
Abstract
Saccharina angustata and S. longissima are ecologically and economically important seaweeds. So far, no microsatellite marker was available to S. angustata and S. longissima due to a lack of genomic data. Here, we developed polymorphic simple sequence repeats (SSRs) in S. longissima and S. angustata and applied these markers for further population genetic analysis. Cross-amplification tests showed that 11 SSR loci exhibited high amplification rate in S. angustata and S. longissima. Polymorphic information content values (PIC) of 11 loci showed 10 loci (PIC = 0.552–0.908), except for SJ31 (PIC = 0.482), have high polymorphism (PIC > 0.5). Polymorphism tests indicated that 10 loci have high polymorphism in three varieties of S. japonica and two related species. Genetic diversity analysis confirmed the validation of these markers in accessing the genetic diversity level of these kelps. STRUCTURE and NJ tree results indicated these 10 microsatellites are effective in evaluating the genetic relationship among different kelp populations. As results, these 10 polymorphic microsatellite markers are valid to Saccharina genetic analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Saccharina Stackhouse is mainly distributed along northwestern coasts of the Pacific Ocean and has approximately 20 species (Kawashima 2012; Lane et al. 2006; Yotsukura et al. 2010a). These kelps are the primary producer in the marine ecosystem and constitute the marine forest to provide marine animal habitats (Yotsukura et al. 2010a). In addition, most of these kelps could be used for food, alginate manufacture, and other economical usages.
Saccharina japonica (Areschoug) Lane, Mayes, Druehl, and Saunders is a typical intertidal kelp, and S. religiosa, S. ochotensis, and S. diabolica are regarded as varieties of S. japonica (Yotsukura et al. 2008, 2010b). Saccharina angustata and S. longissima are closely related to S. japonica. Recently, inter-specific hybridization was used for elite cultivar breeding, such as Rongfu, Dongfang No. 2, and Dongfang No. 3 (Li et al. 2007, 2008; Zhang et al. 2007). Nevertheless, there is less knowledge on the genetic background of S. longissima and S. angustata, and it is necessary to develop more efficient markers for genetic study of these kelps.
Simple sequence repeats (SSRs) are 1–6 nucleotide tandem repeats randomly distributed throughout the genome and have become the most popular markers for population genetic studies (Kalia et al. 2011). There are many reports on the SSRs developed for S. japonica (Liu et al. 2010; Li et al. 2016; Peng et al. 2016; Yotsukura et al. 2016; Zhang et al. 2014, 2018); nevertheless, limited SSR loci are available for other Saccharina species. Cross-species amplification of the SSRs has been reported in many seaweeds (Martinez et al. 2005; Liu et al. 2010; Akita et al. 2018), and it could save time and resources to develop SSRs. It is feasible to screen polymorphism SSRs for S. longissima and S. angustata with SSR loci from S. japonica.
To enhance the efficiency of genetic analysis for these kelps, we aimed to screen polymorphism SSRs in S. longissima and S. angustata and assess the suitability of these markers in further genetic analysis.
Materials and methods
Sample collection and DNA extraction
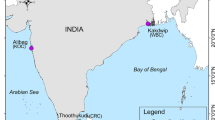
We sampled 10 populations (200 individuals), including three varieties of Saccharina japonica (S. japonica var. religiosa, S. japonica var. ochotensis, and S. japonica var. diabolica) and two related species (S. longissima and S. angustata) from Hokkaido, Japan (Table 1; Fig. 1). We also collected four S. japonica populations from China, Korea, and Russia (Table 1; Fig. 1). All samples were preserved with silica gel. Genomic DNA was extracted as indicated in Zhang et al. (2015). The purity and quality of genomic DNA were checked by the A260/A280 ratio and visualized by gel electrophoresis.

Geographic locations of 10 Saccharina populations used in this study. The abbreviation has been explained in Table 1
SSR loci selection and screening
After primarily screening for the 24 polymorphic microsatellites (Table S1), which were originally from S. japonica (Li et al. 2016; Zhang et al. 2017), we tested these markers on three varieties of S. japonica and two related species (S. longissima and S. angustata). PCR amplification was followed as previously (Zhang et al. 2017). We selected the successful cross-amplification SSR loci to validate polymorphism in 10 populations (200 individuals) of Saccharina (Table 1).
Data analysis
Micro-checker 2.2.3 was applied for checking the potential genotyping errors caused by stuttering or large allele dropout and the presence of null alleles (Van Oosterhout et al. 2004). Linkage disequilibrium (LD) and deviations from Hardy–Weinberg equilibrium (HWE) were estimated for each SSR locus and kelp population using genepop 4.2.2 with 20 batches with 5000 iterations per batch (Raymond and Rousset 1995; Rousset 2008). For polymorphism evaluation of each SSR locus, the number of alleles (NA), the effective number of alleles (NE), the mean observed and expected heterozygosity (HE and HO), and the numbers of private alleles (NP) were calculated using genalex 6.41 (Peakall and Smouse 2006). Polymorphic information content (PIC) was calculated using popgene 1.3.1 (Yeh et al. 1999). Allelic richness (AR) across all loci based on the minimal sample size with the rarefaction method was calculated in fstat 2.9.3.2 (Goudet 1995). Microsatellite data files were converted into the formats for the various analysis software using pgdspider 2.0.1.0 (Lischer and Excoffier 2012) and convert 1.3.1(Glaubitz 2004).
Population genetic differentiation was estimated by calculating the FST in Arlequin 3.5 (Excoffier and Lischer 2010). The significance of the FST value was tested by 10,000 permutations for each pairwise comparison. A hierarchical molecular variance analysis (AMOVA) was conducted to partition the genetic variance using Arlequin 3.5.
The phylogenetic tree was generated based on Nei’s standardized genetic distance Da using the neighbor-joining (NJ) algorithm with 1000 bootstrap replicates in poptree (Takezaki et al. 2010). Tree topologies were viewed and adjusted with figtree 1.4.2 (available at http://tree.bio.ed.ac.uk/software/figtree/). To visualize the pattern of genetic clustering of populations, we used structure 2.3.4 (Pritchard et al. 2000) to assess genetic relatedness of 10 populations. Twenty independent analyses were performed for K = 1–10 based on 1,000,000 Markov chain Monte Carlo iterations following a burn-in period of 500,000 steps. An admixture model was used after assuming correlated allele frequencies among the populations. The best K value was determined by the delta K (ΔK) method (Evanno et al. 2005) in structure harvester (Earl and Vonholdt 2012). The final results were summarized in clumpp 1.1.2 (Jakobsson and Rosenberg 2007) and displayed with distruct 1.1 (Rosenberg 2004).
Results
Evaluation of 11 SSR loci
In the amplification tests, 24 SSR loci exhibited high applicability in three varieties of S. japonica, but 13 SSR loci have a low amplification rate (below 75%) in the two relatives of S. japonica (Table S2). Hence, only 11 SSR loci (SJ3, SJ20, SJ24, SJ31, SJ80, SJ93, SJ101, SJ106, SJ110, SJ133, and SJ167) showed high cross-amplification efficiency to each individual (Table S2). We therefore applied these 11 SSR loci to validate polymorphism for the 10 kelp populations (200 individuals). The percentage of successful amplification (PA) of all loci is above 98.5% in 10 populations (Table 2). microchecker detected no evidence for scoring error due to stuttering, large allele dropout, and null alleles. Linkage disequilibrium (LD) tests for each pair of loci indicated that 11 pairs (3.3%) were significantly in disequilibrium after false discovery rate (FDR) correction. Because these loci did not share corresponding disequilibria in all samples, we speculated that none of the loci were physically linked.
The number of the alleles per locus ranged from 2.8 to 7.8 (Table 2). Polymorphic information content values (PIC) of 11 loci showed SJ31 (PIC = 0.482) has moderate polymorphism (0.25 < PIC < 0.5), and other 10 loci (PIC = 0.552–0.908) has high polymorphism (PIC > 0.5). The allelic richness (AR), number of all alleles (NA), and effective number of all alleles (NE) in SJ167 have lower values; however, PIC and AR indices indicated SJ167 has high polymorphism (PIC = 0.797; AR = 7.136) (Table 2). The successful rate of amplification in SJ20 was 85% in S. angustata; however, SJ20 has higher polymorphism in S. angustata (NA = 20; NE = 13.762; AR = 20) than that in other five populations (NA = 1–8; NE = 1.000–3.791; AR = 1–7.391) (Table S3). In summary, the 11 loci, except for SJ31, have high applicability with high polymorphism (Table 2) and 10 loci were finally screened for futher population genetic analysis (Table 3).
Genetic diversity and population differentiation
Genetic diversity was evaluated for 10 populations (Table S3). At the population level, the mean number of alleles across loci (NA) varied from 2.455 for C (S. japonica populations from China) to 7.455 for SA (S. angustata populations). Allelic richness (AR) based on 17 samples per population was highest (AR = 7.236) in the SA population and lowest (AR = 2.407) in the C population. The mean observed heterozygosity across loci (HO) ranged from 0.173 for SD (S. japonica var. diabolica) to 0.614 for SR (S. japonica var. religiosa) and expected heterozygosity across loci (HE) ranged from 0.175 for SD to 0.668 for SA. We compared the mean values of all genetic diversity indices (NA, AR, HO, and HE) at the population level (Table S3) and found that population SA has higher genetic diversity than other populations. On the contrary, population SD has lower genetic diversity than other populations.
Pairwise FST test exhibited that all the 10 populations have significant genetic differentiation from each other (Table S4; Fig. 2). FST values ranged from 0.087 (SR and K) to 0.732 (R and SD) (Table S4). SD has a deep divergence from other populations (FST > 0.452; P < 0.001). FST values indicated that R from Russia significantly diverged from other populations (except RU) (FST > 0.372; P < 0.001).

The average FST matrix estimated based on SSR loci (lower left). Dark blue and light gray colors indicate high and low FST values, respectively
Genetic structure
STRUCTURE analysis revealed three distinct genetic groups with the highest value of ΔK for K = 3 (Fig. S1; delta K = 3.277) and detected high genetic admixture in several populations (SJ, K, SR, R, and SD) (Fig. 3). Population SD was divided from other populations with K = 4 (delta K = 0.716). Populations RU and R were formed one distinct group with K = 5 (delta K = 2.199). A second highest level of genetic division was identified with K = 6 (delta K = 2.645), indicating that population SJ formed a genetic group and all the genetic groups has little genetic admixture (Fig. 3). NJ tree showed five genetic clades: clade 1 containing populations C and SJ; clade 2 including populations K and SR; clade 3 containing populations SO, RU, and R; clade 4 including population SD; and clade 5 containing populations SL and SA (Fig. 3).

Genetic structure based on STRUCTURE analysis and neighbor-joining tree
AMOVA analysis showed that only 14.55% of the variation occurred among three STRUCTURE groups and as much as 56.43% of the variation occurred within populations (Table S5). In addition, the majority of variation was partitioned within populations (58.34%), with only 20.80% of the variation among the six STRUCTURE groups (Tables S6). All variance components were statistically significant (P < 0.0001).
Discussion
It is proved that the screened 10 universal and polymorphism microsatellites for three varieties of S. japonica, S. longissima, and S. angustata are valid in accessing the genetic diversity. Saccharina angustata population has higher genetic diversity than other kelp populations (Table S3), and it would be potential genetic germplasm for breeding and cultivation. Saccharina japonica var. diabolica is an important cultivar in Japan due to its wide blade (Kawashima 2012), but exhibited lower genetic diversity (Table S3). We infer that selection and cultivation might reduce allelic variation and genetic diversity, and this phenomenon also has been observed in other cultivated seaweeds (Voisin et al. 2005; Zhang et al. 2017). Taxonomically, S. japonica var. diabolica is a variety of S. japonica (Yotsukura et al. 2010b); however, it has deep divergence from S. japonica with a separate genetic lineage (Fig. 2; Table S4).
Based on the NJ tree (Fig. 3), S. longissima and S. angustata formed a distinct genetic cluster and S. japonica and its three varieties clustered together, which was consistent with a previous result with 5S rDNA spacer (Yotsukura et al. 2010c). The wild kelp population from Dalian (C) and S. japonica from Hokkaido (SJ) clustered together and have higher genetic similarity, possibly due to artificial introduction during the cultivation. Likewise, population from Korea (K) and S. japonica var. religiosa from Hokkaido has closer genetic relationship (Fig. 3), which supports the previous assumption that Korean kelp populations might be introduced from Japan (Jang and Gweon 1970; Hwang et al. 2018). Here, the kelp populations from Russia might be the S. japonica var. ochotensis based on the NJ tree (Fig. 3) and genetic differentiation analyses (Fig. 2). These SSR loci exhibited high resolution for discrimination of genetic relatedness between different kelp populations.
In conclusion, the newly screened 10 polymorphic microsatellite markers from S. japonica exhibited high applicability in the two related Saccharina species and are valid in the population genetic analysis. It will be positive to evaluate the genetic background to these kelps in the future.
References
Akita S, Koiwai K, Hanyuda T, Kato S, Nozaki R, Uchino T, Sakamoto T, Kondo H, Hirono I, Fujita D (2018) Development of 11 Ecklonia radicosa (Phaeophyceae, Laminariales) SSRs markers using next-generation sequencing and intra-genus amplification analysis. J Appl Phycol 30:2111–2115
Earl DA, Vonholdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Glaubitz JC (2004) CONVERT: a user-friendly program to reformat diploid genotypic data for commonly used population genetic software packages. Mol Ecol Notes 4:309–310
Goudet J (1995) FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Hwang EK, Ha DS, Park CS (2018) The influences of temperature and irradiance on thallus length of Saccharina japonica (Phaeophyta) during the early stages of cultivation. J Appl Phycol 30:2875–2882
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jang JW, Gweon SH (1970) Studies on Laminaria cultivation. Report of the National Fisheries Research and Development Agency. Vol. 5. National Institute of Fisheries Science, Busan, pp. 63–74
Kalia RK, Rai MK, Kalia S, Singh R, Dhawan AK (2011) Microsatellite markers: an overview of the recent progress in plants. Euphytica 177:309–334
Kawashima S (2012) Morphology and taxonomy of the laminariaceous algae in cold water area of Japan (in Japanese). Oya Nisan Publisher, Tokyo, pp 159–206
Lane CE, Mayes C, Druehl LD, Saunders GW (2006) A multi-gene molecular investigation of the kelp (Laminariales, Phaeophyceae) supports substantial taxonomic re-organization. J Phycol 42:493–512
Li XJ, Cong YZ, Yang GP, Shi YY, Qu SC, Li ZL, Wang GW, Zhang ZZ, Luo SJ, Dai HL, Xie JZ, Jiang GL, Liu JL, Wang TY (2007) Trait evaluation and trial cultivation of Dongfang No. 2, the hybrid of a male gametophyte clone of Laminaria longissima (Laminariales, Phaeophyta) and a female one of L. japonica. J Appl Phycol 19:139–151
Li XJ, Liu JL, Cong YZ, Qu SC, Zhang ZZ, Dai HL, Luo SJ, Han XB, Huang SS, Wang QY, Liang GJ, Sun J, Jin Y, Wang D, Yang GP (2008) Breeding and trial cultivation of Dongfang No. 3, a hybrid of Laminaria gametophyte clones with a more than intraspecific but less than interspecific relationship. Aquaculture 280:76–80
Li QY, Zhang J, Yao JT, Wang XL, Duan DL (2016) Development of Saccharina japonica genomic SSR markers using next-generation sequencing. J Appl Phycol 28:1387–1390
Lischer HEL, Excoffier L (2012) PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28:298–299
Liu FL, Wang XL, Yao JT, Fu WD, Duan DL (2010) Development of expressed sequence tag-derived microsatellite markers for Saccharina (Laminaria) japonica. J Appl Phycol 22:109–111
Martinez EA, Cardenas L, Figueroa C, Vidal RU, Billot C (2005) Microsatellites of Laminaria digitata tested in Lessonia nigrescens: evaluation and improvement of cross amplification between kelps of two different families. J Appl Phycol 17:245–253
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Peng J, Zhang LN, Li XJ, Cui CJ, Wu RN, Tian PP, Li Y, Liu YL (2016) Development of genic SSR markers from an assembled Saccharina japonica genome. J Appl Phycol 28:2479–2484
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Raymond M, Rousset F (1995) GENEPOP (version-1.2) – population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106
Takezaki N, Nei M, Tamura K (2010) POPTREE2: software for constructing population trees from allele frequency data and computing other population statistics with Windows interface. Mol Biol Evol 27:747–752
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Voisin M, Engel CR, Viard F (2005) Differential shuffling of native genetic diversity across introduced regions in a brown alga: aquaculture vs. maritime traffic effects. Proc Natl Acad Sci U S A 102:5432–5437
Yeh FC, Yang RC, Boyle T (1999) POPGENE (version 1.31): Microsoft window-bases freeware for population genetic analysis. University of Alberta and the Centre for International Forestry Research
Yotsukura N, Kawashima S, Kawai T, Abe T, Druehl LD (2008) A systematic re-examination of four Laminaria species: L. japonica, L. religiosa, L. ochotensis and L. diabolica. J Jpn Bot 83:165–176
Yotsukura N, Nagai K, Kimura H, Morimoto K (2010a) Seasonal changes in proteomic profiles of Japanese kelp: Saccharina japonica (Laminariales, Phaeophyceae). J Appl Phycol 22:443–451
Yotsukura N, Shimizu T, Katayama T, Druehl LD (2010b) Mitochondrial DNA sequence variation of four Saccharina species (Laminariales, Phaeophyceae) growing in Japan. J Appl Phycol 22:243–251
Yotsukura N, Kawai T, Kawashima S, Ebata H, Ichimura T (2010c) Nucleotide sequence diversity of the 5s rDNA spacer in the simple blade kelp genera Laminaria, Cymathaere and Kjellmaniella (Laminariales, Phaeophyceae) from northern Japan. Phycol Res 54:269–279
Yotsukura N, Maeda T, Abe T, Nakaoka M, Kawai T (2016) Genetic differences among varieties of Saccharina japonica in northern Japan as determined by AFLP and SSR analyses. J Appl Phycol 28:3043–3055
Zhang QS, Tang XX, Cong YZ, Qu SC, Luo SJ, Yang GP (2007) Breeding of an elite Laminaria variety 90-1 through inter-specific gametophyte crossing. J Appl Phycol 19:303–311
Zhang LN, Peng J, Li XJ, Liu YL, Cui CJ, Wu H, Wu RN, Tian PP, Li Y (2014) Development of 27 trinucleotide microsatellite markers for Saccharina japonica using next generation sequencing technology. Conserv Genet Resour 6:341–344
Zhang J, Yao JT, Sun ZM, Fu G, Galanin DA, Nagasato C, Motomura T, Hu ZM, Duan DL (2015) Phylogeographic data revealed shallow genetic structure in the kelp Saccharina japonica (Laminariales, Phaeophyta). BMC Evol Biol 15:237
Zhang J, Wang XL, Yao JT, Li QY, Liu FL, Yotsukura N, Krupnova TN, Duan DL (2017) Effect of domestication on the genetic diversity and structure of Saccharina japonica populations in China. Sci Rep 7:42158
Zhang J, Liu T, Rui FP (2018) Development of EST-SSR markers derived from transcriptome of Saccharina japonica and their application in genetic diversity analysis. J Appl Phycol 30:2101–2109
Acknowledgements
This study was supported by the Marine Scientific and Technological Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) ( 2018SDKJ0502-1), National Natural Science Foundation of China (31772848 and 31272660), and the Shandong Key Sci-Technology Research Project (2016ZDJS06B2).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 56 kb)
Rights and permissions
About this article

Cite this article
Zhang, J., Wang, X., Yao, J. et al. Screening of polymorphic microsatellites and their application for Saccharina angustata and Saccharina longissima population genetic analysis. J Appl Phycol 31, 3295–3301 (2019). https://doi.org/10.1007/s10811-019-01798-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-019-01798-6