
Abstract
The electrodeposition process of vanadium from LiCl–KCl base electrolytes was investigated by means of cyclic voltammetry, galvanostatic electrolyses and micro analytical analysis of the deposits. It is demonstrated that the valence state of the vanadium ions has a critical influence on the feasibility of performing a reproducible and stable coating process aiming to obtain compact vanadium films. When the electrolyte contained predominantly trivalent vanadium ions, the process was unstable and the deposit consisted of dendrites. In contrast, making use of a comproportionation reaction of metallic vanadium and VCl3 to divalent vanadium ions led to a stable deposition behaviour and allowed to obtain thick deposits with high current efficiencies. The disadvantageous behaviour of melts with mostly trivalent ions is explained by the fact that deposition is interfered by the reduction of trivalent to divalent ions under limiting current conditions.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Vanadium is rarely used in its pure metallic condition. A few applications such as sputter targets or thin vanadium foils to bond titanium to steel are reported [1]. Vanadium alloys like e.g. V–4Cr–4Ti are being developed for nuclear industry and considered to be suitable materials for fusion reactors [2]. However, most of the metallic vanadium (ca. 85% [3]) serves as alloying element for steel production (ferrovanadium). Besides aluminium, it is the most important alloying element for α + β-titanium alloys (e.g. Ti–6Al–V4) and metastable β-titanium alloys (e.g. Ti–15V–3Cr–3Al–3Sn) [4]. Vanadium can be produced by several methods from its compounds such as calciothermic or aluminothermic reduction [1]. Since the products of these processes generally do not exhibit sufficient purity levels, molten salt electrolyses have been studied as refining step [5,6,7,8], e.g. for vanadium prepared by carbothermic reduction of V2O5. Yet it was reported, that through refining electrolysis, high purity vanadium formed at the cathode as large dendrites [5].
The literature on the nature and behaviour of vanadium metal and its ionic species in molten salts is very limited. In one recent publication [9], a study on the electrochemistry of vanadium in NaCl–KCl melts with various methods has been reported, including anodic dissolution, oxidation and reduction reactions of trivalent and divalent vanadium ions as well as the electrodeposition process. Furthermore, the structure of ionic vanadium species has been characterized in different studies [10, 11] by means of absorption spectroscopy.
However, to our knowledge, there is no information published dealing with the electrodeposition of compact and uniform vanadium layers, which is in contrast to other transition metals like titanium [12] or niobium [13]. One relevant application for such a coating process is the deposition of vanadium films on steel parts for bonding steel to titanium, removing the necessity of foils with the required dimensions (see above, [1]).
A further task is the coating of fibres for the production of metal matrix composites (MMC) according to the matrix-coated fibre route [14, 15]. In this context, the co-deposition together with titanium is of special interest and object of ongoing research.
The long-term objective of the work presented here is to establish fundamentals for the development of alloy deposition processes consisting of anodic dissolution [16] and co-deposition of the constituents for titanium alloys like Ti–6Al–4V.
2 Experimental
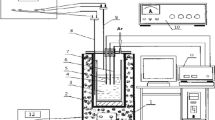
The molten salt experiments were carried out in a glovebox system (Jacomex) under high purity Argon atmosphere (O2, H2O < 1 ppm). All chemicals, electrodes, and crucibles were stored and used within the glovebox in order to minimize contaminations, i.e. especially water pick-up of the highly hygroscopic chemicals or the introduction of oxygen.
The molten salt reactor was attached to the base of the glovebox. A miniaturized experimental setup was employed for this study with a small glassy carbon (GC) crucible (GAT2, HTW, Sigradur G), thin electrodes and small amounts of salts (approx. 10 g per experiment). Within this experimental framework and precautions, it was possible to use high purity anhydrous KCl (Alfa Aesar, ultra dry, 99.95% metals basis) and LiCl (Alfa Aesar, ultra dry, 99.9% metals basis) without further treatment. VCl3 (Alfa Aesar, 99.0%) served as vanadium source and was added to the salt mixture before melting.
Thin tungsten wires (dW = 0.15 mm) were chosen as working electrodes (WE), because they have a similar diameter as the SiC fibres (SCS6, Specialty Materials) so that the suitability for the production of MMC could be verified. Tungsten rods (Ø = 0.5 mm, Alfa Aesar, 99.95%) were also used as counter electrodes (CE). The Glassy carbon crucible served also as quasi-reference electrode (QRE).
Eutectic mixtures of LiCl–KCl were used as electrolyte with additions of 0.34 mol % VCl3 at process temperatures of approx. 700 K. For the experiments with divalent vanadium ions, a metallic vanadium wire (99.5%, Alfa Aesar) was added to the electrolyte and was in contact with the crucible. All electrochemical experiments were carried out with a BANK HC400 potentiostat/galvanostat.
After the experiments, the electrodes were cleaned with deionized water and dried subsequently. The morphologies of the deposits were investigated using Zeiss Ultra 55 scanning electron microscope (SEM) equipped with energy dispersive X-ray spectroscopy (EDS) technique. The diameter (d) and coating length (L) of the plated wires were determined with a stereo microscope (Zeiss Discovery.V12) with the image evaluation software AxioVision SE64.
3 Results
3.1 Electrochemical measurements
In a comparative study cyclic voltammetry (CV) measurements with tungsten working electrodes were carried out in LiCl–KCl melts with 0.34 mol% VCl3, both with and without V metal addition. The investigation covers a large potential window enabling us to study the electrochemical reactions related to vanadium ions occurring in this range. In both systems, the reverse potential (Erev) was chosen positive enough to include the onset of chlorine evolution as internal reference, as proposed e.g. by Berghoute et al. [17].
CV recorded in (LiCl–KCl)(eut.)–0.34 mol% VCl3 melt (Fig. 1, without V metal addition) exhibited current peaks, which can be attributed to the following reactions [9]: The current peaks R1 and O1 correspond to the electrodeposition of divalent ions and its anodic dissolution process (Eq. 1). There is a moderate current increase beginning at − 650 mV (− 1640 mV vs. Cl2/Cl−) before the current increases steeply at − 1030 mV (− 2020 mV vs. Cl2/Cl−).

Cyclic voltammogram recorded in LiCl–KCl(eut.)–0.34 mol% VCl3 using a tungsten wire as WE at ca. 700 K. The dotted (red) curve is the first positive scan, in which no considerable current increase could be observed before chlorine evolution (O3), demonstrating that the vanadium ions in the melt are predominantly in the trivalent state. (Color figure online)
Furthermore, the oxidation of divalent vanadium ions (process O2) as well as the reduction (R2) of trivalent vanadium ions by single-electron transfers was observed (Eq. 2):
The mid-peak potential (Emid) of this reaction is approx. − 126 mV (ca. − 1100 mV vs. Cl2/Cl−), i.e. more negative than the potential of the GC-QRE, because predominately V3+ ions are present in the electrolyte. This is also supported by the fact that during the first positive scan of the CV experiment (Fig. 1, dotted line), which consisted of a total of three cycles, no clear current increase that can be related to oxidation according to Eq. 2 was observed since most of the vanadium ions were already trivalent.
At the positive potential Erev chlorine evolution (O3) occurs. As mentioned above, this reaction served as internal reference to estimate the potential of the reaction (Cl2/Cl−) and allows a comparison with literature data. However, the reproducibility and the current–potential characteristics (i.e. the shapes of the voltammograms in this system), especially of the deposition of vanadium (R1), render further evaluation problematic.
In contrast, reproducible and consistent results were achieved in (LiCl–KCl)(eut.)–VCl3 with V metal addition: The CV measurement shown in Fig. 2 includes 3 cycles which have almost exactly the same curve progressions with two oxidation and reduction processes of vanadium (O1, O2 and R1, R2). The CV measurements confirm that divalent vanadium ions have been generated by comproportionation of V3+ and V(0) (Eq. 3) [11], because in this electrolyte system the potential of the QRE is established by the reaction V/V2+ due to its contact to metallic V and divalent ions. It is close to the stationary potential (− 1955 mV vs. Cl2/Cl−) of vanadium reported for NaCl–KCl at 698 °C [9].

Cyclic voltammogram recorded in LiCl–KCl(eut.)–0.34 mol% VCl3 using a tungsten wire as WE at ca. 700 K. A metallic V wire was added to the electrolyte in order to generate divalent vanadium ions. In contact with the GC crucible, it determines the potential of the reference electrode (V,GC-QRE)
In this system, the same electrochemical reactions V/V2+ and V2+/V3+ as in (LiCl–KCl)(eut.)–VCl3 without V metal addition have been observed. The potential Emid (V3+/V2+) is approx. − 1060 mV and very close to the formal standard potential value (− 1070 mV) reported for 450 °C in LiCl–KCl [18]. The peak separation Ep (O2) − Ep (R2) is approx. 150 mV, which is only slightly higher than the theoretical value for ions having a reversible reaction (ΔE = 2.22 × RT/(1F) = 134 mV [19]), and the peak current densities of reduction and oxidation show similar values (ip (R2)/ip (O2) ≈ 1). The electrodeposition process V2+/V/ (Eq. 1) has been investigated in further experiments (Fig. 3), in which in particular the scan rate was varied. Since ip and v1/2 showed a quite linear relationship (Fig. 3b), electrodeposition (R1) might be a reversible, diffusion controlled reaction. However, there is a distinct shift of Ep when the scan rate exceeds 0.2 V/s (Fig. 3c) and ΔE = Ep (R1) − Ep/2 (R1) increases from ca. − 40 to − 60 mV. This is higher than the theoretical value for reversible reactions involving the formation of insoluble products (ΔE = − 0.77 × RT/(2F) = − 23 mV [20]). Furthermore, the formal standard potential calculated for reversible metal deposition (E´ = Ep − RT/(2F) × ln CV + 0.85 RT/(2F) [8, 21]) is approx. − 1970 mV, which is considerably more negative than the literature value (− 1855 mV [18]). Since not all criteria for reversible reactions are fulfilled, a quasi-reversible behaviour may be assumed for the electrodeposition process.

Voltammetric investigation of electrodeposition process in LiCl–KCl(eut.)–0.34 mol% VCl3 with V metal addition using tungsten wire as WE at ca. 700 K (a). Variation of the peak current as a function of the square root of the scan rate (b). Variation of the peak potential as a function of the scan rate during cyclic voltammetry (c). (Color figure online)
Further reactions such as V3+/V4+ considered by Polovov et al. [9] were not observed in our experiments. As already reported elsewhere, higher oxidation states (V4+ and V5+) can be expected in the presence of oxygen [10, 11].
Altogether we observed in both systems two electrochemical reactions related to the vanadium ions. The reduction of trivalent vanadium ions having a reversible behaviour was found in the electrolyte system with predominantly divalent vanadium ions, while for the electrodeposition not all criteria for reversible reactions were fulfilled.
3.2 Electrodeposition of vanadium
Electrodeposition experiments have been conducted galvanostatically in both electrolyte systems for 30–120 min in order to characterize the stability of the deposition process over elongated periods, finding that the V metal addition leads to a much more stable process. Examples of representative electrolyses are depicted in Fig. 4. In these experiments, the chosen current I = 1 mA corresponded to initial current densities (istart) of ca. 39 mA cm−2 in the case of (LiCl–KCl)(eut.)–0.34 mol% VCl3 and istart = 47 mA cm−2 in the electrolyte system with V metal addition due to slightly different immersion length of the tungsten wires. The evolution of the measured potentials differs strongly between both systems: in the system without V metal addition, there are very strong fluctuations around ca. − 770 mV vs. QRE and the potential shows a shift towards less negative values. This behaviour generally indicates an increase in the surface area due to rougher morphology (e.g. needles or dendrites). As a consequence, the current density and the negative polarization of the electrode decrease. In contrast, the potential of the electrode in the system with V metal addition is very stable throughout the whole deposition process (between − 80 and − 90 mV vs. QRE).

Galvanostatic electrodeposition experiments (I = 1 mA, t = 30 min) on tungsten electrodes at ca. 700 K recorded in (a) LiCl–KCl(eut.)–0.34 mol% VCl3 and (b) LiCl–KCl(eut.)–0.34 mol% VCl3 and V metal addition
A comparison of the potentials related to E (Cl2/Cl−) determined based on the preceding CV measurements (Figs. 1, 2) indicates that the potential of the deposition process in the system with V metal (i.e. with predominantly divalent vanadium ions) is considerably more negative (see additional ordinates on the right side of the diagram in Fig. 4). Without V addition, a more positive potential arises because two competing reactions take place: the reduction V3+/V2+ according to Eq. 2 at considerably more positive potentials (see Sect. 3.1), and the electrodeposition process.
Recapitulating the galvanostatic experiments, we can conclude that the presence of predominantly divalent vanadium ions is crucial for a stable electrodeposition process.
3.3 Characterization of the vanadium deposits
The deposit obtained from (LiCl–KCl)(eut.)–0.34 mol% VCl3 consists mainly of irregularly distributed dendrites (Fig. 5a), i.e. stalks with three-dimensional branch structures [22] (Fig. 5b). The observation of dendrites supports the assumption that material losses by detachment from the cathode may have been responsible for fluctuations of the potential (Fig. 4). Dendrite formation causing an increase of the electrode’s surface area may also explain the potential drift towards less negative potentials observed during the electrodeposition experiment (Fig. 4).

SEM images of vanadium deposited on tungsten cathodes from LiCl–KCl-0.34 mol% VCl3 (a, b) and LiCl–KCl-0.34 mol% VCl3 with V metal addition (c, d)
A thick massive coating was deposited from the electrolyte with vanadium metal addition (Fig. 5c,d). The thickness of the vanadium layer grown within 30 min under galvanostatic conditions (1 mA) is approx. 35 µm (Fig. 5c). Large well-crystallized grains with smooth crystal planes and grain sizes exceeding 50 µm can be recognized (Fig. 5d). In longer deposition experiments (120 min), also coating thicknesses up to more than 100 µm could be achieved.
According to EDS measurements, both deposits consist mainly of vanadium. Small amounts of chlorine have been detected only in the case of the dendritic deposits from (LiCl–KCl)(eut.)–0.34 mol% VCl3 without vanadium metal addition due to electrolyte entrapment.
Altogether, the SEM investigations are consistent with the observations during the electrodeposition experiments, i.e. the stable process conditions, observed in the electrolyte system with predominantly divalent vanadium ions, led to favourable deposits.
3.4 Estimation of the current efficiency
The formation of coherent vanadium films with comparatively uniform coating thicknesses from the electrolyte system with vanadium metal addition, allows us to estimate the current efficiency based on the thickness of the deposit. The diameter of the coated wires varied between ca. 210 and 350 µm depending on the immersion length L of the tungsten wire and the duration of the electrodeposition process (30–120 min). By measuring the diameters d of the coated wires, the volume of the deposited material was determined (\({V_{{\text{coat}}}}=\pi L{{\left( {{d^2} - d_{w}^{2}} \right)} \mathord{\left/ {\vphantom {{\left( {{d^2} - d_{w}^{2}} \right)} 4}} \right. \kern-0pt} 4}\)). The corresponding mass (mcoat) was compared to the mass calculated from Faraday’s law (mcalc) with the assumption that 2 electrons (z = 2) per vanadium ion have been transferred according to Eq. 4.
Current efficiency values between 96 and 110% (Table 1) provide a further strong indication that indeed predominantly divalent vanadium ions are the responsible ion for deposition and prove their existence in the electrolyte. Furthermore, neither material loss due to incoherent deposits [23] nor foil formation, as observed e.g. in electrolytes with titanium ions [14, 24], caused reduced efficiencies.
3.5 Inspection of solidified electrolyte
After the electrodeposition experiments, the solidified salts of the electrolytes were removed from the crucible and crushed within the glovebox under Argon atmosphere (Fig. 6) for chemical analysis. The different colours that were subsequently found can be explained by the oxidation state of the vanadium ions. No information about the colour of electrolytes containing trivalent vanadium ions was found, but it is reported in the literature that trivalent titanium ions cause a purple colour [23] in LiCl–KCl, which would suit to the colour of (LiCl–KCl)(eut.)–0.34 mol% VCl3 (Fig. 6a) taking into account the low concentrations in the present study. According to Polovov et al. [25], divalent vanadium ions caused grey to green colours in NaCl-KCl depending on the concentration. Assuming that a complete conversion of V3+ to V2+ has taken place (Eq. 3) in the system with vanadium addition, the electrolyte contained approx. 0.51 mol% V2+ (ca. 0.75 wt%). For this concentration, a grey colour was found in the aforementioned study [25], whereas we observed a pale yellow to vaguely green colour (Fig. 6b). However, due to the subjectivity of such classifications and the fact that it is not clear whether the colour of the molten or solidified melt was described [25], we cannot determine or resolve a contradiction between both studies. We conclude that the colours of the electrolytes provide a further qualitative indication for the change of the average valence state of vanadium.

Photos of electrolytes after solidification. LiCl–KCl-0.34 mol% VCl3 (a) LiCl–KCl-0.34 mol% VCl3 with added vanadium metal (b)
4 Discussion
The experiments demonstrate the crucial impact of the valence state of the vanadium ions on the quality of the deposits. Due to the presence of the metallic vanadium wire, divalent vanadium ions were generated and stabilized by reaction of vanadium with V3+ (Eq. 3). Furthermore, the depletion of V2+ from the bath due to the cathodic process is compensated by the supply of V2+ from the added vanadium metal which reacts again with the V3+ formed in the anodic reaction (Fig. 7a).

Schematic representation of the conditions during electrodeposition of vanadium from LiCl–KCl with predominantly V2+ ions (a) or V3+ ions (b; x ∈ {2, 3}). The concentration profiles of the ions close to the cathode are represented by straight lines for V3+ and dashed lines for V2+ with vertical arrows indicating the direction of the V2+ concentration changes related to the denoted reactions. In the system with predominantly trivalent vanadium ions competing electrochemical reactions lead to fluctuations of the electrode potential and concentrations of vanadium ions in the vicinity of the electrode (indicated by arrows)
Trivalent ions are less favourable for electrodeposition since the initially dominating reaction that takes place at the cathode is the reduction to divalent ions (Eq. 2). As the limiting current density of this reaction is exceeded, the potential changes drastically towards more negative values and additionally electrodeposition takes place. The deposition process reduces the concentration of V2+ at the electrode (Fig. 7b). If the current density is high enough, the deposition takes place at limiting current from trivalent ions diffusing to the electrode, and the electrolyte is completely depleted of V2+. The observation of two competing reactions (reduction of V3+ and electrodeposition from V2+ or V3+ in Fig. 7, represented by reaction Vx+ + x e− → V) is consistent with the measured potential fluctuations-/oscillations (Fig. 4), as the local current densities constantly change during the electrolysis and also the potential which is enforced by the constant current (1 mA). This may even lead to potentials that are less negative than the potential required for electrodeposition, implicating lacking cathodic protection and, therefore, a partial dissolution of the deposit by comproportionation with V3+ (Eq. 3), until the current density rises again due to the reduced surface area. In the present case, the partial dissolution of vanadium metal may be co-responsible for the material loss (detachment of the dendrites, Fig. 7b). In addition to the discussed disadvantages of the trivalent ions, in the system without vanadium metal chlorine gas is formed at the anode, while the electrolyte is depleted of vanadium ions during the electrodeposition process (Fig. 7b).
5 Conclusions
The results demonstrate the beneficial influence of the divalent oxidation state of the vanadium ions for a stable deposition process: The deposition of thick compact coatings was successfully realized from (LiCl–KCl)(eut.)–0.34 mol% VCl3 after metallic vanadium was added to the electrolyte. Electrodeposition experiments had high current efficiencies (> 95%), which is a further requirement for reproducible coating processes. The results of this study enable developing a fibre coating process for the production for metal matrix composites. Especially, the co-deposition of titanium, vanadium and aluminium is of interest and object of our future investigation.
References
Baroch EF (2000) Vanadium and vanadium alloys. Kirk-Othmer encyclopedia of chemical technology. Wiley, New York
Muroga T (2016) Vanadium for nuclear systems. Reference Module in Materials Science and Materials Engineering, Elsevier, Amsterdam
Moskalyk RR, Alfantazi AM (2003) Processing of vanadium: a review. Miner Eng 16(9):793–805
Peters M, Leyens C (2002) Titan und Titanlegierungen. Wiley, Weinheim
Lei KPV, Sullivan TA (1971) Electrorefining of vanadium prepared by carbothermic reduction of V2O5. Metall Trans 2:2312–2134
Baker DH, Ramsdell JD (1960) Electrolytic vanadium and its properties. J Electrochem Soc 107(12):985–989
Tripathy PK, Sehra JC, Bose DK, Singh RP (1996) Electrodeposition of vanadium from a molten salt bath. J Appl Electrochem 26(8):887–890
Kazakova OS, Kuznetsov SA (2012) Electrochemical behavior and electrorefining of vanadium in melts containing titanium salts. ECS Trans 50(11):181–190
Polovov IB, Tray ME, Chernyshov MV, Volkovich VA, Vasin BD, Rebrin OI (2014) Electrode processes in vanadium-containing chloride melts. In: Gaune-Escard M, Haarberg GM (eds) Molten salt chemistry and technology. Wiley, Hoboken, pp 257–281
Gruen D, McBeth R (1962) Absorbtion spectra of the II, III, IV and V oxidation states of vanadium in LiCl-KCl eutectic octahedral-tetrahedral transformations of V (II) and V (III). J Phys Chem 66(1):57–65
Chernyshov MV, Polovov IB, Volkovich VA, Vasin BD, Rebrin OI, Vonogradov KV, Griffiths TR (2010) Electronic absorption spectra of vanadium species in halide melts. ECS Trans 33(7):287–296
Wei D, Okido M, Oki T (1994) Characteristics of titanium deposits by electrolysis in molten chloride-fluoride mixture. J Appl Electrochem 24(9):923–929
Gillesberg B, Barner JHV, Bjerrum NJ, Lantelme F (1999) Niobium plating processes in alkali chloride melts. J Appl Electrochem 29(8):939–949
Gussone J, Hausmann J (2011) Deposition of titanium on SiC fibres from chloride melts. J Appl Electrochem 41(6):657–662
Chen Z, Li S, Wang Y, Li W, Wei C, Kong W, Jia X, Pei Q, Zhang W (2016) Electrochemical deposition of magnesium on SiC fibers from the LiCl-KCl-MgCl2 molten salt. J Electrochem Soc 9(163):522–525
Milicevic K, Friedrich B, Gussone J, Haubrich J (2017) Anodic dissolution of vanadium in molten LiCl–KCl–TiCl2. J Appl Electrochem 47(5):573–581
Berghoute Y, Salmi A, Lantelme F (1994) Internal reference systems for fused electrolytes. J Electroanal Chem 365(1–2):171–177
Plambeck JA (1967) Electromotive force series in molten salts., J Chem Eng Data 12(1):77–82
Bard AJ, Faulkner LR (2001) Electrochemical methods, fundamentals and applications, 2 edn. Wiley, New York
Mamantov G, Manning DL, Dale JM (1965) Reversible deposition of metals on solid electrodes by voltammetry with linearly varying potential. J Electroanal Chem 9(4):253–259
Berzins T, Delahay P (1953) Oscillographic polarographic waves for the reversible deposition of metals on solid electrodes. J Am Chem Soc 75(3):555–559
Wranglén G (1960) Dendrites and growth layers in the electrocrystallization of metals., Electrochim Acta 2(1–3):130–143
Cordner GDP, Worner HW (1951) Electrolytic preparation of titanium. Aust J Appl Sci 2:358–367
Kühnl H, Ehrlich P, Uihlein RD (1960) Die Abscheidung von Titanmetall durch Schmelzelektrolyse mit löslicher Anode. Z anorg allg Chem 306(5–6):246–259
Polovov IB, Vasin BD, Abakumov AV, Rebrin OI, Chernyshov MV, Volkovich VA, Griffiths TR (2007) Thermodynamics of the formation of vanadium(II) complexes in chloride melts. ECS Trans 3(35):589–597
Acknowledgements
We thank the German Research Foundation (Deutsche Forschungsgesellschaft, DFG) for financially supporting the project (HA 4397/6-1, FR 1713/23-1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gussone, J., Vijay, C.R.Y., Haubrich, J. et al. Effect of vanadium ion valence state on the deposition behaviour in molten salt electrolysis. J Appl Electrochem 48, 427–434 (2018). https://doi.org/10.1007/s10800-018-1165-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-018-1165-7