
Abstract
The red alga Kappaphycus striatus “sacol” brown strain is an economically important species and extensively cultivated at Vanphong Bay, Vietnam. To elucidate the carrageenan quality, the chemical profile of the alkali-modified carrageenan from this alga was investigated by FT-IR, 1H and 13C NMR spectroscopies, and methods of analysis. The structural components of the polysaccharide mainly consisted of 3,6-anhydro-α-D-galactose and β-D-galactose-4-sulfate. FT-IR and 1H and 13C NMR spectra indicated that the polysaccharide was composed predominantly of kappa carrageenan with a small amount of iota carrageenan and 6-O-methylated carrageenan. The red alga K. striatus is good source of carrageenan for application in food or medicine.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Carrageenans are mainly extracted from different species of Rhodophyta cultivated or harvested naturally from the genera Agardhiella, Chondracanthus, Chondrus, Eucheuma, Gigartina, Hypnea, Iridaea, Kappaphycus, Sarconema, and Solieria (Zia et al. 2017). Eucheuma and Kappaphycus are most commonly cultivated across Malaysia and Southeast Asia as a source of carrageenan (Hurtado et al. 2014; Hung et al. 2019). Carrageenans are mainly composed of D-galactose residues linked alternately in 3-linked-β-D-galactopyranose and 4-linked-α-D-galactopyranose units and are classified according to the degree of substitution that occurs on their free hydroxyl groups. There are three main types of carrageenan, which vary in their degree of sulfation. Kappa carrageenan is composed of alternating 3-linked β-D-galactose-4-sulfate and 4-linked 3,6-anhydro-α-D-galactopyranose having one sulfate group per disaccharide repeating unit. Iota carrageenan is composed of alternating 3-linked β-D-galactose-4-sulfate and 4-linked 3,6-anhydro-α-D-galactose-2-sulfate having two sulfate groups per disaccharide repeating unit. Lambda carrageenan has three sulfate groups per disaccharide unit but do not exhibit any 3,6-anhydride bridges contrary to kappa and iota carrageenan. The main differences which effect on the properties of different carrageenans are the number and position of ester sulfate groups and the content of 3,6-anhydro-galactose (Campo et al. 2009). Carrageenans are classically used as agents for the induction of experimental inflammatory pain and have also been shown to have several potential pharmaceutical properties including antitumor, immunomodulatory, antihyperlipidemic, and anticoagulant activities (Campo et al. 2009; Bixler 2017). Recently, carrageenans and their derivatives have been used in controlled drug delivery systems, as well as enhancing dissolution rates of poorly soluble substances (Li et al. 2014; Yegappan et al. 2018).
The red alga, carrageenophyte, Kappaphycus striatus is an economically important food species and extensively cultivated in Vietnam as a source of not only carrageenan but also bioactive compounds for biochemical and medicinal application (Hung et al. 2011, 2015, 2019; Viet et al. 2019; Hung and Trinh 2021). However, little is known about the properties and structure of carrageenans from this alga in Vietnam. Thus, the objective of the present research was to evaluate primarily the properties and structure of kappa carrageenan from the red alga K. striatus “sacol” brown strain cultivated at Vanphong Bay, Vietnam, which will provide more valuable information of the carrageenan group from the red algae, carrageenophytes, for applications in food or medicine.
Materials and methods
Materials
The red alga Kappaphycus striatus “sacol” brown strain (F. Schmitz) Doty ex P. Silva was collected over a cultivation period of 60-day intervals at Vanphong Bay (120°29′ N, 109°10′ E) of Vietnam in 2019, brought to the laboratory, washed with tap water to eliminate sand, debris, and epiphytes, dried by sunlight, and kept at −20°C until used.
Extraction of carrageenan
Alkali-modified carrageenan extraction was carried out according to the method of Ohno et al. (1994). Dry alga (20 g) was incubated in 400 mL 6% KOH at 80°C for 2 h. The algae were then washed and extracted in 400 mL distilled water at 90°C for 2 h. Thereafter, the solution was filtrated and the extract was gellified with 0.2% KCl, frozen overnight and then thawed, washed with fresh water at least twice, and dried at 60°C to constant weight.
Prior to determining the chemical composition and structures, the carrageenan sample was repurified by dialysis using a Spectrapor membrane with Mw cut-off 12,000–14,000 Da, against Milli-Q water for 24 h with frequent changes of water. After dialysis, the sample was filtrated by filter (0.45 μm) and precipitated by absolute ethanol, dried at 60°C, and milled into powder.
3,6-Anhydrogalactose content determination
3,6-Anhydrogalactose content in polysaccharide was determined by the resorcinol method of Yaphe and Arsenault (1965) using D-fructose as a standard. Briefly, 2 mL of each fructose solution at various concentrations, polysaccharide (100 μg mL−1), and distilled water was transferred to a boiling tube and covered with a glass marble. The tubes were placed in an ice bath, and 10 mL of the resorcinol reagent (9 mL of resorcinol 1.36 mM, 1 mL of acetal 2.78 mM, and 100 mL of concentrated HCl) was added to each tube. The contents were mixed and cooled in an ice bath for at least 3 min. The tubes were placed in a 20°C water bath for 4 min and then heated for 10 min at 80°C. It was then cooled for 1.5 min in an ice bath, and the absorbance was measured at 555 nm. 3,6-Anhydrogalactose content was multiplied to a value of 1.087. The experiments were carried out in triplicate and given as average values for each test solution.
Sulfate content determination
Sulfate content was determined according to the method of Terho and Hartiala (1972) using Na2SO4 as a standard. Briefly, 0.5 mL of each Na2SO4 standard solution at various concentrations, polysaccharides (100 μg mL−1), and distilled water was pipetted into test tubes and 2.0 mL of absolute ethanol was added to each tube. One milliliter of BaCl2 buffer (10 mL of acetic acid 2 M, 2 mL of BaCl2 0.005 M, and 8 mL of NaHCO3 0.02 M made up to 100 mL with absolute ethanol) and 1.5 mL of sodium rhodizonate solution (5 mg of rhodizonate natri dissolved in 20 mL distilled water made up to 100 mL with absolute ethanol) were added to each tube and shaken well. The tubes were allowed to stand 10 min in the dark at room temperature, and the absorbance was measured at 520 nm in 30 min. The experiments were carried out in triplicate and are given as average values for each test solution.
Gelling and melting temperature determination
Gelling and melting temperatures were determined according to the method of Craigie and Leigh (1978) using 1.5% carrageenan solution in 0.2% KCl. The gelling temperature was measured by pouring a hot carrageenan solution (10 mL, w/v) into a test tube (diameter 22 mm, height 142 mm). A thermometer (0.1°C divisions) was inserted into the test tube, and then, the solution was allowed to cool at a rate of 0.5°C min−1. Glass beads (3.0 mm) were periodically introduced onto the surface of the solution. The gelling temperature was recorded when the bead failed to sink through the gel. The gelled sample was equilibrated overnight at 20.0°C, and melting temperature was measured by placing a glass bead on the surface of the gel in a test tube clamped in a water bath and warmed at the rate of 0.5°C min−1. Melting temperature was recorded when the glass bead sank to the bottom. The experiments were carried out in triplicate and are given as average values for each test solution.
Viscosity-average molecular weight and gel strength determination
The viscosity-average molecular weight (MW) was obtained from viscometry and extrapolation. The sample was dissolved at an initial concentration of 0.1% in 0.1 M NaCl. Viscosity measurements at different diluted concentrations of carrageenan in 0.1 M NaCl were done at 25 ± 0.1°C in Ubbelohde viscometers. From efflux time of polymer solution (t) and that of solvent 0.1 M NaCl (to), relative viscosity rel = t/to was obtained. Specific viscosity was calculated from the relationship sp = rel − 1. Reduced viscosity for a set of polymer solutions was calculated at different concentrations (g mL−1). Intrinsic viscosity was then obtained from common ordinate intercept on extrapolation of plots of reduced viscosity versus concentration (Pal et al. 2008). MW was calculated according to the Mark–Houwink equation for kappa carrageenan in 0.1 M NaCl at 25°C:
where K = 8.84 × 10−3 and α = 0.86 (Vreeman et al. 1980).
Gel strength was measured according to the method of Ohno et al. (1994). 1.5% solution (1.5 g carrageenan in 100 mL of 0.2% KC1) was heated at 90°C for 30 min. After that, solutions were poured into petri disks (diameter: 94 mm and disk height: 14 mm) and kept at room temperature for 24 h. To measure gel strength, the gel was preincubated at 20°C for approximately 2 h and measured on a rheometer (CR-500DX; Sun Scientific, Japan) with a diameter plunger: 12 mm, gel thickness: 10 mm, and table speed: 60 mm min−1. The assays were carried out in triplicate and are given as average values.
FT-IR spectroscopy
Films were prepared by drying 5 mL (5 mg mL−1) of sample solutions on a polyethylene surface at 60°C, mixed with KBr, pressed into a homogenous pellet and put into the sample holder of the FT-IR spectrometer. The spectral bands were recorded in the range of 4000 to 500 cm−1 on a Bruker mode ALPHA spectrometer.
1H and 13C nuclear magnetic resonance (NMR) spectroscopies
Carrageenan (10 mg) was dissolved in 1 mL of D2O + 1% CD3COOD, and 10 mM of 2,2-dimethyl-2-silapentane-3,3,4,4,5,5-d6-5-sulfonate (DSS) was used as an internal standard. The sample was loaded into a 5 mm O.D. nuclear magnetic resonance tube. The 1H NMR spectrum was recorded at 70°C and operated at 500.24 MHz on a Bruker Avance 500 III spectrometer. Sixty-four scans were accumulated at a pulse angle of 90° with an interpulse delay of 6.50 s and an acquisition time of 2.04 s. The 13C NMR spectrum was measured at 70°C and operated at 125.78 MHz, accumulated over 20,000 scans with an interpulse delay of 6.50 s and an acquisition time of 1.04 s. Chemical shifts were corrected relative to the internal standard of DSS (δ = 0.000 ppm for both 1H and 13C NMR) (Van de Velde et al. 2004).
Results and discussion
Chemical composition and properties of polysaccharide
Carrageenan yield, 3,6-anhydrogalactose and sulfate contents, gelling and melting temperatures, gel strength, and viscosity-average molecular weight of carrageenan are shown in Table 1. The data are in range of those reported for the carrageenans from the genera Kappaphycus and Eucheuma with carrageenan yields from 18.8 to 58.3%, 3,6-anhydrogalactose content from 15 to 40%, ester sulfate from 23.1 to 34.5%, gelling temperature from 32.7 to 34.5°C, melting temperature from 52 to 56°C, molecular weight from 100 to 700 kDa, and gel strength from 505 to 1158 g cm−2 (Santos 1989; Ohno et al. 1996; Mendoza et al. 2002; Hayashi et al. 2007; Hurtado et al. 2008; Periyasamy et al. 2014; Hung et al. 2019).
The differences in the yields, molecular weights, chemical compositions, and qualities between carrageenan samples can be attributed to the extraction methodology used in each study and the time of algal harvest (Periyasamy et al. 2014; Zuldin et al. 2016; Hung et al. 2009, 2019; Viet et al. 2019).
FT-IR analysis of polysaccharide
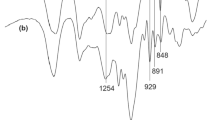
The FT-IR spectral profile of polysaccharide revealed the typical absorption bands related to sulfated polysaccharide of kappa carrageenan (Fig. 1). The absorption bands at 930, 850, and 1260 cm−1 proved the existence of C–O–C of 3,6-anhydro-D-galactose, C4–O–S stretching, and O=S=O symmetric vibrations of β-D-galactose-4-sulfate, respectively, for kappa carrageenans (Santos 1989; Mendoza et al. 2002; Aguilan et al. 2003; Chiovitti et al. 2008; Arman and Qader 2012; Souza et al. 2018). The band at 1640 cm−1 was related to the structural water deformation (Kacurakova and Wilson 2001). The presence of bands at 1159, 1070, 2934, and 3411 cm−1 (Fig. 1) was assigned to the bridge –O stretch, stretch vibrations of C–O, C–H, and O–H existing, respectively (Arman and Qader 2012; Souza et al. 2018).

FT-IR spectrum of carrageenan extracted from Kappaphycus striatus “sacol” brown strain
1H and 13C NMR spectroscopies
The signal assignments of the 1H NMR spectrum (Fig. 2a) of polysaccharide from K. striatus “sacol” brown strain are summarized in Table 2. The signals at 5.10 and 4.64 ppm indicated the α-anomeric protons of 3,6-anhydro-α-D-galactose and β-D-galactose-4-sulfate as reported for kappa carrageenans (Aguilan et al. 2003; Villanueva and Montano 2003; Campo et al. 2009; Cosenza et al. 2014). Additional proton signals at 3.62, 3.98, 4.84, 3.80, and 3.97 ppm of β-D-galactose-4-sulfate and at 4.14, 4.52, 4.53, 4.61, and 4.31 ppm of 3,6-anhydro-α-D-galactose residues matched the 1H NMR assignments for kappa carrageenans (Aguilan et al. 2003; Campo et al. 2009; Cosenza et al. 2014). In addition to the methyl signal, a signal is observed at 3.43 ppm in the 1H NMR spectrum, indicated methylation in the C-6 position of D-galactose-4-sulfate of kappa carrageenans (Mendoza et al. 2002; Aguilan et al. 2003; Villanueva and Montano 2003). Moreover, a weak signal at 5.31 ppm (Fig. 2a) was assigned to the anomeric proton of 4-linked α-D-galactose-6-sulfate residues of the biological precursor to kappa carrageenans (Mendoza et al. 2002; Aguilan et al. 2003). The weak signal at 5.37 ppm was assigned to the α-anomeric proton at C-2 of 3,6-anhydro-α-D-galactose-2-sulfate (Mendoza et al. 2002; Aguilan et al. 2003), indicating the presence of iota carrageenan in K. striatus carrageenan component.

a 1H and b 13C NMR spectra of carrageenan extracted from Kappaphycus striatus “sacol” brown strain
The chemical shifts in the 13C NMR spectrum of polysaccharide (Fig. 2b) corresponded to the signals of the carbon atoms in the disaccharide repeating unit (Table 2) (Aguilan et al. 2003; Chiovitti et al. 2008; Cosenza et al. 2014). The major signals at 102.6 and 95.3 ppm were assigned to the anomeric carbons of β-D-galactose-4-sulfate and 3,6-anhydro-α-D-galactose, respectively (Mendoza et al. 2002; Aguilan et al. 2003; Villanueva and Montano 2003; Chiovitti et al. 2008; Cosenza et al. 2014). On the other hand, the carbon signals at 69.7, 78.9, 74.1, 74.9, and 61.4 ppm of D-galactose-4-sulfate and at 69.9, 79.3, 78.5, 76.9, and 69.4 ppm of 3,6-anhydro-D-galactose corresponded to those of C-2, C-3, C-4, C-5, and C-6 in the 13C NMR spectrum of kappa carrageenans (Aguilan et al. 2003; Chiovitti et al. 2008; Cosenza et al. 2014). A signal at 59.2 ppm is seen in this spectrum, which was assigned to the methoxyl group on C-6 of the β-D-galactose-4-sulfate (Estevez et al. 2000; Mendoza et al. 2002; Aguilan et al. 2003). It has been reported that the carrageenan extracts from Kappaphycus were composed predominantly of kappa carrageenan with trace amounts of iota carrageenan. In fact, the signals at 92.0 and 75 ppm in the 13C NMR spectrum (Fig. 2b) were assigned to the anomeric carbons for C-1 and C-2 of 3,6-anhydro-α-D-galactose-2-sulfate (Mendoza et al. 2002; Aguilan et al. 2003), which showed the presence of iota carrageenan in carrageenan component of this alga. The chemical shift values of NMR spectra in this study were identical to those of kappa carrageenan and are in good agreement with the values reported (Aguilan et al. 2003; Campo et al. 2009).
The polysaccharide from the red alga K. striatus “sacol” brown strain is composed predominantly of kappa carrageenan with a small amount of iota carrageenan. The physicochemical properties and structure of kappa carrageenan were similar to those of carrageenans from the red algae, carrageenophytes, reported, indicating that the red alga K. striatus may promise to be a good source of carrageenans to apply in food or medicine. However, to obtain a better understanding of physicochemical properties and chemical compositions of the red alga K. striatus cultivated, we need further detailed studies.
References
Aguilan J, Broom J, Hemmingson J, Dayrit F, Montano N, Dancel M (2003) Structural analysis of carrageenan from farmed varieties of Philippine seaweed. Bot Mar 46:179–192
Arman M, Qader SAU (2012) Structural analysis of kappa-carrageenan isolated from Hypnea musciformis (red algae) and evaluation as an elicitor of plant defense mechanism. Carbohydr Polym 88:1264–1271
Bixler HJ (2017) The carrageenan controversy. J Appl Phycol 29:2201–2207
Campo VL, Kawano DF, da Silva DB, Carvalho I (2009) Carrageenans: biological properties, chemical modifications and structural analysis - a review. Carbohydr Polym 77:167–180
Chiovitti A, Kraft GT, Bacic A, Craik DJ, Liao ML (2008) Kappa-/beta-carrageenans from Australian red algae of the Acrotylaceae (Gigartinales, Rhodophyta). Phycologia 47:35–40
Cosenza VA, Navarro DA, Fissore EN, Rojas AM, Stortz CA (2014) Chemical and rheological characterization of the carrageenans from Hypnea musciformis (Wulfen) Lamoroux. Carbohydr Polym 102:780–789
Craigie JS, Leigh C (1978) Carrageenans and agars. In: Hellebust JA, Craigie JS (eds) Handbook of phycological methods. Cambrige University Press, Cambrige, pp 109–131
Estevez JM, Ciancia M, Cerezo AS (2000) The system of low-molecular-weight carrageenans and agaroids from the room-temperature-extracted fraction of Kappaphycus alvarezii. Carbohydr Res 325:287–299
Hayashi L, Paula EJD, Chow F (2007) Growth rate and carrageenan analyses in four strains of Kappaphycus alvarezii (Rhodophyta, Gigartinales) farmed in the subtropical waters of São Paulo State, Brazil. J Appl Phycol 19:393–399
Hung LD, Hori K, Nang HQ, Kha T, Hoa LT (2009) Seasonal changes in growth rate, carrageenan yield and lectin content in the red alga Kappaphycus alvarezii cultivated in Camranh Bay, Vietnam. J Appl Phycol 21:265–272
Hung LD, Sato Y, Hori K (2011) High-mannose N-glycan-specific lectins from the red alga Kappaphycus striatum (Carrageenophyte). Phytochemistry 72:855–861
Hung LD, Hirayama M, Ly BM, Hori K (2015) Biological activity, cDNA cloning and primary structure of lectin KSA-2 from the cultivated red alga Kappaphycus striatum (Schmitz) Doty ex Silva. Phytochem Lett 14:99–105
Hung LD, Hoa LT, Hau LN, Trung DT (2019) The lectin accumulation, growth rate, carrageenan yield, and quality from the red alga Kappaphycus striatus cultivated at Camranh Bay, Vietnam. J Appl Phycol 31:1991–1998
Hung LD, Trinh PTH (2021) Structure and anticancer activity of a new lectin from the cultivated red alga, Kappaphycus striatus. J Nat Med 75:223–231
Hurtado AQ, Critchley AT, Trespoey A, Bleicher-Lhonneur G (2008) Growth and carrageenan quality of Kappaphycus striatum var. sacol grown at different stocking densities, duration of culture and depth. J Appl Phycol 20:551–555
Hurtado AQ, Gerung GS, Yasir S, Critchley AT (2014) Cultivation of tropical red seaweeds in the BIMP-EAGA region. J Appl Phycol 26:707–718
Kacurakova M, Wilson R (2001) Developments in mid-infrared FT-IR spectroscopy of selected carbohydrates. Carbohydr Polym 44:291–303
Li L, Ni R, Shao Y, Mao S (2014) Carrageenan and its applications in drug delivery. Carbohydr Polym 103:1–11
Mendoza WC, Montano NE, Ganzon-Fortes ET, Villanueva RD (2002) Chemical and gelling profile of ice-ice infected carrageenan from Kappaphycus striatum (Schmitz) Doty “sacol” strain (Solieriaceae, Gigartinales, Rhodophyta). J Appl Phycol 14:409–418
Ohno M, Largo DB, Ikumoto T (1994) Growth rate, carrageenan yield and gel properties of cultured kappa-carrageenan producing red alga Kappaphycus alvarezii (Doty) Doty in the subtropical waters of Shikoku, Japan. J Appl Phycol 6:1–5
Ohno M, Nang HQ, Hirase S (1996) Cultivation and carrageenan yield and quality of Kappaphycus alvarezii in the waters of Vietnam. J Appl Phycol 8:431–437
Pal S, Sen G, Mishra S, Dey RK, Jha U (2008) Carboxymethyl tamarind: synthesis, characterization and its application as novel drug-delivery agent. J Appl Polym Sci 110:392–400
Periyasamy C, Anantharaman P, Balasubramanian T, Subba Rao PV (2014) Seasonal variation in growth and carrageenan yield in cultivated Kappaphycus alvarezii (Doty) Doty on the coastal waters of Ramanathapuram district, Tamil Nadu. J Appl Phycol 26:803–810
Santos GA (1989) Carrageenans of species of Eucheuma J. Agardh and Kappaphycus Doty (Solieriaceae, Rhodophyta). Aquat Bot 36:55–67
Souza RB, Frota AF, Silva J, Alves C, Neugebauer AZ, Pinteus S, Rodrigues JAG, Cordeiro EMS, de Almeida RR, Pedrosa R, Benevides NMB (2018) In vitro activities of kappa-carrageenan isolated from red marine alga Hypnea musciformis: antimicrobial, anticancer and neuroprotective potential. Int J Biol Macromol 112:1248–1256
Terho TT, Hartiala K (1972) Method for determination of sujphate content of glycosaminoglycans. Anal Biochem 41:471–476
Van de Velde F, Pereirac L, Rollema HS (2004) The revised NMR chemical shift data of carrageenans. Carbohydr Res 339:2309–2313
Viet TNTB, Bach TN, Renou F, Nicolai T (2019) Structure and rheological properties of carrageenans extracted from different red algae species cultivated in Cam Ranh Bay, Vietnam. J Appl Phycol 31:1947–1953
Villanueva RD, Montano MNE (2003) Fine chemical structure of carrageenan from the commercially cultivated Kappaphycus striatum (sacol variety) (Solieriaceae, Gigartinales, Rhodophyta). J Phycol 39:513–518
Vreeman HJ, Snoeren THM, Payens TAJ (1980) Physicochemical investigation of κ-carrageenan in the random state. Biopolym 19:1357–1374
Yaphe W, Arsenault GP (1965) Improved resorcinol reagent for the determination of fructose, and of 3,6-anhydrogalactose in polysaccharides. Anal Biochem 13:143–148
Yegappan R, Selvaprithiviraj V, Amirthalingam S, Jayakumar R (2018) Carrageenan based hydrogels for drug delivery, tissue engineering and wound healing. Carbohydr Polym 198:385–400
Zia KM, Tabasum S, Nasif M, Sultan N, Aslam N, Noreen A, Zuber M (2017) A review on synthesis, properties and applications of natural polymer based carrageenan blends and composites. Int J Biol Macromol 96:282–301
Zuldin WH, Yassir S, Shapawi R (2016) Growth and biochemical composition of Kappaphycus (Rhodophyta) in customized tank culture system. J Appl Phycol 28:2453–2458
Funding
This research was supported by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 106-YS.06-2015.16.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 293 kb)
Rights and permissions
About this article

Cite this article
Hung, L.D., Nguyen, H.T.T. & Trang, V.T.D. Kappa carrageenan from the red alga Kappaphycus striatus cultivated at Vanphong Bay, Vietnam: physicochemical properties and structure. J Appl Phycol 33, 1819–1824 (2021). https://doi.org/10.1007/s10811-021-02415-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-021-02415-1