4-day culture). Before fixation, follicles were assessed for morphology, diameter, and viability. After fixation, the follicles were stained to visualize Cx43 and TZPs and cell nuclei with Hoechst staining
Freezing and warming
The IrvineScientific® Vitrification Kit for human oocytes and embryos (Alere Health BV. Tilburg, NL) was used for vitrification and warming following slight adaptations to the manufacturer’s guidelines. On day 2, category 1 follicles were transferred to a mini cell strainer (25 μm, nylon, Funakoshi, Japan), immersed in culture medium (Fig. 2). Subsequently, the mini cell strainer with PAFs was first transferred to equilibration solutions 1 and 2 (ES1 and ES2; 7.5% DMSO, 7.5% EG) for 1 min each, then to vitrification solution (VS; 15% DMSO, 15% EG, 0.5 M sucrose) for 30 s and then directly plunged into liquid nitrogen (LN2). To eliminate the remaining solution from the mini cell strainer, it was briefly placed on an absorbent paper towel after each step. The vitrification procedure was carried out at room temperature (RT). Follicles were warmed the same day. After plunging the mini cell strainer immediately in a large volume (10 mL) of warming solution (38.5 °C; 1 M sucrose) for 1 min, mini cell strainers were transferred to dilution solution (0.5 M sucrose) for 4 min at RT. Next, PAFs were washed in the washing medium twice during 4 min. PAFs stayed in the mini cell strainer and were immersed in fresh culture medium in a 35-mm petri dish (38.5 °C, 5% CO2). Follicles were finally evaluated following a 24-h recovery period.

PAFs are placed in a mini cell strainer immersed in cryoprotective agents (ES and VS) in a 4-well plate. Mini cell strainers are transferred with tweezers between different media and equally serve as cryo-container when they are plunged in LN2
Follicle quality and viability assessment
Neutral Red (NR) staining
To assess follicle survival and immediate viability, follicles were stained with the non-toxic viability indicator Neutral Red [51]. One hundred microliters of Neutral Red solution (3.3 g/L, Sigma) was added to the culture medium. A follicle was considered to be positively stained when both the oocyte and at least ¾ of the granulosa cells were colored red. When the dye is incorporated in the lysosomes of the follicles, they are considered metabolically active and thus viable. Following 20 min of incubation (38.5 °C, 5% CO2), follicle staining was evaluated [51].
Cx43 and TZP staining
The protocol for immunostaining of connexins was adapted from Ortiz-Escribano et al. [52]. Follicles were fixed in 4% paraformaldehyde (PFA) for 20 min at RT. They were washed in PBS + 1 mg/mL PVP and permeabilized with 1% Triton X-100 (T8787, Sigma) and 0.05% Tween 20 (P1379, Sigma) in PBS for 20 min at RT. Subsequently, follicles were blocked with a solution consisting of 10% normal goat serum and 0.05% Tween 20 prepared in PBS, at 4 °C for 1 h. Follicles were then incubated with rabbit anti-Cx43 polyclonal antibody (1:500, C6219, Sigma) diluted in blocking solution at 4 °C overnight. Subsequently, follicles were washed and incubated with goat anti-rabbit secondary antibody conjugated with FITC (1:250, A16105, Invitrogen) for 2 h at RT. Next, to visualize filamentous actin (F-actin) and consequently TZPs because they are composed of F-actin, follicles were washed and incubated with Alexa Fluor™ 568 Phalloidin (1:40, A12380, Thermo Fisher Scientific) for 1 h at RT. Follicles were counterstained with Hoechst 10 μg/mL for 10 min, washed, and mounted with Antifade mounting medium (Life Technologies) on microscope slides. Images were obtained using a Leica SP8 confocal microscope equipped with a 405-nm diode laser (to detect the Hoechst nuclear stain, blue) and a white laser source (Leica WLL) used at 488 nm (connexins, green) and at 577 nm (to visualize TZP staining, red). For Cx43 and TZP analysis, a × 60water immersion (N.A. 1.20) objective was used and image acquisition settings were kept constant for the recordings of all follicles. Z-stacks were taken with a 0.36-μM interval through the complete follicle. The expression of Cx43 was classified as positive (present) or negative (absent) (Fig. 3). The follicles that showed connexin expression in at least ¾ of GCs through the complete follicle were evaluated as Cx43-positive. The organization of the TZPs was classified as (0) total absence (no physical connections between the oocyte and granulosa cells [no TZPs visible]); (1) partial absence (gaps between the oocyte and granulosa cells [no dense staining of F-actin]); and (2) complete (no contact loss between oocyte and granulosa cells [i.e., dense F-actin staining]) (categorization of TZPs was adapted from [53]) (Fig. 4). To avoid that the quantification of the expression of Cx43 and TZPs could be attributed to differences in granulosa cell number, the expression of Cx43 and TZPs was qualified and not quantified. Negative controls were obtained by omission of the primary antibodies for Cx43 and omission of TZP staining.

Follicle staining negative (a) and positive (b) for Cx43

The organization of the TZPs was classified as (0) total absence (no physical connections between the oocyte and granulosa cells [no TZPs visible]); (1) partial absence (gaps between the oocyte and granulosa cells [no dense staining of F-actin]); and (2) complete (no contact loss between oocyte and granulosa cells [i.e., dense F-actin staining]) (categorization of TZPs was adapted from [53]). O oocyte, GC granulosa cell. TZPs are indicated with arrows; interrupted TZPs are indicated with an arrow head
Statistical analysis
Different statistical models were used to assess the differences in follicle diameter and the presence of Cx43 between groups. The procedure to isolate the follicles from the ovaries, as described in “Collection of ovaries, ovarian follicle isolation, and culture,” was carried out 36 times. For each isolation procedure, follicles were derived from 10 ovaries that originated from 10 different cows. The ovaries were selected as described in “Collection of ovaries, ovarian follicle isolation, and culture.” Follicle isolation was carried out as outlined in “Collection of ovaries, ovarian follicle isolation, and culture” on all 10 ovaries together. Two isolation procedures were performed subsequently on different ovaries, from the same slaughterhouse batch. After two isolation procedures, approximately 160 intact follicles were selected for culture. No separate follicle isolations were performed for each separate ovary. As a consequence, there is no specific information on which follicle originated from which ovary. Therefore, all follicles within one replicate are considered independent statistical units. To account for possible replicate effects, the replicate number was entered as a random effect in the subsequent statistical analysis. Treatment was entered as a fixed effect. Interaction between the treatment and replicate was also entered, but if this latter term was not significant, it was omitted from the model. Follicle diameter was considered a dependent continuous variable. Cx43 expression (binary, present vs. absent) was considered a dependent categorical variable. Treatment group was considered an independent categorical variable (categorical: fresh control, 2-day culture, 4-day culture, vitrified). Potential differences in follicle diameter between groups were analyzed using analysis of variance (ANOVA). Prior to ANOVA, data were analyzed for normal distribution and homogeneity of variance by performing the Kolmogorov-Smirnov and Levene tests, respectively. Potential differences in Cx43 expression between groups were analyzed using a Fisher exact test. Differences between groups were considered to be significant when the P value is < 0.05 [54]. Data are presented as means ± standard deviation (S.D.). All statistical analyses were performed using IBM SPSS version 24® (New York, USA).
Results
Follicle gross morphology
In total, about 2200 primary follicles were cultured, of which it usually took good-quality follicles 2 days to develop until the secondary stage. From day 2 of culture onwards, the outline of the basal membrane was more pronounced and the morphological distinction between the granulosa cells could no longer clearly be made (Fig. 5). At day 2 of culture, 244 follicles were classified as category 1, meaning that only 11% of all follicles were suitable to use for the experiments according to their gross morphology. Follicles were collected from 36 isolation procedures. However, 5 replicates contained no category 1 follicles following 2 days of culture and these replicates were not taken into account for further analyses.

Light microscopic morphological aspects of different follicle stages upon isolation, on 2 and 4 days of culture and following vitrification. a Primordial follicle upon isolation (NR-positive, Ø: 45 μm). b Transitory follicle upon isolation (NR-positive, Ø: 42 μm). c Primary follicle upon isolation (NR-positive, Ø: 48 μm). d Secondary follicle on day 2 of culture (non-stained, Ø: 60 μm). e Secondary follicle on day 4 of culture (non-stained, Ø: 63 μm). f Vitrified follicle 1 day post warming (NR-positive, Ø: 45 μm). White arrow heads indicate flattened granulosa cells, white arrows indicate cuboidal granulosa cells, black arrows indicate the oocyte. NR, Neutral Red; Ø, follicular diameter, reported as an average of two perpendicular measures. Magnification: × 400
Follicle diameter
The follicle diameter of PAFs was measured on day 0 (n = 60), day 2 (n = 201), day 3 (post vitrification; n = 40), and day 4 (n = 89). The mean follicle diameter (μm) ± S.D. for category 1 follicles pooled over all replicates was 46.8 ± 4.6 upon isolation, 48.5 ± 7.0 on day 2, and 50.5 ± 7.7 on day 4 (Fig. 6). On day 3, 1 day post vitrification, the mean follicle diameter (μm) ± S.D. was 51.0 ± 7.7. Mean follicular diameters were significantly different between day 0 and day 2 (P = 0.041) and between day 0 and day 4 (P = 0.001).

Boxplot showing average follicular diameter (μm) upon isolation (n = 60), and following 2 (n = 201), respectively, 4 days of culture (n = 89)
Neutral Red staining
To evaluate immediate follicle survival and instant viability, PAFs were stained with Neutral Red (NR). Upon isolation and on days 2 and 4 of culture, only category 1 follicles were taken into account. Follicles stained positive for the vital dye NR were considered viable. Ninety-six of all category 1 follicles on day 2 and day 4 of culture were stained Neutral Red–positive. Out of 96 follicles that were vitrified, 48 were successfully retrieved and 70.8% (n = 34) of them were stained NR-positive after warming and 24-h culture (Fig. 7).

a Light microscopic (LM) image of two non-stained follicles (black arrows) on the mesh of the mini cell strainer. b LM image of NR-positive follicles on the mesh of the mini cell strainer. c Detailed LM image of NR-positive follicle. d NR-positive follicle removed from the mesh
Presence of Cx43 and TZPs in different follicle stages
Cx43 was detected in granulosa cells of all pre-antral follicle stages, at the site of TZPs (Fig. 8). Cx43 staining started with distinct spots in resting primordial follicles. As seen in Fig. 8, the Cx43-positive dots per granulosa cell, and hence, the number of gap junctions increased notably with follicle development.

Confocal images (single confocal plane) of expression and localization of Cx43 and TZPs in PAFs in different stages. Images show a single confocal plane at the median level of the follicle. a Primordial follicle upon isolation: oocyte surrounded by one layer of flattened granulosa cells. b Primary follicle upon isolation: oocyte surrounded by one layer of cuboidal granulosa cells. c Secondary follicle after 2-day culture: oocyte surrounded by several layers of cuboidal granulosa cells. Hoechst nuclear staining in blue, Cx43 staining in green, and Alexa Fluor™ 568 Phalloidin staining actin-rich TZPs in red; merged picture shows an overlay of the three stains. White dotted line indicates the oocyte. GC granulosa cells. TZPs are indicated by arrows
Cx43 and TZPs before and following vitrification
Cx43 and TZPs could be detected in PAFs in all groups (Fig. 9), before and following vitrification. In Fig. 9, from each group, one follicle was depicted as representative for the whole group.

Confocal images (single confocal plane) of expression and localization of Cx43 and TZPs in PAFs in all treatment groups. D0: primary follicle fixed upon isolation; D2: follicle following 2 days of culture; D4: follicle following 4 days of culture; VIT: follicle fixed after vitrification and warming and a 24-h recovery period in culture. Hoechst nuclear staining in blue, Cx43 staining in green, and Alexa Fluor™ 568 Phalloidin staining actin-rich TZPs in red; merged picture shows an overlay of the three stains. White dotted line indicates the oocyte. GC granulosa cells. TZPs are indicated by arrows
From the NR-positive follicles that were fixed immediately upon isolation and stained for Cx43 (n = 48), 91.7% (n = 44) were stained positive for Cx43 (Fig. 10). From the NR-positive follicles that were stained for TZPs (n = 48), 16.7% (n = 8) showed a complete TZP network. 8.3% (n = 4) showed a complete absence of TZPs, and 75% (n = 36) showed a partial absence of TZPs (Fig. 11). In 16.7% (n = 8) of cases, both TZPs and Cx43 were preserved. From NR-positive follicles that were fixed following 2 days of culture, 23 follicles were stained for Cx43 and all of them (100%) were stained positive for Cx43. From the NR-positive follicles that were stained for TZPs (n = 23), 30.4% (n = 7) showed a complete TZP network. 52.2% (n = 12) showed a total absence of TZPs, and 17.4% (n = 4) showed a partial absence of TZPs. In 30.4% (n = 7) of cases, both TZPs and Cx43 were preserved. From the NR-positive follicles that were fixed after 4 days of culture and were stained for Cx43 (n = 38), 65.8% (n = 25) were stained positive for Cx43. This is significantly different from follicles assessed after isolation and on day 2 (P = 0.002). From the NR-positive follicles that were stained for TZPs (n = 38), 10.5% (n = 4) showed a complete TZP network. 42.1% (n = 16) showed a total absence of TZPs, and 47.4% (n = 18) showed a partial absence of TZPs. In none of the cases, both TZPs and Cx43 were preserved. From the vitrified NR-positive follicles that were stained for Cx43 (n = 31), 77.4% (n = 24) were stained positive for Cx43. From the NR-positive follicles that were stained for TZPs (n = 31), 35.5% (n = 11) showed a complete TZP network. Twenty-nine percent (n = 9) showed a total absence of TZPs, and 35.5% (n = 11) showed a partial absence of TZPs. In 19.4% (n = 6) of cases, vitrification resulted in the preservation of both TZPs and Cx43.

Percentage of Cx43-positive follicles per group. Columns with the same letters do not differ significantly from each other at the P < 0.05 level

The percentage of pre-antral follicles allocated to a particular category of TZP organization per group. (0) Total absence (no physical connections between the oocyte and granulosa cells [no TZPs visible]). (1) Partial absence (gaps between the oocyte and granulosa cells [no dense staining of F-actin]). (2) Complete (no contact lost between oocyte and granulosa cells [i.e., dense staining of F-actin])
Discussion
These data are the first to demonstrate that it is possible to visualize Cx43 and TZPs in isolated bovine PAFs before and following vitrification and warming, using a simplified and more efficient cryopreservation protocol while using mini cell strainers. The detection and descriptive analysis of immunofluorescent-labeled connexins and TZPs can provide an indication about their future developmental capacity post warming. Immunofluorescent staining of connexins in ovine-isolated follicles was performed by da Silva et al. [43], on follicles embedded in paraffin. We were not able to detect Cx37 protein expression in isolated PAFs, potentially due to the absence of Cx37 or the deficiency of reactivity of the antibody for bovine Cx37. As follicles shrink after fixation, the even smaller size hampers follicle manipulation. Due to many transfer steps between different media, a considerable loss of follicles was difficult to prevent. A vital Neutral Red staining was used to improve follicle visibility for handling and assessment.
For the first aim of this study, we demonstrated Cx43 and TZPs in category 1 freshly fixed and cultured follicles. These membrane proteins constitute cell-cell connections and mediate intercellular communication. The identification of Cx43 and TZPs indicates the presence of these proteins which are essential for follicle development. Freshly fixed follicles (91.7%) showed a slightly decreased percentage of Cx43-positive follicles compared to follicles that were cultured for 2 days (100%). Follicles that were cultured for 2 days showed a higher percentage of Cx43-positive follicles; this may indicate that follicles need a recovery period after the isolation procedure. Or, as the selection of good-quality follicles based on morphology is more adequate on day 2 than immediately upon isolation, the batch of follicles selected on day 0 may contain follicles that were incorrectly qualified as category 1. Follicles that were cultured for 4 days showed the lowest percentage of Cx43-positive follicles (65.8%), which may indicate that current culture conditions were not optimal for follicle development. In each group and pre-antral follicle stage, we saw follicles with a complete TZP network. However, the percentages of the presence or (partial) absence of the TZP network varied between follicles in different treatment groups. This may be due to variability in follicle quality; although when examining morphology and viability, the follicles appeared to be quite similar (category 1 and Neutral Red–positive). In this perspective, we suggest that gross morphology can predict immediate individual cell viability [49] but not the presence of cell-cell connections. To start the experiment with a homogenous group of follicles on day 2 of culture, only category 1 secondary follicles at the right size range (40–80 μm) were selected for future use. In our opinion, only when these good-quality follicles are used, an experiment can succeed and be reproducible. On the downside, this strict use of category 1 follicles makes it more difficult to obtain a sufficient number of follicles to complete a full experiment.
Secondly, we aimed to visualize Cx43 and TZPs in PAFs following vitrification and warming. Several studies have been conducted on the expression of connexins and TZPs and most of them demonstrated that cryopreservation might cause damage to these membrane proteins, although the results are not consistent. A reason for cryodamage may be that hemichannels ((HC) consist of six Cx proteins and 2 HCs create gap junctions between adjacent cells) are normally closed but open in response to stress conditions such as cryopreservation. Excessive HC opening is detrimental for cell function and may lead to cell death [52]. Our results show that Cx43 expression in vitrified isolated follicles (77.4%) was slightly lower when compared to follicles that were freshly fixed (91.7%) or cultured for 2 days (100%). 35.5% showed a complete TZP network after vitrification, the highest percentage from all groups. Also, Tanpradit et al. [55] showed a reduced Cx43 expression pattern after both slow freezing and vitrification of feline PAFs enclosed in ovarian tissue. In a study of da Silva et al. [43], it was shown that Cx43 was reduced in ovine secondary follicles after vitrification and in vitro culture of ovarian tissue. In contrast, Donfack et al. [28] found that in vitro culture of the vitrified goat, ovarian cortex did not alter the expression of Cx43. Barrett et al. [31] showed that immediately after slow freezing and thawing, mouse-isolated follicles were unable to transfer Lucifer Yellow into surrounding granulosa cells, indicating that the gap junctions were not functional. However, after 2 days of culture, cryopreserved follicles were able to reestablish gap junctions and transport the dye from the oocyte into the surrounding somatic cells. Trapphoff et al. [56] also reported the recovery of contacts by TZPs between mouse oocytes and granulosa cells from vitrified pre-antral follicles during the first 4 days of culture. They suggest that the thick cortical actin layer in oocytes is maintained and may contribute to the rapid reestablishment of membrane organization at the cortex and restoration of contacts between somatic cells and the oocyte. Thus, cryopreservation of follicles does not necessarily damage cell-cell connections in the long term when a sufficient recovery period is provided. However, most studies were performed using slow freezing and ovarian tissue, and data on the survival of cell-cell connections in isolated PAFs after vitrification is very scarce.
Processing isolated PAFS remains a huge challenge in terms of follicular retrieval and manipulation; therefore during this study, we tried to optimize the vitrification protocol and increase the efficiency of follicle recovery using mini cell strainers. In a previous study [23], we reported that bovine PAFs can be successfully cryopreserved by a simple two-step vitrification method using HSV straws. However, transferring individual PAFs between droplets using Stripper tips is time-consuming while these manipulations can cause osmotic damage to the follicles. The disadvantage of straws and cryovials is that they only permit relatively low cooling and warming rates, while the ability to survive vitrification is also highly dependent on the rate of warming [57]. Vitrification of alginate embedded PAFs in 20-μm metal mesh cell strainers resulted in a higher efficiency, but lower viability [23]. Therefore, in this study, we aimed for better permeation of cryoprotectants combined with presupposed higher cooling and warming rates and used a mini cell strainer to freeze non-embedded PAFs. Multiple follicles can be vitrified at the same time, as a mini cell strainer can hold many more follicles, whereas only a few follicles can be loaded into one straw. It was a much easier, quicker, and simpler way to freeze PAFs. The mini cell strainer has a small handle that can easily be held by tweezers. The step-by-step transfer of the cell strainer loaded with PAFs between freezing and warming media and the passage through LN2 can be carried out with tweezers. After being submerged in LN2, the cells cool down very quickly. The visibility of non-stained PAFs on the mesh of the cell strainer is, however, limited and hampers follicle retrieval. Positive staining with Neutral Red improves the visibility and makes the retrieval of PAFs from the cell strainer easier. Another difficulty is that PAFs tend to settle on the rim of the cell strainer, which makes it impossible to visualize them. Therefore, it is necessary to carefully search for them using a hand pipette with glass capillary along the whole rim of the cell strainer. When visible follicles were retrieved, the cell strainer was rinsed with culture medium in a 35-mm petri dish, always yielding extra follicles. In view of the relative high follicle loss (the recovery rate varied between 33.3 and 69.2%), we think it is beneficial to use cell strainers with an even smaller mesh size, taking into account follicle shrinkage.
Martino et al. [58] were the first to use EM grids for the cryopreservation of bovine oocytes and reported a significantly higher survival rate for vitrified-warmed oocytes using grids as compared to oocytes frozen in straws. These results may be explained by the physical characteristics such as the ultra-thermo-conductivity of the metal grid that results in about threefold higher cooling rates than that obtained with straws [59]. In addition, Kim et al. [60] showed that the developmental capacity of immature bovine oocytes vitrified and warmed using EM grids was not hampered. Park et al. [61] successfully cryopreserved in vitro produced bovine blastocysts using EM grids. More than a decennium later, Nakashima et al. [62] showed that vitrification using a nylon mesh container for human embryo ultrarapid vitrification is an easy and inexpensive method that improves the reliability of human embryo cryopreservation. Due to the small mesh size and convenient size of the cell strainer, we chose to work with mini cell strainers. A commercially available vitrification kit for human oocytes and embryos was used in this experiment with a slightly modified protocol. We reduced the number of transfer steps and shortened the exposure time as it did not hamper the survival rate of isolated bovine pre-antral follicles. We found this modification beneficial because of the reduced exposure time of PAFs to cryoprotectants, the reduced risk of losing follicles, and the decreased working time.
In conclusion, we visualized Cx43 and TZPs in isolated bovine PAFs during short-term culture and following vitrification and warming, suggesting the maintenance of communication between the oocyte and the somatic companion cells. The aim of this study was limited to the visualization of cell-cell connections in isolated PAFs. Future research should further confirm the presence and functionality of these cell-cell connections at the gene transcription and protein levels, using real-time quantitative PCR and Western blot analyses. In addition, these extra insights will help to gain a greater understanding of the variations in expression of the TZP network that were observed within our study groups. It is also important to consider that in early pre-antral follicles, TZPs have only just started to develop and they are continuously remodeling during the formation of the zona pellucida, which starts to develop from the secondary stage onwards in bovines and humans. Furthermore, in view of the challenges that still need to be overcome regarding follicular retrieval and manipulation, we were able to use a simpler and much more workable cryopreservation method, by means of mini cell strainers. Vitrification in cell strainers has important advantages, as manipulation is much easier and time is saved because follicles are vitrified in group.
Change history
15 February 2021
A Correction to this paper has been published: https://doi.org/10.1007/s10815-020-02040-w
References
Hewitt M, Weiner SL, Simone JV. Childhood cancer survivorship: improving care and quality of life. Washington, D.C.: National Academies Press; 2003.
Robison LL, Hudson MM. Survivors of childhood and adolescent cancer: life-long risks and responsibilities. Nat Rev Cancer. 2014;14(1):61–70.
Rodriguez-Wallberg KA, Oktay K. Options on fertility preservation in female cancer patients. Cancer Treat Rev. 2012;38(5):354–61.
Hyman JH, Tulandi T. Fertility preservation options after gonadotoxic chemotherapy. Clin Med Insights Reprod Health. 2013;7:61–9.
Green DM, Kawashima T, Stovall M, Leisenring W, Sklar CA, Mertens AC, et al. Fertility of female survivors of childhood cancer: a report from the childhood cancer survivor study. J Clin Oncol. 2009;27(16):2677–85.
Seli E, Agarwal A. Fertility preservation in females: emerging technologies and clinical applications. first ed. New York: Springer; 2012.
Jeruss JS, Woodruff TK. Preservation of fertility in patients with cancer. N Engl J Med. 2009;360(9):902–11.
Donnez J, Dolmans M-M, Diaz C, Pellicer A. Ovarian cortex transplantation: time to move on from experimental studies to open clinical application. Fertil Steril. 2015;104(5):1097–8.
Forman EJ. Ovarian tissue cryopreservation: still experimental? Fertil Steril. 2018;109(3):443–4.
Silber S. Ovarian tissue cryopreservation and transplantation: scientific implications. J Assist Reprod Genet. 2016;33(12):1595–603.
von Wolff M, Sänger N, Liebenthron J. Is ovarian tissue cryopreservation and transplantation still experimental? It is a matter of female age and type of cancer. J Clin Oncol. 2018;36(33):JCO1800425.
Jadoul P, Guilmain A, Squifflet J-L, Luyckx M, Votino R, Wyns C, et al. Efficacy of ovarian tissue cryopreservation for fertility preservation: lessons learned from 454 cases. Hum Reprod. 2017;32(5):1046–54.
Donnez J, Manavella DD, Dolmans M-M. Techniques for ovarian tissue transplantation and results. Minerva Ginecol. 2018;70(4):424–31.
Rosendahl M, Greve T, Andersen CY. The safety of transplanting cryopreserved ovarian tissue in cancer patients: a review of the literature. J Assist Reprod Genet. 2013;30(1):11–24.
Dolmans M-M, Masciangelo R. Risk of transplanting malignant cells in cryopreserved ovarian tissue. Minerva Ginecol. 2018;70(4):436–43.
Fisch B, Abir R. Female fertility preservation: past, present and future. Reproduction. 2018;156(1):F11–27.
Smitz J, Dolmans MM, Donnez J, Fortune JE, Hovatta O, Jewgenow K, et al. Current achievements and future research directions in ovarian tissue culture, in vitro follicle development and transplantation: implications for fertility preservation. Hum Reprod Update. 2010;16(4):395–414.
Telfer EE, McLaughlin M. In vitro development of ovarian follicles. Semin Reprod Med. 2011;29(1):15–23.
Telfer EE, Zelinski MB. Ovarian follicle culture: advances and challenges for human and nonhuman primates. Fertil Steril. 2013;99(6):1523–33.
Vanacker J, Dolmans MM, Luyckx V, Donnez J, Amorim CA. First transplantation of isolated murine follicles in alginate. Regen Med. 2014;9(5):609–19.
Luyckx V, Dolmans MM, Vanacker J, Legat C, Fortuno Moya C, Donnez J, et al. A new step toward the artificial ovary: survival and proliferation of isolated murine follicles after autologous transplantation in a fibrin scaffold. Fertil Steril. 2014;101(4):1149–56.
Bus A, Langbeen A, Martin B, Leroy J, Bols P. Is the pre-antral ovarian follicle the ‘holy grail’ for female fertility preservation? Anim Reprod Sci. 2019;207:119–30.
Bus A, van Hoeck V, Langbeen A, Leroy J, Bols PEJ. Effects of vitrification on the viability of alginate encapsulated isolated bovine pre-antral follicles. J Assist Reprod Genet. 2018;35(7):1187–99.
Rienzi L, Gracia C, Maggiulli R, LaBarbera AR, Kaser DJ, Ubaldi FM, et al. Oocyte, embryo and blastocyst cryopreservation in ART: systematic review and meta-analysis comparing slow-freezing versus vitrification to produce evidence for the development of global guidance. Hum Reprod Update. 2017;23(2):139–55.
Carabatsos MJ, Sellitto C, Goodenough DA, Albertini DF. Oocyte-granulosa cell heterologous gap junctions are required for the coordination of nuclear and cytoplasmic meiotic competence. Dev Biol. 2000;226(2):167–79.
Matzuk MM, Burns KH, Viveiros MM, Eppig JJ. Intercellular communication in the mammalian ovary: oocytes carry the conversation. Science. 2002;296(5576):2178–80.
Gilchrist RB, Lane M, Thompson JG. Oocyte-secreted factors: regulators of cumulus cell function and oocyte quality. Hum Reprod Update. 2008;14(2):159–77.
Donfack NJ, Alves KA, Alves BG, Rocha RMP, Bruno JB, Bertolini M, et al. Stroma cell-derived factor 1 and connexins (37 and 43) are preserved after vitrification and in vitro culture of goat ovarian cortex. Theriogenology. 2018;116:83–8.
Anderson E, Albertini DF. Gap junctions between the oocyte and companion follicle cells in the mammalian ovary. J Cell Biol. 1976;71(2):680–6.
Kidder GM, Mhawi AA. Gap junctions and ovarian folliculogenesis. Reproduction. 2002;123(5):613–20.
Barrett SL, Shea LD, Woodruff TK. Noninvasive index of cryorecovery and growth potential for human follicles in vitro. Biol Reprod. 2010;82(6):1180–9.
Carabatsos MJ, Elvin J, Matzuk MM, Albertini DF. Characterization of oocyte and follicle development in growth differentiation factor-9-deficient mice. Dev Biol. 1998;204(2):373–84.
Motta PM, MAKABE S, NAGURO T, CORRER S. Oocyte follicle cells association during development of human ovarian follicle. A study by high resolution scanning and transmission electron microscopy. Arch Histol Cytol. 1994;57(4):369–94.
Fair T, Hulshof S, Hyttel P, Greve T, Boland M. Oocyte ultrastructure in bovine primordial to early tertiary follicles. Anat Embryol. 1997;195(4):327–36.
Armstrong D, Baxter G, Hogg C, Woad K. Insulin-like growth factor (IGF) system in the oocyte and somatic cells of bovine preantral follicles. Reproduction. 2002;123(6):789–97.
Eppig JJ. A comparison between oocyte growth in coculture with granulosa cells and oocytes with granulosa cell-oocyte junctional contact maintained in vitro. J Exp Zool. 1979;209(2):345–53.
Brower PT, Schultz RM. Intercellular communication between granulosa cells and mouse oocytes: existence and possible nutritional role during oocyte growth. Dev Biol. 1982;90(1):144–53.
Albertini DF, Combelles CM, Benecchi E, Carabatsos MJ. Cellular basis for paracrine regulation of ovarian follicle development. Reproduction. 2001;121(5):647–53.
Larsen W. Biological implications of gap junction structure, distribution and composition: a review. Tissue Cell. 1983;15(5):645–71.
Grazul-Bilska AT, Reynolds LP, Redmer DA. Gap junctions in the ovaries. Biol Reprod. 1997;57(5):947–57.
Kidder GM, Vanderhyden BC. Bidirectional communication between oocytes and follicle cells: ensuring oocyte developmental competence. Can J Physiol Pharmacol. 2010;88(4):399–413.
Nuttinck F, Peynot N, Humblot P, Massip A, Dessy F, Flechon JE. Comparative immunohistochemical distribution of connexin 37 and connexin 43 throughout folliculogenesis in the bovine ovary. Mol Reprod Dev. 2000;57(1):60–6.
da Silva AMS, Bruno JB, de Lima LF, de Sá NAR, Lunardi FO, Ferreira ACA, et al. Connexin 37 and 43 gene and protein expression and developmental competence of isolated ovine secondary follicles cultured in vitro after vitrification of ovarian tissue. Theriogenology. 2016;85(8):1457–67.
Albertini DF, Rider V. Patterns of intercellular connectivity in the mammalian cumulus-oocyte complex. Microsc Res Tech. 1994;27(2):125–33.
Vanhoutte L, Cortvrindt R, Nogueira D, Smitz J. Effects of chilling on structural aspects of early preantral mouse follicles. Biol Reprod. 2004;70(4):1041–8.
Langbeen A, Bartholomeus E, Leroy JL, Bols PE. Bovine in vitro reproduction models can contribute to the development of (female) fertility preservation strategies. Theriogenology. 2015;84(4):477–89.
Adams G, Pierson R. Bovine model for study of ovarian follicular dynamics in humans. Theriogenology. 1995;43(1):113–20.
Campbell B, Souza C, Gong J, Webb R, Kendall N, Marsters P, et al. Domestic ruminants as models for the elucidation of the mechanisms controlling ovarian follicle development in humans. Reproduction. 2003;61:429–43.
Jorssen EP, Langbeen A, Marei WF, Fransen E, De porte HF, Leroy JL, et al. Morphologic characterization of isolated bovine early preantral follicles during short-term individual in vitro culture. Theriogenology. 2015;84(2):301–11.
Braw-Tal R, Yossefi S. Studies in vivo and in vitro on the initiation of follicle growth in the bovine ovary. J Reprod Fertil. 1997;109(1):165–71.
Langbeen A, Jorssen EP, Granata N, Fransen E, Leroy JL, Bols PE. Effects of neutral red assisted viability assessment on the cryotolerance of isolated bovine preantral follicles. J Assist Reprod Genet. 2014;31(12):1727–36.
Ortiz-Escribano N, Szymanska KJ, Bol M, Vandenberghe L, Decrock E, Van Poucke M, et al. Blocking connexin channels improves embryo development of vitrified bovine blastocysts. Biol Reprod. 2017;96(2):288–301.
Lierman S, Tilleman K, Cornelissen M, De Vos WH, Weyers S, T’Sjoen G, et al. Follicles of various maturation stages react differently to enzymatic isolation: a comparison of different isolation protocols. Reprod BioMed Online. 2015;30(2):181–90.
Dohoo I, Martin S, Stryhn H. Vet Epidemiol Res. Second ed. Charlottetown: VER inc; 2009.
Tanpradit N, Comizzoli P, Srisuwatanasagul S, Chatdarong K. Positive impact of sucrose supplementation during slow freezing of cat ovarian tissues on cellular viability, follicle morphology, and DNA integrity. Theriogenology. 2015;83(9):1553–61.
Trapphoff T, El Hajj N, Zechner U, Haaf T, Eichenlaub-Ritter U. DNA integrity, growth pattern, spindle formation, chromosomal constitution and imprinting patterns of mouse oocytes from vitrified pre-antral follicles. Hum Reprod. 2010;25(12):3025–42.
Jin B, Mazur P. High survival of mouse oocytes/embryos after vitrification without permeating cryoprotectants followed by ultra-rapid warming with an IR laser pulse. Sci Rep. 2015;5:9271.
Martino A, Songsasen N, Leibo S. Development into blastocysts of bovine oocytes cryopreserved by ultra-rapid cooling. Biol Reprod. 1996;54(5):1059–69.
Steponkus P, Myers S, Lynch D, Gardner L, Bronshteyn V, Leibo S, et al. Cryopreservation of Drosophila melanogaster embryos. Nature. 1990;345(6271):170–2.
Kim E-Y, Kim N-H, Yi B, Yoon S, Park S, Chung K, et al. Developmental capacity of bovine follicular oocytes after ultra-rapid freezing by electron microscope grid I. cryopreservation of bovine immature oocytes. Korean J Fertil Steril. 2001;25(1):71–6.
Park S-P, Kim EY, Kim DI, Park NH, Won YS, Yoon SH, et al. Simple, efficient and successful vitrification of bovine blastocysts using electron microscope grids. Hum Reprod. 1999;14(11):2838–43.
Nakashima A, Ino N, Kusumi M, Ohgi S, Ito M, Horikawa T, et al. Optimization of a novel nylon mesh container for human embryo ultrarapid vitrification. Fertil Steril. 2010;93(7):2405–10.
Acknowledgments
The authors thank Silke Andries and Els Merckx for their excellent technical assistance and the local slaughterhouses for their cooperation in sample collection. The authors thank Dr. Bronwen Martin for reading and editing the article. The Leica SP8 (Hercules grant AUHA.15.12) confocal microscope was funded by the Hercules Foundation of the Flemish Government.
Funding
All (co-)authors state that the funding of this research was provided by the independent Operational Costs of the University of Antwerp.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Prof. Luc Leybaert was not included in the author group.
Rights and permissions
About this article

Cite this article
Bus, A., Szymanska, K., Pintelon, I. et al. Preservation of connexin 43 and transzonal projections in isolated bovine pre-antral follicles before and following vitrification. J Assist Reprod Genet 38, 479–492 (2021). https://doi.org/10.1007/s10815-020-01993-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-020-01993-2