
Abstract
Truncating variants of the MAGEL2 gene, one of the protein-coding genes within the Prader-Willi syndrome (PWS) critical region on chromosome 15q11, cause Schaaf-Yang syndrome (SYS)—a neurodevelopmental disorder that shares several clinical features with PWS. The current study sought to characterize the neurobehavioral phenotype of SYS in a sample of 9 patients with molecularly-confirmed SYS. Participants received an assessment of developmental/intellectual functioning, adaptive functioning, autism symptomatology, and behavioral/emotional functioning. Compared to individuals with PWS, patients with SYS manifested more severe cognitive deficits, no obsessions or compulsions, and increased rates of autism spectrum disorder.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Paternal genes on chromosome 15q11-q13 have been indicated in the development of Prader-Willi syndrome (PWS; Cassidy et al. 2012). PWS is characterized by severe hypotonia, feeding difficulties, failure to thrive, and developmental delays in infancy (Holm et al. 1993). As children with PWS age, common neurobehavioral presentations of the syndrome include: intellectual disability, stubbornness, temper tantrums, as well as manipulative and compulsive behaviors (Dykens et al. 2011; Rice and Einfeld 2015).
Recent research on truncating variants on the paternal allele of the MAGEL2 gene, housed within the PWS-critical region of chromosome 15q11-q13, has led to the discovery of a genetic syndrome similar to PWS (Fountain et al. 2016; Mejlachowicz et al. 2015; Schaaf et al. 2013; Soden et al. 2014) that was initially referred to as “Prader-Willi-like syndrome.” However, differences in phenotypes between individuals with PWS and individuals with truncating variants of the paternal allele of the MAGEL2 gene on chromosome 15q11 have led to classification of a distinct syndrome, named Schaaf-Yang syndrome (OMIM 615547). While the two syndromes present with genetic variation in the same region of chromosome 15q11-q13, PWS is the result of paternal deletion of the entire 15q11-q13 locus or maternal uniparental disomy. In contrast, Schaaf-Yang syndrome is the result of truncating point mutations on the paternal allele of a single gene (MAGEL2) on chromosome 15q11. PWS and Schaaf-Yang syndrome share several clinical phenotypes, particularly in infancy, including: neonatal hypotonia, feeding difficulties, and hypogonadism (Fountain et al. 2016; Fountain and Schaaf 2016). However, only 35–50% of individuals with Schaaf-Yang syndrome go on to develop hyperphagia and associated clinical obesity, which are characteristic features of PWS (Fountain et al. 2016; Fountain and Schaaf 2016). A majority of individuals with Schaaf-Yang syndrome present with joint contractures, which are rarely observed in children with PWS (Fountain et al. 2016; Fountain and Schaaf 2016). With regard to cognitive and behavioral phenotypes, PWS and Schaaf-Yang syndrome again show some overlap, as both syndromes present with developmental delay, intellectual disability, as well as stubborn, compulsive and manipulative behaviors (Dykens et al. 2011; Rice and Einfeld 2015; Fountain et al. 2016). Perhaps the most striking phenotypic difference between PWS and Schaaf-Yang syndrome is the difference in prevalence rates of autism spectrum disorder (ASD) between the two syndromes. The prevalence of ASD in children with PWS is approximately 12.3–27% (Bennett et al. 2015; Dykens et al. 2017). In contrast, one study reported all four individuals with molecularly confirmed Schaaf-Yang syndrome met DSM-IV-TR criteria for a clinical diagnosis of ASD (Schaaf et al. 2013). Additionally, 10 of 13 patients in another study carried a diagnosis of ASD (Fountain et al. 2016). However, neither of these studies included a rigorous, systematic assessment of ASD in this cohort.
The primary goal of the current study was to characterize the neurocognitive and neurobehavioral phenotype of this disorder. A second goal was to examine the distinctiveness of Schaaf-Yang syndrome from what has been described in previous research about individuals with PWS, given that the genes responsible for each disorder are located in the same region (i.e., chromosome 15q11-q13). In the current study, a sample of 9 patients with molecularly confirmed Schaaf-Yang syndrome were recruited from a clinical database. Participants received a comprehensive neurocognitive evaluation, including assessment of developmental/intellectual functioning, adaptive functioning, autism symptomatology, and parent-reported behavioral/emotional functioning.
Methods
Participants
Since the syndrome’s initial description in 2013, multiple families of children with molecularly confirmed Schaaf-Yang syndrome have contacted the senior author for inclusion in the Schaaf-Yang syndrome registry in order to be informed of upcoming research studies. Eligible individuals from this registry were contacted via phone and/or email by the research coordinator to inform them of the study; families were contacted up to two times about this opportunity. Inclusion criteria were: (1) previously identified truncating mutation in the paternal allele of MAGEL2; (2) age 5–17 years at enrollment, because participation in the study required endocrine testing [results of which have been reported separately (McCarthy et al. 2018)] and normal ranges were not established for some tests for children under the age of five; (3) reside in the United States; and (4) English speaking. Exclusion criteria included: (1) dependence on mechanical ventilation; (2) inability to travel to Texas Children’s Hospital; and (3) history of not tolerating fasting for up to seven hours for the required endocrine testing. The study was approved by the Institutional Review Board at Baylor College of Medicine. A total of 9 patients (56% female) consented to participate in this study out of a possible 19 known individuals with Schaaf-Yang syndrome at the time of recruitment who were living in the United States. Their average age was 10.3 years old (SD = 3.9 years), with a range from 5 to 17 years of age.
Materials
Cognitive and psychological assessments administered as a part of this research study included: Stanford-Binet Intelligence Scales, 5th Edition (SB-5; Roid 2003), Autism Diagnostic Observation Schedule, 2nd Edition (ADOS-2; Lord et al. 2011; 6 participants received Module 1 and 3 participants received Module 3), and Autism Diagnostic Interview—Revised (ADI-R; Rutter et al. 2008). Additionally, parents were asked to complete validated questionnaires to further assess behavioral functioning across multiple areas, including the Social Responsiveness Scale—Second Edition (SRS-2; Constantino and Gruber 2012), Yale-Brown Obsessive Compulsive Scale (Y-BOCS; Goodman et al. 1989), Behavior Assessment System for Children—Third Edition (BASC-3; Reynolds and Kamphaus 2015), and the Adaptive Behavior Assessment System, Third Edition (ABAS-3; Harrison and Oakland 2015). Participants who were unable to complete the SB-5 (n = 3) because of functional limitations were instead administered the Mullen Scales of Early Learning (MSEL; Mullen 1995), which was the most appropriate test given their developmental level. No standard score could be calculated for participants who completed the MSEL (Participants 1, 2, and 7) because they were outside of the normative age range. As such, a ratio IQ [i.e., (mental age ÷ chronological age) * 100] was obtained for all participants based on each test’s provided age equivalents.
Procedure
Consent forms were mailed to interested families 3–4 days ahead of their scheduled research visit for their review. Parents provided written consent in person at the onset of the initial appointment, following a review of the study and participation requirements and had the opportunity to ask questions with the study physician or research coordinator. Following consent, families participated in two, half-day, comprehensive medical and behavioral assessments by a team of providers at Texas Children’s Hospital. Participants were administered the previously described battery of standardized cognitive and psychological assessments, administered by psychologists and licensed psychological associates who were research reliable on the ADOS-2 and ADI-R.
Within 3 months of the conclusion of participation, families received a brief letter summarizing the findings of the neurocognitive testing, including test scores and whether participants met criteria for intellectual disability and/or ASD.
Results
Eight participants had complete evaluation data for the ADOS-2, ADI-R, BASC-3, ABAS-3, SRS-2, and Y-BOCS. Cognitive testing could not be completed with one participant due to issues with compliance. Key results are presented in Table 1.
Developmental History
Based on reports during the ADI-R, 8 of 9 parents indicated concerns regarding their child’s development from birth, while the remaining participant’s parents endorsed concerns at 1 year of age (i.e., delayed crawling). Common responses from the parents who reported concerns beginning at birth (n = 8) included abnormal/minimal crying (n = 8), hand contractures (n = 7), low muscle tone (n = 3), respiratory difficulties/cyanosis (n = 3), poor oral intake (n = 2), poor skin color (n = 2), and unstable temperature (n = 2). At the time they were evaluated for this study, four participants had not yet begun walking independently. For those who were walking independently, onset occurred between 2 years, 9 months and 7 years of age. Two participants had not yet spoken their first words at the time of evaluation for this study. For those who had, first words occurred between 2 and 12 years of age. No parents reported a loss in skills at any time.
Intellectual Ability
Intellectual ability was measured with the SB-5 for five participants and with the MSEL for three participants. The average nonverbal ratio IQ (NVRIQ) score across the entire sample of cognitive data (n = 8) was 30.6 (SD = 17.4; range = 4.0–58.1). The average verbal ratio IQ (VRIQ) score across the sample with available cognitive data was 23.9 (SD = 18.1; range = 1.0–51.6). The average full scale ratio IQ (FSRIQ) score across the participants with cognitive data was 27.0 (SD = 17.4; range = 3.0–53.5).
For the participants who were able to complete the SB-5 (n = 5), (Fig. 1) presents the histograms of the IQ domains, with a superimposed curve of the normal distribution (M = 100; SD = 15). The average nonverbal deviation IQ (NVIQ) for this subsample was 54.2 (SD = 16.1; range = 42–78), while the average verbal deviation IQ (VIQ) for the subsample was 52.6 (SD = 14.9; range = 43–77). The average full scale deviation IQ (FSIQ) for this subsample was 51.0 (SD = 15.9; range = 40–76).

Histograms of the Stanford Binet, 5th Edition (SB-5) composite scores with a curve of the normative scores using a normal distribution with a mean of 100 and standard deviation of 15 for nonverbal deviation IQ (NVIQ), verbal deviation IQ (VIQ), and full scale deviation IQ (FSIQ)
Intellectual disability (ID) diagnoses were made for all participants according to DSM-5 criteria, based on intellectual assessment data (n = 8), parent ratings of adaptive functioning (n = 9; results reported below) and information obtained via clinical interview (n = 9). For the participant with missing cognitive data due to noncompliance, diagnosis was made based on parent ratings of adaptive functioning, information obtained via clinical interview, and clinical observations of the child’s level of functioning during the evaluation. A majority of participants (4 of 9) presented with severe to profound ID, while 2 of 9 presented with moderate ID and 2 of 9 participants presented with mild ID. Of note, the one participant who was unable to complete a measure of intellectual/developmental functioning likely qualified for a diagnosis of moderate ID based on diagnostic interview, clinical observations, and parent ratings of adaptive functioning.
Behavioral Phenotypes
Behavioral phenotypes of the sample were assessed using the Y-BOCS, BASC-3, and ABAS-3. None of the participants presented with significant ratings on the Y-BOCS (7 of 9 participants reported zero symptoms on the Y-BOCS), revealing an absence of comorbid obsessions and compulsions in this sample.
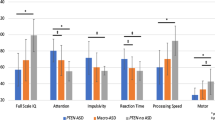
On the BASC-3 clinical scales [left side of Fig. 2], the average Internalizing T score for the sample was 47.4 (SD = 5.3; range = 39–56), which falls within the normative average. The average Externalizing T score for the sample was 59.0 (SD = 16.4, range = 38–80), which falls within the normative average. However, 1 of 9 participants (11%) had scores within the “At-Risk” range and 3 of 9 participants (33%) had scores in the “Clinically Significant” range on the Externalizing domain. The average Behavioral Symptoms T score for the sample was 65.7 (SD = 12.7, range = 48–81), which is higher than the normative average, with 2 of 9 participants (22%) having scores within the “At-Risk” range and 4 of 9 participants (44%) with scores in the “Clinically Significant” range. The sample had elevated average scores (ranging from at-risk to clinically significant) relative to the normative sample on the Hyperactivity (M = 62.8; SD = 16.9; range = 40–84), Attention Problems (M = 64.6; SD = 8.0; range = 54–77), Atypicality (M = 75.2; SD = 17.6; range = 48–105), and Withdrawal (M = 66.9; SD = 4.6; range = 62–74) subscales. On the BASC-3 adaptive scales [right side of Fig. 2] the average Adaptive Skills T score for the sample was 26.9 (SD = 6.4, range = 19–40), which is well-below the normative average. Three of 9 participants (33%) had Adaptive Skills T scores within the “At-Risk” range and 6 of 9 participants (67%) with scores in the “Clinically Significant” range. Participants had decreased scores (ranging from at-risk to clinically significant) relative to the normative sample on the Social Skills (M = 28.7; SD = 8.6; range = 19–44), Leadership (M = 28.1; SD = 4.5; range = 22–33), Activities of Daily Living (M = 25.1; SD = 7.6; range = 16–39), and Functional Communication (M = 24.9; SD = 8.9; range = 10–41) subscales, while the mean score for the sample on Adaptability was within the normative range (M = 44.0; SD = 8.8; range = 34–60). The average scores for Atypicality, Social Skills, Leadership, Activities of Daily Living, and Functional Communication were all clinically significantly different from the normative population (above 70 for Atypicality and below 30 for Social Skills, Leadership, Activities of Daily Living, and Functional Communication), representing particular clinical deficits in the Schaaf-Yang cohort. In addition, while the elevations on the Hyperactivity, Attention Problems, and Withdrawal scales did not reach clinical significance, these represent subclinical deficits that may reach clinical significance with a larger sample size.

Behavior Assessment Scale for Children, Third Edition (BASC-3) average clinical and adaptive subscale T scores. Average normative score of 50 is indicated by the black line. Clinical and adaptive subscales are delineated by vertical dotted line
All ABAS-3 average scores were significantly below the comparable normative averages, indicating clinically significant adaptive-functioning deficits in the Schaaf-Yang cohort. Figure 3 presents histograms of each ABAS-3 domain with a superimposed curve of the normal distribution (M = 100; SD = 15). The average General Adaptive Composite (GAC) score was 54.1 (SD = 10.7; range = 44 to 77)—well-below the normative average of 100, with 8 of 9 scores (89%) falling in the “Extremely Low” range and 1 of 9 scores (11%) falling in the “Low” range. The average score on the Conceptual Composite was 57.3 (SD = 11.8; range = 47–78)—again, well-below the normative average of 100, with 7 of 9 scores (78%) falling in the “Extremely Low” range and 2 of 9 scores (22%) falling in the “Low” range. The average score on the Social Composite was 60.4 (SD = 7.8; range = 51–70), which is well-below the normative average of 100, with 9 of 9 scores (100%) falling in the “Extremely Low” range. The average score on the Practical Composite was 55.9 (SD = 11.1; range = 48–83), which is well-below the normative average of 100, with 8 of 9 scores (89%) falling in the “Extremely Low” range and 1 of 9 scores (11%) falling in the “Below Average” range.

Histograms of the Adaptive Behavior Assessment System, Third Edition (ABAS-3) composite scores with a curve of the normative scores using a normal distribution with a mean of 100 and standard deviation of 15
ASD Symptomatology
ASD symptoms were assessed via parent ratings on the SRS-2, parent report during the ADI-R, clinical evaluation on the ADOS-2, and expert clinical judgment. On the SRS-2, all participants presented with elevated symptoms on the Total Score (M = 81.4; SD = 10.8) and Social Communication and Interaction domain (M = 80.6; SD = 10.8), while 8 of 9 participants presented with elevated symptoms on the Restricted Interests and Repetitive Behavior domain (M = 83.2; SD = 11.6). On the ADOS-2, all participants’ scores were consistent with an ADOS-2 classification of Autism. Comparison scores for the entire sample were ≥6, indicating moderate to severe clinical presentation for all participants.
On the ADI-R, the diagnostic algorithm could not be reliably completed for one participant (Participant 1) because she was too low functioning. 100% of the sample for which the diagnostic algorithm could be completed (n = 8) exceeded the cutoff for Domain A: Qualitative Abnormalities in Reciprocal Social Interaction; 87.5% of the sample exceeded the cutoff for Domain B: Qualitative Abnormalities in Communication; and 87.5% of the sample exceeded the cutoff for Domain C: Restricted, Repetitive, and Stereotyped Patterns of Behavior. Across the full sample for which the diagnostic algorithm could be completed, 6 of 8 participants (75%) exceeded all three diagnostic cutoffs on the ADI-R. Notably, three parents reported that it was difficult to tell if their child was upset/crying or happy/laughing in past, and two additional parents reported that their child presented with a limited range of facial expressions/emotional expression in the past. Two parents reported current abnormal laugh and cry, two parents reported current difficulty interpreting child’s emotional expressions, and one parent reported current concerns regarding lack of emotional expression from their child.
Clinical diagnosis of ASD was determined jointly for each child by the two ASD expert clinicians (L.N.B. and R.P.G.-K.) after review of results from the cognitive/developmental assessment, ADOS-2, SRS-2, and ADI-R. 89% of participants received a diagnosis of ASD. The one participant (Participant 1) who did not receive a diagnosis presented with clinically significant ASD symptoms throughout the evaluation and per parent report; however, general intellectual functioning was too low to reliably make an ASD diagnosis.
Discussion
Differences in clinical phenotypes between individuals with PWS and individuals with truncating variants of the paternal allele of the MAGEL2 gene led to classification of a distinct syndrome, named Schaaf-Yang syndrome (OMIM 615547). The current study sought to characterize the neurocognitive and neurobehavioral phenotype of this disorder. Additionally, this study sought to examine the distinctiveness of the Schaaf-Yang syndrome phenotype from what has been previously published about the neurocognitive and neurobehavioral phenotype of PWS.
Cognitive functioning of the sample was lower than expected, given that children with PWS typically present with mild to moderate ID. One study found that the distribution of intellectual functioning in individuals with PWS is approximately normal, though the mean is 40 points below the general population mean (Whittington et al. 2004). This finding translates to a mean IQ standard score of 60, which falls within the classification of mild ID. Several participants in the current study were so low functioning that they were unable to complete a standardized assessment of intellectual functioning. As such, a deviation IQ score was unable to be computed for the entire sample. The mean full scale ratio IQ score for the sample was 27.0, indicative of much lower average cognitive functioning than that seen in PWS samples. Additionally, average adaptive functioning ratings for the current sample were calculated, as diagnosis of ID is based on both intellectual functioning and adaptive functioning, with the Diagnostic and Statistical Manual of Mental Disorders—5th Edition (American Psychiatric Association 2013) placing particular emphasis on adaptive functioning when determining the severity level. Results indicated “extremely low” functioning across all adaptive domains compared to the normative average.
Taken together, results from the Schaaf-Yang sample indicate 44% of the sample presented with severe/profound ID, 33% presented with moderate ID, and 22% presented with mild ID. Comparatively, a previous review paper reported intelligence levels of participants with PWS from 57 studies and noted 5.6% with severe/profound ID, 27.3% with moderate ID, 34.4% with mild ID, and 32.7% with borderline/age-appropriate cognitive functioning (Curfs and Fryns 1992). Taken together, results of the current study highlight that intellectual functioning differentiates PWS from Schaaf-Yang syndrome, such that both populations are likely to present with ID, though individuals with Schaaf-Yang syndrome appear to present with more severe ID.
Another difference between PWS and Schaaf-Yang syndrome highlighted by the current study is the difference in prevalence of obsessive thoughts and compulsive behaviors between the two syndromes. No parents in the current sample reported significant symptoms of obsessive thoughts or compulsive behaviors, which is in direct contrast to studies of children with PWS. Specifically, a study by Dykens et al. (1996) reported high rates of symptoms on the YBOCS in children and adults with PWS, with 45–80% of the sample reporting that symptoms were time consuming, distressful, or negatively impacting functioning, which suggests a diagnosis of obsessive–compulsive disorder in a large portion of the sample rather than just obsessive–compulsive traits.
Strikingly, our sample of patients with Schaaf-Yang syndrome presented with a high prevalence of ASD, with 89% of the sample receiving a diagnosis of ASD based on clinical observation, diagnostic interview, and ratings from the ADOS-2 and ADI-R. Furthermore, the one participant who did not receive a diagnosis presented with significant ASD symptomatology, though was too low functioning to reliably make an ASD diagnosis. The prevalence of ASD in this Schaaf-Yang syndrome sample represents a stark difference from PWS, where the prevalence of ASD is approximately 27% (Bennett et al. 2015). Furthermore, the clinical presentation of ASD in this sample is indicative of a more severe presentation, as the average comparison score from the ADOS-2, a measure of clinical severity, fell within the moderate to high level of ASD symptoms. Additionally, average ratings on the SRS-2 across the entire sample were within the “severe” range of symptoms for both Social Communication and Interaction and Restricted Interests and Repetitive Behavior domains.
Other neurobehavioral features were present in the sample, including a clinically significant increase in concerns related to atypicality, and subclinical concerns related to hyperactivity, attention problems, and social withdrawal compared to the normative population (per parent ratings on the BASC-3). Atypicality and withdrawal are not surprising, given the high rates of ASD diagnoses within the current sample. Additionally, the subclinical concerns related to hyperactivity and attention problems are often seen in children with ASD and/or ID. Several studies of the neurobehavioral phenotype of PWS were reviewed. No studies were available that utilized the BASC-3, though several studies were identified that utilized a measure with similar domains, the Child Behavior Checklist (CBCL; Dykens et al. 1992, 1999; Dykens and Kasari 1997; Dykens and Roof 2008; Kim et al. 2005; Skokauskas et al. 2011). Results of these studies indicated increased externalizing behaviors, social problems, and attention problems. Additionally, no studies were found with a PWS sample that used the ABAS-3 to assess adaptive functioning, though one study was identified that used a measure with similar domains, the Vineland Adaptive Behavior Scale (Dykens and Roof 2008). Results of this study indicated significantly impaired adaptive functioning compared to the measure’s normative sample, consistent with what was found in the current study of individuals with Schaaf-Yang syndrome. However, comparison of results is limited by the different measures used in PWS research compared to the current sample.
It is currently unclear why individuals with Schaaf-Yang syndrome may represent subsets of phenotypes that are more severe than those seen among individuals with Prader-Willi syndrome. This seems counterintuitive, as the entire paternal copy of MAGEL2 is missing among cases with PWS, both as a result of paternal deletion of the 15q11q13 locus and maternal uniparental disomy. Why would deletion of the entire gene have less severe phenotypic consequences than truncating point mutations? The MAGEL2 gene is a one-exon gene, which means that nonsense or frameshifting mutations are expected to lead to a truncated protein rather than undergoing nonsense-mediated decay. A truncated protein could have a neomorphic function or could cause some kind of dominant-negative effect. This may be relevant, given that MAGEL2 functions as part of a protein complex, along with TRIM27 and USP7 (Hao et al. 2015). Alternatively, the less severe consequences of MAGEL2 deletion could have to do with the imprinted nature of the 15q11 chromosome locus. Deletion of the paternal allele of MAGEL2 may cause leaky expression of the maternal allele of MAGEL2, as has been suggested in studies of the mouse brain (Matarazzo and Muscatelli 2013). On the other hand, patients with Schaaf-Yang syndrome have truncating point mutations of MAGEL2, leaving the paternal promoter intact, and consequently no leaky expression from the maternal copy would be expected.
Strengths, Limitations, and Future Directions
One strength of the current study is the comprehensive, phenotypic assessment that was standardized across the sample. This afforded clear characterization of intellectual and adaptive functioning, ASD symptomatology, and other parent-reported psychiatric features, including the previously unreported identification of common first concerns (e.g., abnormal/absent crying at birth, hand contractures) and difficulties with emotional expression (e.g., inability to distinguish laughing from crying). Another strength is the lack of ascertainment bias that is common in studies examining the rates of ASD in various samples. Participants were recruited through a genetic disorder registry, rather than an autism clinic or foundation, which prevented the effect of a recruitment bias on conclusions regarding ASD prevalence in the sample. In contrast, the study is limited by ascertainment bias from a genetic standpoint. Specifically, individuals often receive genetic testing because of significantly impaired levels of functioning that prompt a genetic work-up. Thus, individuals who have significant developmental delays and neurocognitive deficits are potentially overrepresented in this sample. It is unclear at this time whether there are individuals with this genetic mutation in the general population who remain undiagnosed because of a lack of significant impairment motivating further evaluation.
The small sample size is another limitation of the current study. However, at the time of recruitment, only 19 individuals had been diagnosed with Schaaf-Yang syndrome in the United States, and 9 of these individuals completed the current study. At the time this manuscript was written, over 100 individuals had confirmed Schaaf-Yang syndrome globally, affording the opportunity for more in depth examinations of this population in the future with larger samples. Additionally, the comparisons made between the phenotypes of Schaaf-Yang syndrome and PWS were made based on the results of the current study in comparison with previously published research on samples of youth with PWS. These conclusions would be stronger had the current study included a PWS comparison group.
In conclusion, this study highlighted significant differences between the clinical presentations of individuals with PWS and individuals with Schaaf-Yang syndrome. Specifically, compared to individuals with PWS, patients with Schaaf-Yang syndrome were more likely to present with more severe cognitive deficits/ID, less likely to present with obsessions or compulsions, and more likely to qualify for a diagnosis of ASD. Results reflect that the Schaaf-Yang population is at significant risk for impaired functioning across multiple domains as a result of their genetic condition. Further evaluation of treatments and services that would be effective for use with this population specifically is important, considering that traditional methods may not be appropriate, given their broad functional limitations.
References
American Psychiatric Association. (2013). Diagnostic and statistical manual of mental disorders (5th ed.). Arlington: American Psychiatric Association.
Bennett, J. A., Germani, T., Haqq, A. M., & Zwaigenbaum, L. (2015). Autism spectrum disorder in Prader-Willi syndrome: A systematic review. American Journal of Medical Genetics A, 167(12), 2936–2944.
Cassidy, S. B., Schwartz, S., Miller, J. L., & Driscoll, D. J. (2012). Prader-Willi syndrome. Genetics in Medicine, 14(1), 10–26.
Constantino, J. N., & Gruber, C. P. (2012). Social Responsiveness Scale (2nd ed.). Los Angeles: Western Psychological Services.
Curfs, L. M. G., & Fryns, J.-P. (1992). Prader-Willi syndrome: A review with special attention to the cognitive and behavioral profile. Birth Defects: Original Article Series, 28(1), 99–104.
Dykens, E. M., Cassidy, S. B., & King, B. H. (1999). Maladaptive behavior differences in Prader-Willi syndrome due to paternal deletion versus maternal uniparental disomy. American Journal on Mental Retardation, 104(1), 67–77.
Dykens, E. M., Hodapp, R. M., Walsh, K., & Nash, L. J. (1992). Adaptive and maladaptive behavior in Prader-Willi syndrome. Journal of the American Academy of Child & Adolescent Psychiatry, 31(6), 1131–1136.
Dykens, E. M., & Kasari, C. (1997). Maladaptive behavior in children with Prader-Willi syndrome, Down syndrome, and nonspecific mental retardation. American Journal of Mental Retardation, 102(3), 228–237.
Dykens, E. M., Leckman, J. F., & Cassidy, S. B. (1996). Obsessions and compulsions in Prader-Willi syndrome. Journal of Child Psychology and Psychiatry, 37, 995–1002.
Dykens, E. M., Lee, E., & Roof, E. (2011). Prader-Willi syndrome and autism spectrum disorders: An evolving story. Journal of Neurodevelopmental Disorders, 3(3), 225–237.
Dykens, E. M., & Roof, E. (2008). Behavior in Prader-Willi syndrome: Relationship to genetic subtypes and age. The Journal of Child Psychology and Psychiatry, 49(9), 1001–1008.
Dykens, E. M., Roof, E., Hunt-Hawkins, H., Danker, N., Lee, E. B., Shivers, C. M., Daniell, C., & Kim, S.-J. (2017). Diagnoses and characteristics of autism spectrum disorders in children with Prader-Willi syndrome. Journal of Neurodevelopmental Disorders, 9, 18–30.
Fountain, M. D., Aten, E., Cho, M. T., Juusola, J., Walkiewicz, M. A., Ray, J. W.,… Schaaf, C. P. (2016). The phenotypic spectrum of Schaaf-Yang syndrome: 18 new affected individuals from 14 families. Genetics in Medicine. https://doi.org/10.1038/gim.2016.53.
Fountain, M. D., & Schaaf, C. P. (2016). Prader-Willi syndrome and Schaaf-Yang syndrome: Neurodevelopmental diseases intersecting at the MAGEL2 gene. Diseases, 4(1), 2.
Goodman, W. K., Price, L. H., Rasmussen, S. A., Mazure, C., Fleischmann, R. L., Hill, C. L., Heninger, G. R., & Charney, D. S. (1989). The Yale-Brown Obsessive Compulsive Scale I: Development, use, and reliability. Archives of General Psychiatry, 46(11), 1006–1011.
Hao, Y. H., Fountain, M. D. Jr., Fon Tacer, K., Xia, F., Bi, W., Kang, S. H., Patel, A., Rosenfeld, J. A., Le Caignec, C., ISidor, B., Krantz, I. D., Noon, S. E., Pfotenhauer, J. P., Morgan, T. M., Moran, R., Pedersen, R. C., Saenz, M. S., Schaaf, C. P., & Potts, P. R. (2015). USP7 acts as a molecular rheostat to promote WASH-dependent endosomal protein recycling and is mutated in a human neurodevelopmental disorder. Molecular Cell, 59(6), 956–969.
Harrison, P. F., & Oakland, T. (2015). Adaptive behavior assessment system (3rd ed.). Torrance: Western Psychological Services.
Holm, V. A., Cassidy, S. B., Butler, M. G., Hanchett, J. M., Greenswag, L. R., Whitman, B. Y., & Greenberg, F. (1993). Prader-Willi syndrome: Consensus diagnostic criteria. Pediatrics, 91(2), 398–402.
Kim, J. W., Yoo, H. J., Cho, S. C., Hong, K. E., & Kim, B. N. (2005). Behavioral characteristics of Prader-Willi syndrome in Korea: Comparison with children with mental retardation and normal controls. Journal of Child Neurology, 20(2), 134–138.
Lord, C., Rutter, M., DiLavore, P. C., Risi, S., Gotham, K., & Bishop, S. (2011). Autism diagnostic observation schedule (2nd ed.). Torrance: Western Psychological Services.
Matarazzo, V., & Muscatelli, F. (2013). Natural breaking of the maternal silence at the mouse and human imprinted Prader-Willi locus: A whisper with functional consequences. Rare Diseases, 1, e27221–e272287.
McCarthy, J. M., McCann-Crosby, B. M., Rech, M. E., Yin, J., Chen, C. A., Ali, M. A., Nguyen, H. N., Miller, J. L., & Schaaf, C. P. (2018). Hormonal, metabolic and skeletal phenotype of Schaaf-Yang syndrome: A comparison to Prader-Willi syndrome. Journal of Medical Genetics, 55(5), 307–315.
Mejlachowicz, D., Nolent, F., Maluenda, J., Ranjatoelina-Randrianaivo, H., Giuliano, F., Gut, I., Sternberg, D., Laquerrière, A., & Melki, J. (2015). Truncating mutations of MAGEL2, a gene within the Prader-Willi locus, are responsible for severe arthrogyposis. American Journal of Human Genetics, 97(4), 616–620.
Mullen, E. M. (1995). Mullen Scales of Early Learning. Circle Pines: American Guidance Service Inc.
Reynolds, C. R., & Kamphaus, R. W. (2015). Behavior Assessment System for Children (3rd ed.). Bloomington: NCS Pearson, Inc.
Rice, L. J., & Einfeld, S. L. (2015). Cognitive and behavioural aspects of Prader-Willi syndrome. Current Opinion in Psychiatry, 28(2), 102–106.
Roid, G. H. (2003). Stanford-Binet Intelligence Scales, Fifth Edition. Itasca: Riverside Publishing.
Rutter, M., LeCouteur, A., & Lord, C. (2008). Autism diagnostic interview—Revised. Los Angeles: Western Psychological Services.
Schaaf, C. P., Gonzalez-Garay, M. L., Xia, F., Potocki, L., Gripp, K. W., Zhang, B., … Yang, Y. (2013). Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nature Genetics, 45(11), 1405–1408.
Skokauskas, N., Sweeny, E., Meehan, J., & Gallagher, L. (2011). Mental health problems in children with Prader-Willi syndrome. Journal of the Canadian Academy of Child and Adolescent Psychiatry, 21(3), 194–203.
Soden, S. E., Saunders, C. J., & Willig, L. K. (2014). Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Science Translational Medicine, 6(265), 265ra168.
Whittington, J., Holland, A., Webb, T., Butler, J., Clarke, D., & Boer, H. (2004). Cognitive abilities and genotype in a population-based sample of people with Prader-Willi syndrome. Journal of Intellectual Disability Research, 48(2), 172–187.
Funding
This study was funded by the Foundation for Prader-Willi Research.
Author information
Authors and Affiliations
Contributions
CPS conceived of the study and participated in its design and coordination. LNB and RPGK performed neurobehavioral assessments. LRD assisted with neurobehavioral assessments, scoring, and data collection. JM helped with patient recruitment and data collection. MMT drafted the manuscript, performed statistical analyses, and created figures presented in the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
R.P.G.-K. would like to disclose that she recently entered into an agreement with Yamo Pharmaceuticals, LLC, to provide consulting services in clinical trial development. This relationship is unrelated to the current project and was initiated following completion of data collection. The remaining authors declare that they have no conflict of interest.
Ethical Approval
All procedures performed in the study involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Thomason, M.M., McCarthy, J., Goin-Kochel, R.P. et al. Neurocognitive and Neurobehavioral Phenotype of Youth with Schaaf-Yang Syndrome. J Autism Dev Disord 50, 2491–2500 (2020). https://doi.org/10.1007/s10803-018-3775-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10803-018-3775-7