
Abstract
Purpose
To assess the role of the genetic background, the culture medium supplements, and the presence of modulators of signaling pathways on mouse embryonic stem cell derivation from single blastomeres from 8-cell embryos.
Methods
Mice from permissive and non-permissive genetic backgrounds, different culture media supplements, knockout serum replacement (KSR) and N2B27, and the presence or absence of 2i treatment were used to derive mouse embryonic stem cells (mESC) from single blastomeres isolated from 8-cell embryos and from control embryos at the blastocyst stage. After the sixth passage, the putative mESC were analyzed by immunofluorescence to assess their pluripotency and, after in vitro differentiation induction, their ability to differentiate into derivatives of the three primary germ layers. Selected mESC lines derived from single blastomeres in the most efficient culture conditions were further characterized to validate their stemness.
Results
In control embryos, high mESC derivation efficiencies (70–96.9%) were obtained from permissive backgrounds or when embryos were cultured in medium complemented with 2i regardless of their genetic background. By contrast, only blastomeres isolated from embryos from permissive background cultured in KSR-containing medium complemented with 2i were moderately successful in the derivation of mESC lines (22.9–24.5%). Moreover, we report for the first time that B6CBAF2 embryos behave as permissive in terms of mESC derivation.
Conclusions
Single blastomeres have higher requirements than whole blastocysts for pluripotency maintenance and mESC derivation. The need for 2i suggests that modulation of signaling pathways to recreate a commitment towards inner cell mass could be essential to efficiently derive mESC from single blastomeres.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Embryonic stem cells (ESC) are pluripotent cells established from preimplantation embryos, which retain their ability to self-renew or differentiate into specific cell types [1]. Due to these properties, ESC have a unique potential for regenerative medicine, disease modeling, and animal engineering [2]. Traditionally, ESC have been derived from the inner cell mass of blastocysts [3,4,5] or from embryos at earlier stages of development [6,7,8]. However, the use of whole embryos to establish ESC in humans entails some controversy due to the need for embryo destruction. To circumvent the ethical concerns, ESC can be alternatively established from single blastomeres [9,10,11,12]. Using this approach, autologous ESC lines could be produced for children born from biopsied embryos in preimplantation genetic testing programs. On the other hand, the separate use of all the blastomeres from an embryo to initiate the ESC derivation process, though still involving embryo destruction, could increase the probability of obtaining an ESC line from a particular embryo [13]. This, in turn, could also lead to a reduction in the number of embryos used for ESC derivation, a relevant point not only for human embryos but also for highly valuable embryos from other mammalian species.
Although ESC lines have been successfully derived from both mouse and human single blastomeres, the derivation efficiencies are typically much lower than those achieved from whole embryos [8, 14,15,16,17]. In the mouse, where the majority of studies have been performed, derivation of ESC from single blastomeres biopsied from embryos at the 2-, 4-, and 8-cell stages indicated that the derivation efficiency gradually declines with increasing embryo developmental age, due to a progressive reduction in cytoplasmic volume and/or increase in the degree of cell fate commitment [14]. The best derivation efficiencies obtained so far from a single blastomere from an 8-cell embryo (1/8 blastomere) cultured under standard conditions have reached only 14% [14]. Therefore, derivation of ESC lines from single blastomeres must be optimized.
ESC derivation is a complex process depending on several factors, which have been extensively studied in the derivation of mouse ESC (mESC) from whole blastocysts [18]. Among them, the genetic background (strain) of the embryos, the supplements added to the culture media and the use of modifiers of signaling pathways seem to play a major role, aside from the use of feeder cells.
According to their ability to yield mESC, mouse strains can be classified into permissive strains, such as 129S2 and C57BL, and non-permissive strains, like CBA, NOD, and FVB [19, 20]. Despite the wide use of hybrid strains like B6D2F1 and B6CBAF1 in procedures such as somatic cell nuclear transfer [21,22,23], these strains have seldom been used for mESC derivation [24,25,26] and their permissiveness for mESC derivation is yet unclear.
Regarding culture media supplements, the addition of serum or a serum substitute is required to efficiently support mESC derivation. Fetal calf serum (FCS) contains growth factors that support self-renewal [19, 26]. However, it may also contain potential differentiation factors and, consequently, only validated batches of FCS can be used for stem cell culture [18, 26]. Knockout serum replacement (KSR), a more defined FCS-free formulation, also presents batch-to-batch variability and needs to be tested [27, 28]. To circumvent this limitation, the protein complex N2B27 has been used as a serum substitute, though it only supports derivation and self-renewal of mESC in the presence of certain signaling modulators. Otherwise, N2B27-supplemented medium induces the differentiation of stem cells to neural precursors [29].
Indeed, the modulation of signaling pathways has also proved to be essential for successful mESC derivation and culture. The first modulator used was leukemia inhibitory factor (LIF), which supports self-renewal and improves mESC maintenance [30, 31]. More recently, the cocktail of inhibitors known as 2i, comprising the glycogen synthase kinase 3 (GSK3) inhibitor CHIR99021 and the mitogen-activated protein kinase (MAPK) kinase (MEK) inhibitor PD0325901, has allowed the derivation of mESC from various mouse strains in several culture media without the need for LIF or feeder cells [32]. Yet, the combination of LIF with 2i and feeder cells has been shown to further enhance mESC derivation rates and clonal expansion [33]. On the other hand, the dual inhibition of MEK by PD0325901 and of transforming growth factor β (TGFβ) by SB431542, known as R2i, has also allowed the derivation of mESC from both permissive and non-permissive strains [34].
Despite the wide knowledge about the conditions to efficiently derive mESC from whole blastocysts, little is known about the impact of these conditions when mESC are established from isolated blastomeres. The majority of studies in which mESC are derived from 1/8 blastomeres use 129S2 inbred or hybrid embryos cultured in medium supplemented with FCS or KSR and in the presence of feeder cells. But, in the absence of signaling modulators other than LIF, the derivation rates are extremely low [9]. Addition of MEK inhibitor or the adrenocorticotropic hormone (ACTH), which supports clonal propagation of mESC [35], led to an increase in derivation efficiencies to 4–14% [11, 14, 16]. Similarly, the combination of LIF and 2i in medium supplemented with KSR resulted in derivation rates of 14% from 1/8 blastomeres from the C57BL permissive strain [8]. Addition of a chimeric E-cadherin to medium supplemented with KSR, LIF, ACTH [13], or the R2i cocktail to N2B27 medium supplemented with LIF [36] has allowed the highest derivation efficiencies obtained so far from 1/8 blastomeres (33.6% and 46–50%, respectively), using either 129S2 × C57BL blastomeres [13] or blastomeres from the non-permissive strains NMRI and BALB [36].
The scarcity of papers dealing with the derivation of mESC from 1/8 blastomeres, as well as the variation in mouse strains, media supplements, and signaling modulators between studies often hinder the comparison of the results obtained. On the other hand, it is not clear whether the requirements for pluripotency maintenance and self-renewal are the same for single blastomeres and whole embryos and whether the effect of the genetic background and culture conditions is similar in both types of samples. In this context, in the present study, we have followed a systematic approach to analyze the impact of the genetic background, culture media supplements, and signaling modulators on the derivation of mESC from 1/8 blastomeres and blastocysts, used as the control group. Specifically, we used embryos from three different strains of mice (permissive hybrid 129B6F1, non-permissive inbred CBA, and hybrid B6CBAF2, of unknown permissiveness), which were cultured in the most common conditions for mESC derivation from whole blastocysts, i.e., medium containing either KSR or N2B27 and in the presence or absence of 2i.
Materials and methods
Feeder cells culture
Because single blastomeres require feeder cells for their proper development into outgrowths [36, 37], in this study, human foreskin fibroblasts (HFF-1; ATCC®SCRC-1041™) were used as feeder cells. HFF-1 were inactivated with 10 μg/ml mitomycin C (Serva) for 3 h to produce feeder cells. The medium used for HFF-1 inactivation and feeder cell culture was Dulbecco’s modified Eagle’s medium (DMEM; BioWest) supplemented with 10% FCS (BioWest).
Feeder cells were cultured on 4-well plates for the derivation of mESC from whole embryos and for stem cell culture maintenance. Instead, feeder cells were cultured in 50 μl microdrops covered with mineral oil (Sigma) in a 60-mm Petri dish for the derivation of mESC from single blastomeres.
Embryo collection
Embryos were collected from 6- to 12-week-old females from three different strains: 129S2 females mated with C57BL males (129B6F1 embryos), B6CBAF1 females mated with males from the same hybrid strain (B6CBAF2 embryos), and CBA females mated with males from the same strain. All the mice were obtained from Charles River Laboratories, except those of the CBA strain that were purchased from Harlan Laboratories. Due to their poor breeding efficiency and an unresponsive nature [38], 129S2 females were mated with C57BL males in order to improve embryo production and quality by hybrid vigor [39].
Prior to mating, females’ superovulation was induced by intraperitoneal injection of 5 IU pregnant mare’s serum gonadotropin (Foligon) followed by the injection of 5 IU human chorionic gonadotropin (hCG; Divasa-Farmavic) 48 h later.
Embryos were collected at the 2-cell stage, 48 h after the hCG injection, by flushing the oviducts with HEPES-buffered CZB medium [40]. They were then cultured in equilibrated KSOMaag Evolve® medium (Zenith Biotech) supplemented with 4 mg/ml bovine serum albumin (BSA; Sigma) at 37 °C and 5% CO2 until the 8-cell or blastocyst stage.
Blastomere isolation
Blastomeres of 8-cell embryos were isolated by micromanipulation in PBS (Sigma) supplemented with 1% BSA. Using a 10-μm-diameter pipette containing acidic Tyrode’s solution [39], the zona pellucida was drilled and blastomeres were individually aspirated with a 20-μm-diameter biopsy pipette.
In order to avoid biased results due to a possible blastomere commitment at the 8-cell stage or to embryo origin, all the blastomeres from each embryo were isolated and blastomeres from different embryos were pooled together and distributed randomly among the different groups before starting the derivation process.
Establishment of embryonic stem cell lines and culture maintenance
Mouse ESC lines were derived using medium containing either KSR or N2B27. The KSR medium consisted of DMEM supplemented with 100 μM 2-β-mercaptoethanol (Gibco), 1× non-essential amino acids (Gibco), 50 U/ml penicillin and 50 μg/ml streptomycin (Gibco), 20% KSR (Life Technologies), and 103 U/ml leukemia inhibitory factor (LIF; Merk Millipore). Since it has been reported that different batches of KSR may result in significantly different derivation efficiencies [27, 28], each batch of KSR was tested prior to use and only those batches yielding derivation efficiencies higher than 65% from 129B6F1 blastocysts were selected. The N2B27 medium was composed of a 1:1 mixture of DMEM-F12 (Gibco) and neurobasal medium plus 100× N2 (Gibco), 50× B27 (Gibco), and 1 mM L-glutamine (Gibco) and was supplemented with 50 μM 2-β-mercaptoethanol, 1× non-essential amino acids, 103 U/ml LIF, 50 U/ml penicillin, and 50 μg/ml streptomycin. When indicated, these media were also supplemented with the 2i inhibitor cocktail [32], a combination of the MEK inhibitor PD0325901 (Axon Medchem; 1 μM) and the GSK3 inhibitor CHIR99021 (Axon Medchem; 3 μM).
Blastocysts were seeded on a monolayer of feeder cells in 4-well plates after removing their zona pellucida with acidic Tyrode’s solution. They were cultured at 37 °C and 5% CO2 in KSR medium or N2B27 medium, with or without 2i treatment, changing the medium every 48 h. Stem cell lines were weekly subcultured and maintained for 6 passages.
Single blastomeres were seeded on a monolayer of feeder cells in 50 μl microdrops of KSR or N2B27 medium with or without the 2i treatment. In all cases, the medium was supplemented with 0.1 mg/ml ACTH (Prospec). Blastomeres were cultured at 37 °C and 5% CO2, changing the medium every 24–48 h for 7–9 days until outgrowths were observed. At the first subculture, outgrowths were seeded on feeder cells in 4-well plates with KSR or N2B27 medium alone or in the presence of 2i. Mouse ESC lines were cultured for six passages at 37 °C and 5% CO2 changing the medium every 48 h and weekly subculturing them.
Immunofluorescence analysis of lines pluripotency
Putative mESC stem cell lines were first selected according to their morphology, choosing only the lines containing colonies with defined edges. At the seventh passage, mESC lines cultured on glass coverslips were fixed and immunostained for the detection of OCT4 and SOX2 pluripotency markers. Some selected lines were also immunostained for NANOG pluripotency marker.
Cells were fixed in 4% paraformaldehyde (Sigma) during 20 min at room temperature (RT) and washed three times with PBS for 5 min/each. Samples were blocked and permeabilized with a PBS solution containing 0.2% sodium azide, 0.5% Triton X-100, and 3% goat serum for 30 min at 37 °C. Cells were incubated with the primary antibodies overnight at 4 °C, washed three times with PBS for 5 min, and incubated with secondary antibodies for 2 h at RT. The primary antibodies were mouse monoclonal anti-OCT4 (Santa Cruz, sc-5279, 1:50 dilution, antibody registry AB_628051), rabbit polyclonal anti-SOX2 (Merck Millipore, AB5603, 1:200 dilution, antibody registry AB_2286686), and rabbit polyclonal anti-NANOG (Abcam, ab80892, 1:100 dilution, antibody registry AB_2150114). The secondary antibodies were Alexa Fluor 488 chicken anti-mouse IgG (Molecular Probes - Invitrogen, A-21200, 1:500 dilution) and Alexa Fluor 594 goat anti-rabbit IgG (Molecular Probes - Invitrogen, A-11037, 1:500 dilution). All the antibodies were diluted in a PBS-based solution containing 0.2% sodium azide (Sigma), 0.1% Triton X-100 (Sigma), and 3% goat serum (BioWest).
After secondary antibody incubation, samples were washed again three times with PBS for 5 min, and the nuclear material was counterstained with 10 μg/ml Hoescht 33258 (Molecular Probes - Invitrogen) diluted in Vectashield (Vector Laboratories). Finally, samples were mounted and analyzed with an epifluorescence microscope (Olympus BX61) and the Cytovision software (Applied Imaging, Inc.).
In vitro differentiation
At the seventh passage, all the derived mESC lines were subjected to in vitro spontaneous differentiation by culturing them in DMEM supplemented with 10% FCS in feeder-free conditions for 10 days. Due to the resistance of mESC lines cultured in presence of 2i to undergo spontaneous differentiation, 2i was removed from the culture medium a week before starting the differentiation process in order to diminish the strong pluripotency signaling and ease colonies differentiation.
Differentiated cells were fixed, immunostained, and analyzed using the same protocol described for pluripotency analysis. Mouse monoclonal anti-Tubulin β 3 (TUJ1; Biolegend, MMS-435P, 1:500 dilution, antibody registry AB_2313773), mouse monoclonal anti-α smooth muscle actin (αSMA; Sigma, A5228, 1:200 dilution, antibody registry AB_262054), and mouse monoclonal anti-alpha-fetoprotein (AFP; R&D Systems, MAB1368, 1:50 dilution, antibody registry AB_357658) antibodies were used to detect ectoderm, mesoderm, and endoderm differentiation markers, respectively. Secondary antibody was Alexa Fluor 488 chicken anti-mouse IgG (Molecular Probes - Invitrogen, A-21200, 1:500 dilution).
Alkaline phosphatase staining and modal karyotype
Alkaline phosphatase (ALP) activity was determined in some selected blastomere-derived lines (three lines per mouse strain) using a two-component buffered alkaline phosphatase substrate containing a 5-bromo-4-chloro-3-indolyl phosphate (BCIP) analogue and nitro blue tetrazolium (NBT) (Sigma). Briefly, mESC colonies were fixed with 4% paraformaldehyde (Sigma) during 1 min at RT, washed with PBS, and incubated with a 1:1 mixture of BCIP and NBT for 10 min at RT. Samples were observed under an inverted microscope (Olympus IX71).
To obtain the modal karyotype, passage 16–18 mESC were arrested at metaphase with 0.02 μg/ml colcemid (Gibco) for 7 h at 37 °C. Then, they were resuspended in a pre-warmed 0.075 M KCl hypotonic solution and incubated for 10 min at 37 °C. Finally, cells were fixed with chilled freshly prepared methanol (Merck)/acetic acid (Merck) (3:1 ratio) and dropped on chilled slides. Chromosomes were counterstained with DAPI (Abbot Molecular) and analyzed with an epifluorescence microscope (Olympus BX61) and the Cytovision software (Applied Imaging, Inc.). The number of chromosomes was counted in approximately 60 metaphases per sample.
RNA extraction and real-time quantitative PCR
The expression of pluripotency genes (Oct4, Nanog, Esrrb, and Tfcp2l1) was assessed by real-time quantitative PCR (qPCR) on some selected blastomere-derived lines (three lines per mouse strain). Commercially available E14 mESC line and STO mouse fibroblasts were used as a positive and negative controls, respectively. For RNA extraction, cells were trypsinized and centrifuged and pellets were washed with PBS. Total RNA was extracted with Maxwell RSC SimplyRNA Tissue Kit (Promega) and its concentration and quality were assessed using a Nanodrop spectrophotometer (ThermoFisher). One microgram of the extracted RNA was used as a template for the reverse transcriptase reaction using the iScript cDNA Synthesis Kit (Bio-Rad). The qPCR reactions were performed in triplicate using iTaq Universal SYBR Green Supermix (Bio-Rad), on a CFX96 Real-Time System thermocycler (Bio-Rad). The amplification program consisted on a first denaturation step of 3 min at 95 °C followed by 40 cycles of 10 s at 95 °C (denaturing) and 30 s at 60 °C (annealing and extension). Finally, to assess the specificity of the amplification product, an additional thermal denaturizing cycle was performed to obtain the melt curve of the qPCR products. Validated PrimePCR SYBR Green Assays (Bio-Rad) for Pou5f1 (Oct4, qMmuCED0046525), Nanog (qMmuCID0005399), Esrrb (qMmuCED0039638), and Tfcp2l1 (qMmuCID0013329) were used to assess pluripotency and Gapdh (qMmuCED0027497) was used to normalize gene expression between samples. A no template control (NTC) was added for each primer. The cycle quantification value (Cq value) was determined for each sample.
Statistical analysis
In the derivation experiments from whole embryos, 30 blastocysts from each mouse strain were used for each treatment and culture condition, with at least three experimental replicates. In the derivation experiments from single blastomeres, the same criteria were applied but every group consisted of a minimum of 140 blastomeres. In both cases, the derivation efficiency was calculated as the number of mESC lines obtained divided by the number of whole blastocysts or isolated blastomeres seeded under the different conditions described. Only the mESC lines that were positive for OCT4 and SOX2 pluripotency markers and that, after in vitro differentiation induction, were also positive for the three differentiation markers analyzed were considered as true mESC lines and were used for the calculation of derivation efficiencies. Results were analyzed with χ2 and Fisher exact test using GraphPad Prism software. p < 0.05 was considered statistically significant.
In the qPCR experiments, normalization of Cq values was performed against the expression of Gadph housekeeping gene and relative expression was calculated using the ΔΔCq method by comparing to E14 control. Results were analyzed with an ANOVA test with the Bonferroni correction using the CFX Maestro software (Bio-Rad). p < 0.05 was considered statistically significant.
Results
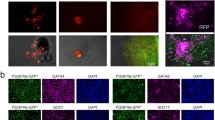
In this study, we generated a total of 345 lines with a stem cell-like morphology after 6 weeks of culture: 233 lines from whole blastocysts (control group) and 112 lines from single 1/8 blastomeres. All the putative mESC lines generated were positive for the pluripotency (OCT4 and SOX2) and differentiation markers (Fig. 1) and were considered true mESC lines, except one line derived from a single B6CBAF2 blastomere cultured in KSR medium. This line was discarded and was not considered in the statistical analyses.

Immunofluorescence detection of pluripotency and differentiation markers in putative mESC lines derived from single blastomeres. a Putative mESC line cultured for six passages, showing a stem cell-like morphology and defined edges. b, c mESC colonies expressing OCT4 (b, green) and SOX2 (c, red). d Morphology of a mESC line after spontaneous in vitro differentiation induction for 10 days. e, f, g Differentiated mESC lines expressing TUJ1 (e, green), αSMA (f, green), and AFP (g, green). In all immunofluorescence images, nuclei are counterstained with Hoechst (blue). The scale bar corresponds to 100 μm
To start the derivation process, 1/8 blastomeres were seeded into culture medium microdrops containing feeder cells (Fig. 2a) and their development was monitored until outgrowths were observed. Blastomeres required 24 h to undergo the first division into 2 cells (Fig. 2b). On the second day of culture, blastomeres had divided into 8–16 cells (Fig. 2c) and, from the third day of culture onward, they attached to the feeder layer and formed a small clump of cells (Fig. 2d). The clump kept growing until forming an outgrowth on day 8, whose size and shape varied depending on the genetic background of the blastomere and the culture conditions.

Division of 129B6F1 single blastomeres. a Single blastomere seeded in KSR medium. b Single blastomere cultured for 24 h in KSR medium and divided into 2 cells. c Single blastomere cultured for 2 days in KSR medium and divided into 8–16 cells. d Single blastomere cultured for 4 days in KSR medium, forming a small cell clump. The scale bar indicates 100 μm
KSR medium does not efficiently support the derivation of mESC lines from 1/8 blastomeres
Results from the control group showed that blastocysts from the permissive strain 129B6F1 cultured in KSR medium yielded 74.3% (26/35) of mESC lines, similar to the 77.4% (24/31) achieved from B6CBAF2 blastocysts (Fig. 3a). Both efficiencies significantly differed from the 46.9% (15/39) of mESC lines derived from non-permissive CBA blastocysts, indicating that B6CBAF2 must be considered a permissive strain in terms of mESC derivation.

Derivation efficiencies of mESC from whole blastocysts (a) and single 1/8 blastomeres (b) in different media in the presence or absence of 2i. Significant differences are denoted with *p ≤ 0.05, **p ≤ 0.01, and ****p ≤ 0.0001 (χ2 and Fisher exact test)
When mESC were established from single blastomeres under the same culture conditions, the derivation efficiencies were extremely lower than those obtained with whole blastocysts. Nevertheless, blastomeres from 129B6F1 and B6CBAF2 strains produced equivalent derivation rates (8/165 = 4.9% and 5/169 = 3.0%, respectively), whereas CBA blastomeres did not produce any mESC line (0/144; Fig. 3b). These results agree with B6CBAF2 being a permissive strain for mESC derivation. They also indicate that KSR medium alone is not suitable for mESC derivation from single 1/8 blastomeres.
Regarding morphology, blastocysts cultured in KSR medium produced outgrowths surrounded by swollen refractive cells (Fig. 4a), whereas outgrowths emerged from 1/8 blastomeres presented a flat shape and defined edges (Fig. 4b). For all strains, mESC lines established from either blastocysts or blastomeres presented a mixture of flattened colonies with defined edges (Fig. 4c) and colonies with peripheral differentiation signs (Fig. 4d), requiring a careful selection in every passage to discard differentiated cells.

Morphology of outgrowths and mESC colonies derived from whole blastocysts and single 1/8 blastomeres in KSR medium without treatment. a Outgrowth from a CBA blastocyst cultured in KSR medium consisting of a clump of pluripotent cells surrounded by swollen refractive cells highlighted with arrowheads. b Outgrowth from a 129B6F1 blastomere cultured in KSR medium. c, d mESC colonies at passage 4 cultured in KSR medium, presenting either defined edges (c) or peripheral differentiation signs, highlighted with dashed ellipses (d). The scale bar corresponds to 100 μm
N2B27 medium impairs the establishment of mESC lines from both whole blastocysts and single blastomeres
As expected, blastocysts cultured in N2B27 medium without treatment resulted in lower derivation efficiencies than those obtained in KSR medium (1/32 = 3.1% for 129B6F1, 3/32 = 9.4% for B6CBAF2, and 1/31 = 3.2% for CBA embryos), with no statistical differences among strains (Fig. 3a). In the case of 1/8 blastomeres, derivation efficiencies were also extremely low irrespectively of the genetic background (7/197 = 3.6% for 129B6F1, 1/144 = 0.7% for B6CBAF2, and 3/162 = 1.9% for CBA embryos) and did not differ from those of their whole blastocysts counterparts, except in the case of the B6CBAF2 strain. Moreover, no significant differences were observed for any of the strains in the rates of mESC derivation between single blastomeres cultured in KSR or N2B27 medium.
As in KSR medium, blastocysts produced outgrowths with peripheral swollen refractive cells (Fig. 5a), whereas the outgrowths produced from single blastomeres were large and presented defined edges, but often showed regions of varying thicknesses (Fig. 5b). Despite the prominent number of blastomere-derived outgrowths initially emerged under this culture condition (from 34.7% for B6CBAF2 blastomeres to 43.2% for 129B6F1 blastomeres), the vast majority of them tended to differentiate during the following passage resulting in a final low mESC derivation rate. The few mESC lines established from blastocysts and blastomeres presented both defined edges and peripheral differentiation signs (Fig. 5c).

Morphology of outgrowths and mESC colonies derived from whole blastocysts and single 1/8 blastomeres in N2B27 medium without treatment. a Outgrowth from a B6CBAF2 blastocyst cultured in N2B27 medium consisting of a clump of pluripotent cells and swollen refractive cells around, highlighted with arrowheads. b Outgrowth from a B6CBAF2 blastomere cultured in N2B27 medium. c mESC colonies at passage 5 cultured in N2B27 medium, presenting defined edges and peripheral differentiation signs, highlighted with dashed ellipses. The scale bar corresponds to 100 μm
Addition of 2i after outgrowth formation is not sufficient to compensate for their massive differentiation in N2B27 medium
To further investigate the origin of the massive differentiation at low passages of outgrowths derived from blastomeres cultured in N2B27 medium without 2i treatment, the biggest outgrowths were split in two with the help of a scalpel before the first passage. One half was maintained in N2B27 medium without treatment while the other half was moved to N2B27 medium with 2i. From a total of 42 129B6F1, 35 B6CBAF2, and 10 CBA outgrowths in this new group, only 5%, 5.7%, and 0%, respectively, progressed to establish a mESC line. These derivation efficiencies were equivalent to those obtained in the non-treated group, suggesting that 2i addition after outgrowth formation is not sufficient to prevent the massive differentiation underwent by outgrowths from isolated blastomeres in N2B27 medium.
Addition of 2i abolishes the effect of the genetic background and medium supplements in mESC derivation from whole blastocysts, but not from single blastomeres
Addition of 2i to KSR medium did not significantly alter derivation rates from 129B6F1 (22/29 = 75.9%) and B6CBAF2 (29/34 = 85.3%) blastocysts. However, it produced a significant increase in the derivation rate from blastocysts from the non-permissive CBA strain (27/31 = 87.1%) when compared with the use of KSR medium alone (Fig. 3a). As a result, derivation rates in KSR medium containing 2i were similar among the three mouse strains used, indicating that the use of 2i is able to compensate for the differences due to the embryos genetic background. This was further confirmed with the addition of 2i to N2B27 medium, which also produced similar rates of mESC derivation regardless of the strain used (28/34 = 82.4% for 129B6F1, 31/32 = 96.9% for B6CBAF2, and 26/31 for CBA embryos; Fig. 3a). Moreover, the significant increase in derivation efficiencies in comparison with the N2B27 medium without treatment and the equivalence with those obtained in KSR medium with 2i indicated that addition of 2i is also able to compensate for medium deficiencies during the derivation of mESC from whole blastocysts.
In sharp contrast, the addition of 2i to KSR medium significantly increased derivation rates from single blastomeres in relation to their non-treated counterparts, but only in the two permissive strains, reaching derivation efficiencies of 24.5% (38/155) for 129B6F1 blastomeres and 22.9% (35/153) for B6CBAF2 blastomeres (Fig. 3b). Even though 3 mESC lines could be established from CBA blastomeres in KSR medium with 2i, the derivation rate (3/162 = 1.9%) was not significantly different than in KSR medium alone and was significantly lower than for blastomeres of the two permissive strains cultured in the same 2i conditions. Thus, contrary to our observations with whole blastocysts, it seems that the use of 2i cannot abolish the strong effect of the genetic background when mESC are derived from single blastomeres.
In a similar way, 2i was unable to compensate for culture medium deficiencies during the derivation of mESC from single blastomeres. Derivation rates in 2i-containing N2B27 medium remained extremely low (5/178 = 2.8% for 129B6F1, 1/143 = 0.7% for B6CBAF2, and 5/162 = 3.1% for CBA embryos; Fig. 3b), and similar to those obtained with their non-treated counterparts. Consequently, derivation rates from single blastomeres cultured in the two different media containing 2i were significantly different for the two permissive strains, while similar for the non-permissive CBA strain.
Contrary to their non-treated counterparts, outgrowths produced from control blastocysts cultured in both media containing 2i presented defined edges (Fig. 6a). Nonetheless, outgrowths derived from single blastomeres differed depending on the medium used. Blastomeres cultured in KSR medium with 2i produced flat outgrowths with defined edges (Fig. 6b, c), which were bigger in the case of permissive (Fig. 6b) than of non-permissive strains (Fig. 6c). Instead, blastomeres cultured in N2B27 medium supplemented with 2i formed two types of outgrowths, flat outgrowths with defined edges (Fig. 6d), which progressed and gave rise to mESC lines, and the more abundant (83.9% to 97.9%) small and extremely flattened outgrowths that presented non-defined edges and vacuolated cells (Fig. 6e) and were non-progressive. As the number of passages increased, mESC colonies cultured in N2B27 medium supplemented with 2i were more dome shaped (Fig. 6f) than colonies observed in KSR medium supplemented with 2i, which showed a uniform morphology with a flat shape and defined edges (Fig. 6g).

Morphology of outgrowths and mESC colonies derived from whole blastocysts and single 1/8 blastomeres in either KSR medium or N2B27 medium with 2i. a Outgrowth from a 129B6F1 blastocyst cultured in N2B27 medium containing 2i presenting defined edges. b Outgrowth from a 129B6F1 blastomere cultured in KSR medium with 2i. c Outgrowth from a CBA blastomere cultured in KSR medium with 2i. d Outgrowth from a B6CBAF2 blastomere cultured in N2B27 medium with 2i. e Non-progressive outgrowth from a CBA blastomere cultured in N2B27 medium with 2i. f mESC colony at passage 6 cultured in N2B27 medium with 2i, presenting defined edges and a dome shape. g mESC colony cultured in KSR medium with 2i (passage 4), showing defined edges and a flat shape. The scale bar indicates 100 μm
Characterization of mESC derived from single blastomeres
To further characterize the pluripotency of the blastomere-derived mESC lines, we selected three lines of each genetic background (hybrid B6CBAF2, non-permissive CBA, and permissive 129B6F1) derived in the most efficient conditions (KSR medium containing 2i) and included the commercially available E14 mESC line (derived from a 129/Ola blastocyst) as a control. ALP activity was detected in 100% of the colonies analyzed (Fig. 7a) and all the lines showed NANOG immunostaining (Fig. 7b). E14 mESC, as well as the three lines established from CBA blastomeres, retained an euploid modal karyotype of 40 acrocentric chromosomes. On the contrary, mESC lines established from 129B6F1 and B6CBAF2 blastomeres displayed aneuploid karyotypes. One B6CBAF2 mESC line had a modal karyotype of 42 chromosomes. The remaining two B6CBAF2 mESC lines and the three 129B6F1 mESC lines had a modal karyotype of 39 chromosomes, though in the four lines the second more represented population had 40 chromosomes. One of the B6CBAF1 lines with a modal karyotype of 39 chromosomes showed a chromosomal fusion in one third of the metaphases analyzed. Regarding the expression of pluripotency genes, equivalent levels of Oct4, Nanog, Esrrb, and Tfcp2l1 were found by qPCR among the blastomere-derived mESC lines of different genetic background and between these lines and the E14 control line, except for significant lower levels of Oct4 expression in 129B6F1 and B6CBAF2 lines than in the E14 control (Fig. 7c).

Extended pluripotency characterization from mESC lines derived from single blastomeres cultured in KSR medium containing 2i. a mESC colonies stained for alkaline phosphatase. b mESC colonies expressing NANOG (red). In both images, the scale bar indicates 100 μm. c Expression of pluripotency markers in blastomere-derived mESC lines measured by qPCR. Gapdh was used for normalization of cDNA amount and expression levels relative to E14 control mESC line are shown. Data represent the mean of three biological and three technical replicates ± SEM. Significant differences among blastomere derived from different genetic backgrounds and between them and the control E14 line are denoted with ***p ≤ 0.001 and ****p ≤ 0.0001 (ANOVA with Bonferroni correction)
Discussion
In this study, we have shown that the genetic background of the embryos, the culture medium supplements, and the presence of modifiers of the activity of signaling pathways do not equally affect the derivation efficiency of mESC established from whole blastocysts and single blastomeres.
Control experiments performed with whole blastocysts from the permissive 129B6F1 and the non-permissive CBA strains confirmed previous observations that the genetic background of the embryos has a profound effect on mESC derivation efficiencies and that this effect can be abolished by the addition of 2i [18, 32]. The differential permissiveness of mouse strains for the establishment of mESC has been recently attributed to their distinct inherent ability to modulate intracellular signaling pathways in response to LIF. Specifically, permissive strains strongly activate the Jak-Stat3 pathway, which promotes pluripotency and, in turn, causes the repression of the mitogen-activated protein kinase (MAPK) pathway, which promotes differentiation. Conversely, non-permissive strains show the opposite activity pattern of signaling pathways [41]. Because of these differences, blastocysts from permissive strains do not require any additional modulation of signaling pathways, other than LIF, to efficiently produce mESC in a supportive medium, like KSR medium. By contrast, blastocysts from non-permissive strains require the additional presence of 2i in order to suppress MAPK signaling and promote pluripotency maintenance and efficient mESC generation. Similarly, 2i is key to efficiently derive mESC from both permissive and non-permissive strains when blastocysts are cultured in a suboptimal medium like N2B27.
With regard to the hybrid B6CBAF2 embryos, here we show, for the first time, that they must be considered a permissive strain in terms of mESC derivation. As B6CBAF1 animals result from a cross between a female from the permissive C57BL strain and a male from the non-permissive CBA strain, this result could suggest that the permissive background is dominant over the non-permissive one. Alternatively, permissiveness to mESC derivation could be determined by the oocyte cytoplasm. However, this second hypothesis seems less probable because the derivation process takes place at the blastocyst stage, when almost all maternal inherited mRNAs are already degraded [42, 43], and after the expression of the paternal genome has already begun at the G2 phase of the 1-cell embryo [44, 45]. Therefore, further experiments including exchanging the origin of the oocytes and spermatozoa between permissive and non-permissive strains would be required to elucidate which mechanism determines the permissiveness of the embryos from hybrid strains.
The effect of the genetic background was also evident during the derivation of mESC from single blastomeres but, contrary to our observations in whole blastocysts, the addition of 2i was not able to suppress this effect or to compensate for medium deficiencies (as in N2B27 medium). Indeed, as we demonstrate in this study, improved mESC derivation from 1/8 blastomeres only occurred when blastomeres from permissive strains were cultured in KSR medium in the presence of 2i. Thus, unlike with whole blastocysts, KSR medium alone is not sufficient to support mESC derivation from single blastomeres, even from permissive strains.
The differential effect of 2i on mESC derivation from single 1/8 blastomeres and whole blastocysts cultured in KSR medium might be related to an effect of the 2i treatment on blastomere fate and their potential to yield mESC. In this sense, it has been reported that ERK, the downstream effector of the MAPK pathway, is expressed only at the apical surface of 8-cell stage mouse embryos, and that Ras-MAPK signaling promotes trophectoderm (TE) differentiation. Inhibition of ERK by the MEK inhibitor PD98059 in 8-cell stage embryos attenuated Cdx2 expression, delayed blastocyst formation, and reduced TE outgrowth from embryo explants, suggesting that Ras-MAPK signaling may have a role in the position-dependent segregation of TE and inner cell mass (ICM) at the 8-cell to morula transition [46]. Similarly, it has been reported that embryos cultured in 2i or in PD0352901 alone exhibited a decrease in TE cell numbers compared with controls [47]. More recently, 2i has been suggested to block the commitment of totipotent blastomeres to embryonic or extraembryonic lineages [48]. According to all these observations, the group of non-treated 1/8 blastomeres in our study could be more prone to TE formation than the group of 2i-treated ones, since the addition of 2i and the consequent inactivation of MEK and ERK could increase the number of 1/8 blastomeres diverted towards an ICM fate or retaining totipotency and potentially capable of generating mESC. In line with diverting blastomeres fate, it has been reported that isolated 1/8 blastomeres cultured in the presence of chimeric E-cadherin molecules yield significantly higher numbers of mESC lines than control blastomeres. The authors hypothesized that the binding of chimeric E-cadherins to the native E-cadherins present on the blastomeres surface mimics the naturally occurring adherens junctions and triggers a signaling pattern equivalent to that induced by the neighboring blastomeres in intact 8-cell embryos [13]. Altogether, the previous and the present results seem to reinforce the idea that mimicking the signaling of the blastomeres committed to become ICM could improve the derivation of mESC lines from isolated blastomeres.
Nevertheless, in contrast to the results with whole blastocysts, the 2i-induced improvement of the derivation efficiency was only observed when blastomeres from permissive strains were cultured in KSR medium. These results indicate that the potential effect of the 2i treatment on blastomere fate described above may not be sufficient to sustain pluripotency and to allow mESC derivation under the suboptimal N2B27 medium culture or in a non-permissive genetic background, which displays an inherent strong activation of MAPK pathway. Recently, Gonzalez and co-workers reported that mESC cultured in KSR medium show an overexpression of genes related with cellular development and a repression of genes related with migration and differentiation, resulting in expression patterns similar to those of the ICM of early blastocysts. These changes were stronger in 2i conditions than in KSR medium alone [49]. Therefore, changes in gene expression induced by components of the KSR medium and the 2i treatment, together with potential changes in blastomere fate induced by MEK inhibition and a suitable genetic background, appear to be key factors for the derivation of mESC from single blastomeres.
In contrast to our failure to efficiently obtain mESC from single blastomeres from the non-permissive CBA strain, Hassani et al. [36] were able to establish around 25% of mESC lines from non-permissive BALB and NMRI 1/8 blastomeres cultured in N2B27 medium with 2i. However, they used an enriched N2B27 medium, containing a higher concentration of N2 (2×) and 5 mg/ml BSA. Although our N2B27 medium was highly effective, when combined with 2i treatment, for the derivation of mESC from whole blastocysts, it was ineffective both for the derivation of mESC from 1/8 blastomeres and for the prevention of the massive differentiation occurring during the second passage. Therefore, it is tempting to speculate that single blastomeres, which are conditioned by the reduction in cytoplasmic volume and isolation, would require an extra enriched medium to grow. On the other hand, in the same study, Hassani and co-workers report the best derivation rates obtained so far (46–50%) from single blastomeres culturing them with R2i [36]. Given these promising results, the use of an enriched N2B27 medium and the addition of R2i could be considered for future studies.
Despite the importance of 2i in mESC derivation from single blastomeres, and from whole blastocysts of non-permissive strains, we observed that the constant presence of 2i during mESC culture hindered their subsequent spontaneous differentiation. Gonzalez and co-workers have recently reported that, after a long culture in 2i, ESC retain the signaling pattern acquired during culture and, therefore, the capacity to self-renew even when they are subjected to differentiation conditions [49]. In view of these observations, 2i should be removed from the culture medium at least 1 week before inducing mESC differentiation. Removing 2i just after establishing mESC lines or at low passages might also help to maintain chromosomal stability, since it has been reported that prolonged inhibition of GSK3 [50] or of MEK [51] induce chromosome instability. Thus, the prolonged culture in 2i could be the cause of the aneuploidy detected in some of our blastomere-derived lines. Alternatively, some lines could be more prone to chromosomal instability regardless of the culture conditions or the culture length, as has been reported by other authors [52, 53].
All in all, single blastomeres seem to be subjected to higher requirements than whole blastocysts to yield mESC lines, and only the combination of blastomeres from permissive strains (including B6CBAF2) cultured in KSR medium containing 2i enabled a significant improvement of mESC derivation rates. Moreover, our results suggest that the modulation of signaling pathways to recreate a commitment towards ICM could be essential to efficiently derive mESC lines from single blastomeres.
References
Biswas A, Hutchins R. Embryonic stem cells. Stem Cells Dev. 2007;16:213–21.
Weinberger L, Ayyash M, Novershtern N, Hanna JH. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat Rev Mol Cell Biol Nature Publishing Group. 2016;17:155–69.
Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–6.
Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. PNAS. 1981;78:7634–8.
Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–7.
Eistetter HR. Pluripotent embryonal stem cell lines can be established from disaggregated mouse morulae. Development. 1989;31:275–82.
Tesar PJ. Derivation of germ-line-competent embryonic stem cell lines from preblastocyst mouse embryos. PNAS. 2005;102:8239–44.
Lee K-H, Chuang C-K, Guo S-F, Tu C-F. Simple and efficient derivation of mouse embryonic stem cell lines using differentiation inhibitors or proliferation stimulators. Stem Cells Dev. 2012;21:373–83.
Delhaise F, Bralion V, Schuurbiers N, Dessy F. Establishment of an embryonic stem cell line from 8-cell stage mouse embryos. Eur J Morphol. 1996;34:237–43.
Klimanskaya I, Chung Y, Becker S, Lu SJ, Lanza R. Human embryonic stem cell lines derived from single blastomeres. Nature. 2006;444:481–5.
Chung Y, Klimanskaya I, Becker S, Marh J, Lu S-J, Johnson J, et al. Embryonic and extraembryonic stem cell lines derived from single mouse blastomeres. Nature. 2006;439:216–9.
Chung Y, Klimanskaya I, Becker S, Li T, Maserati M, Lu SJ, et al. Human embryonic stem cell lines generated without embryo destruction. Cell Stem Cell. 2008;2:113–7.
González S, Ibáñez E, Santaló J. Influence of E-cadherin-mediated cell adhesion on mouse embryonic stem cells derivation from isolated blastomeres. Stem Cell Rev Rep. 2011;7:494–505.
Wakayama S, Hikichi T, Suetsugu R, Sakaide Y, Bui H-T, Mizutani E, et al. Efficient establishment of mouse embryonic stem cell lines from single blastomeres and polar bodies. Stem Cells. 2007;25:986–93.
Lorthongpanich C, Yang SH, Piotrowska-Nitsche K, Parnpai R, Chan AWS. Development of single mouse blastomeres into blastocysts, outgrowths and the establishment of embryonic stem cells. Reproduction. 2008;135:805–13.
González S, Ibáñez E, Santaló J. Establishment of mouse embryonic stem cells from isolated blastomeres and whole embryos using three derivation methods. J Assist Reprod Genet. 2010;27:671–82.
Taei A, Hassani SN, Eftekhari-Yazdi P, Rezazadeh Valojerdi M, Nokhbatolfoghahai M, Masoudi NS, et al. Enhanced generation of human embryonic stem cells from single blastomeres of fair and poor-quality cleavage embryos via inhibition of glycogen synthase kinase b and Rho-associated kinase signaling. Hum Reprod. 2013;28:2661–71.
Czechanski A, Byers C, Greenstein I, Schrode N, Donahue LR, Hadjantonakis A-K, et al. Derivation and characterization of mouse embryonic stem cells from permissive and nonpermissive strains. Nat Protoc. 2014;9:559–74.
Kawase E, Suemori H, Takahashi N, Okazaki K, Hashimoto K, Nakatsuji N. Strain difference in establishment of mouse embryonic stem (ES) cell lines. Int J Dev Biol. 1994;38:385–90.
Brook FA, Gardner RL. The origin and efficient derivation of embryonic stem cells in the mouse. PNAS. 1997;94:5709–12.
Wakayama T, Perry ACF, Zuccotti M, Johnson KR, Yanagimachi R. Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature. 1998;394:369–74.
Gao S, McGarry M, Priddle H, Ferrier T, Gasparrini B, Fletcher J, et al. Effects of donor oocytes and culture conditions on development of cloned mice embryos. Mol Reprod Dev. 2003;66:126–33.
Mallol A, Santaló J, Ibáñez E. Improved development of somatic cell cloned mouse embryos by vitamin C and latrunculin A. PLoS One. 2015;10:e0120033.
Wakayama T, Tabar V, Rodriguez I, Perry ACF, Studer L, Mombaerts P. Differentiation of embryonic stem cell lines generated from adult somatic cells by nuclear transfer. Science. 2001;292:740–3.
Costa-Borges N, Gonzalez S, Santaló J, Ibáñez E. Effect of the enucleation procedure on the reprogramming potential and developmental capacity of mouse cloned embryos treated with valproic acid. Reproduction. 2011;141:789–800.
Lee K-H. Conditions and techniques for mouse embryonic stem cell derivation and culture. In: Bhartiya D, editor. Pluripotent stem cells. InTech; 2013. p. 85–115.
Cheng J, Dutra A, Takesono A, Garrett-Beal L, Schwartzberg PL. Improved generation of C57BL/6J mouse embryonic stem cells in a defined serum-free media. Genesis. 2004;39:100–4.
Chaudhry MA, Vitalis TZ, Bowen BD, Piret JM. Basal medium composition and serum or serum replacement concentration influences on the maintenance of murine embryonic stem cells. Cytotechnology. 2008;58:173–9.
Ying QL, Smith AG. Defined conditions for neural commitment and differentiation. Methods Enzymol. 2003;365:327–41.
Smith AG, Heath JK, Donaldson DD, Wong GG, Moreau J, Stahl M, et al. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature. 1988;336:688–90.
Williams RL, Hilton DJ, Pease S, Willson TA, Stewart CL, Gearing DP, et al. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature. 1988;336:684–7.
Ying Q-L, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, et al. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–23.
Tamm C, Galitó SP, Annerén C. A comparative study of protocols for mouse embryonic stem cell culturing. PLoS One. 2013;8:e81156.
Hassani SN, Totonchi M, Farrokhi A, Taei A, Larijani MR, Gourabi H, et al. Simultaneous suppression of TGF-β and ERK signaling contributes to the highly efficient and reproducible generation of mouse embryonic stem cells from previously considered refractory and non-permissive strains. Stem Cell Rev Rep. 2012;8:472–81.
Ogawa K, Matsui H, Ohtsuka S, Niwa H. A novel mechanism for regulating clonal propagation of mouse ES cells. Genes Cells. 2004;9:471–7.
Hassani S-N, Pakzad M, Asgari B, Taei A, Baharvand H. Suppression of transforming growth factor β signaling promotes ground state pluripotency from single blastomeres. Hum Reprod. 2014;29:1739–48.
Boroviak T, Loos R, Bertone P, Smith A, Nichols J. The ability of inner-cell-mass cells to self-renew as embryonic stem cells is acquired following epiblast specification. Nat Cell Biol. 2014;16:516–28.
Batlle-Morera L, Smith A, Nichols J. Parameters influencing derivation of embryonic stem cells from murine embryos. Genesis. 2008;46:758–67.
Nagy A, Vintersten K, Behringer R. Manipulating the mouse embryo: a laboratory manual. 3rd ed. New York: Cold Spring Harb. Lab Press; 2003.
Chatot CL, Ziomek CA, Bavister BD, Lewis JL, Torres I. An improved culture medium support development of random-bred 1-cell mouse embryos in vitro. J Reprod Fertil Ltd. 1989;86:679–88.
Ohtsuka S, Niwa H. The differential activation of intracellular signaling pathways confers the permissiveness of embryonic stem cell derivation from different mouse strains. Development. 2015;142:431–7.
Bachvarova R, De Leon V. Polyadenylated RNA of mouse ova and loss of maternal RNA in early development. Dev Biol. 1980;74:1–8.
Telford NA, Watson AJ, Schultz GA. Transition from maternal to embryonic control in early mammalian development: a comparison of several species. Mol Reprod Dev. 1990;26:90–100.
Bouniol C, Nguyen E, Debey P. Endogenous transcription occurs at the 1-cell stage in the mouse embryo. Exp Cell Res. 1995;218:57–62.
Aoki F, Worrad DM, Schultz RM. Regulation of transcriptional activity during the first and second cell cycles in the preimplantation mouse embryo. Dev Biol. 1997;181:296–307.
Lu C-W, Yabuuchi A, Chen L, Viswanathan S, Kim K, Daley GQ. Ras-MAPK signaling promotes trophectoderm formation from embryonic stem cells and mouse embryos. Nat Genet. 2008;40:921–6.
Nichols J, Silva J, Roode M, Smith A. Suppression of Erk signalling promotes ground state pluripotency in the mouse embryo. Development. 2009;136(19):3215–22.
Morgani SM, Canham MA, Nichols J, Sharov AA, Portero R, Ko SHM, et al. Totipotent embryonic stem cells arise in ground-state culture conditions. Cell Rep. 2013;3:1945–57.
Gonzalez JM, Morgani SM, Bone RA, Bonderup K, Abelchian S, Brakebusch C, et al. Embryonic stem cell culture conditions support distinct states associated with different developmental stages and potency. Stem Cell Reports. 2016;7:177–91.
Tighe A, Ray-Sinha A, Staples OD, Taylor SS. GSK-3 inhibitors induce chromosomal instability. BMC Cell Biol. 2007;8:34.
Choi J, Huebner AJ, Clement K, Walsh RM, Savol A, Lin K, et al. Prolonged MEK1/2 suppression impairs the developmental potential of embryonic stem cells. Nature. 2017;548:219–23.
Weissbein U, Benvenisty N, Ben-David U. Genome maintenance in pluripotent stem cells. J Cell Biol. 2014;204:153–63.
Gaztelumendi N, Nogues C. Chromosome instability in mouse embryonic stem cells. Sci Rep. 2014;4:1–8.
Acknowledgments
We thank Jonatan Lucas for his technical assistance with feeder cell culture, the staff from Servei Estabulari from Universitat Autònoma de Barcelona for animal care, and Salvador Bartolomé for his assistance and advice in the design of the qPCR experiments.
Funding
This work has been supported by Ministerio de Economia y Competitividad (AGL2014-52408-R) and Generalitat de Catalunya (2014 SGR-524). MVC was beneficiary of a PIF-UAB fellowship and OM is beneficiary of a FI fellowship from the Generalitat de Catalunya.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
Mouse care and procedures were conducted according to the protocols approved by the Ethics Committee on Animal and Human Research of the Universitat Autònoma de Barcelona and by the Departament d’Agricultura, Ramaderia, Pesca I Alimentació of the Generalitat de Catalunya (ref. 8741).
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Vila-Cejudo, M., Massafret, O., Santaló, J. et al. Single blastomeres as a source of mouse embryonic stem cells: effect of genetic background, medium supplements, and signaling modulators on derivation efficiency. J Assist Reprod Genet 36, 99–111 (2019). https://doi.org/10.1007/s10815-018-1360-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-018-1360-9