
Abstract
In this study, we present a novel anode architecture for high-performance lithium-ion batteries based on a Silicon/3-aminosilane-functionalized CNT/Carbon (Si/A-CNT/C) composite. A high-yield, low-cost approach has been developed to stabilize and support silicon as an active anode material. Silicon (Si) nanoparticles synthesized in a hot-wall reactor and aminosilane-functionalized carbon nanotubes (A-CNT) were dispersed in styrene and divinylbenzene (DVB) and subsequently polymerized forming a porous Si/A-CNT/C composite. Transmission electron microscopy showed that this method enables the interconnection and a uniform encapsulation of Si nanoparticles within a porous carbon matrix especially using aminosilane-functionalized CNT (A-CNT). Electrochemical characterization shows that this material can deliver a delithiation capacity of 2293 mAh g−1 with a capacity retention of more than 90 % after 200 cycles at lithiation and delithiation rate of 0.5 C. We conclude that the porous Si/A-CNT/C composite material can accommodate sufficient space for Si volume expansion and extraction and improve the electronic and ionic conduction. Excellent electrochemical performance during repeated cycling can thus be achieved.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Lithium-ion batteries are widely used in portable electronics and have been recognized as the most promising technology for efficient energy storage with high energy and power densities [1]. One of the most promising anode materials for high-performance Li-ion batteries is silicon, the second most abundant element on earth. It possesses the highest known lithium storage capacity of ~4200 mAh g−1, which is almost ten times higher than the capacity of commercial graphite anodes currently used [2]. The use of Si anodes in Li-ion batteries is challenging due to its low conductivity and its high volume change (~300 %) during lithiation and delithiation. Accordingly, cracking and pulverization is observed resulting in a rapid fading of the anode capacity [3]. To solve the problems originating from such large volume changes, research has been conducted on employing nanostructured Si (e.g., mesoporous core–shell structures [4], nanotubes [5], nanofibers [6], and nanowires [7]) because it can tolerate the stress during cycling and can better accommodate the repeated volume expansion and contraction. While many of the nanostructured silicon materials are available only in minute quantities, the gas-phase synthesis of Si nanopowder has shown the potential for low-cost production with high yield [8].
To overcome the drawbacks with respect to conductivity and cracking, Si/C composite materials consisting of nanosized Si dispersed in a carbon matrix were prepared. Such materials show an improved performance and conductivity compared to pure Si electrodes [9]. Despite all these efforts, the electrode stability remains limited which is attributed to an insufficient stability of the interconnection between the Si particles and the carbon matrix. Several recent studies have focused on enhancing the cycling stability by adding carbon nanotube (CNT) networks into their systems [10]. The results showed a significant improvement of the cycling stability of electrodes compared to Si-based electrodes without CNTs (capacity retention of 70 % after 40 cycles). Such results were however based on mostly one-dimensional (1D) CNT or expensive single-walled CNTs (SWCNTs) [11]. Also in these systems, the high capacity was maintained for a double-digit number of cycles only [10].
In this work, we report on a unique structure of an Si-based electrode based on nanosized silicon particles embedded in a porous carbon matrix and three dimensionally linked with multi-wall carbon nanotubes (MWCNTs). This synthetic approach takes advantage of CNT additives in Li-ion batteries that provide additional mechanical strength and linking. Moreover, functionalized MWCNTs allow for good integration of Si particles, preventing the deterioration of the electrode structure during repeated volume change. The architecture of this material improves Si-based anodes through the formation of a 3D porous framework in combination with good electrical properties, which effectively improve the transport of both electrons and ions due to shortened diffusion pathways and 3D interconnectivity [10]. The use of APTS (3-amino-propyl-triethoxysilane) surface-modified MWCNTs with aminosilane groups (A-CNTs) helps maintain the integrity of the Si electrodes and the electrical conductive network eventually leading to a better cycling performance with improved rate capability and cyclability of the battery.
2 Experimental
2.1 Sample preparation
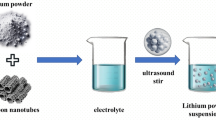
The synthesis of the Si/A-CNT/C composite was performed in three steps: preparation of a homogeneous dispersion of Si particles and A-CNT, polymerization, and subsequent pyrolysis. Figure 1 shows the detailed preparation process of Si/A-CNT/C composite.

Schematic of the synthesis procedure toward Si/A-CNT/C
Si nanoparticles were obtained from a hot-wall reactor (primary particle sizes of 30–120 nm in diameter; details on the synthesis of Si particles can be found in our previous work [8]). BET measurement shows that Si NPs possess a specific surface area of around 22 m2 g−1, which corresponds to an average particle size of 117 nm (assuming spherical particles). A Rietveld refinement of the diffraction pattern indicates a mean crystallite size of 21 nm (Figure S1 in the supplementary material). MWCNTs (FloTubeTM9000 MWCNT) were supplied from CNano Inc and have an average diameter and a length of 11 nm and 10 µm, respectively. The surface modification of A-CNTs was done using 3-amino-propyl-triethoxysilane (APTS) following the procedure described by Yuen et al. [12] (see also TEM images of A-CNTs in Fig. 3a). Si (10 g) and A-CNT (0.5 g) particles were dispersed in co-porogenic solvents of acetonitrile and toluene and sonicated for 30 min prior to polymerization. The A-CNT and Si were homogeneously distributed in a porogen solution prior to any further synthesis. The dispersion was mixed with a monomer solution (35 g of styrene and DVB, 10/90, v/v) and kept in an ultrasonic bath for 30 min. The mixture was then polymerized at 80 °C for 24 h. Finally, the resulting polymer composite was carbonized at 850 °C under nitrogen flow for 3 h and yielded the Si/A-CNT/C composite. After the carbonization, the composite material is grounded to fine powder. For comparison, Si/CNT/C composite was prepared under the same conditions.
2.2 Material characterization
Powder X-ray diffraction patterns were collected with an X-ray powder diffractometer (PANalytical X-Ray Diffractometer X´Pert PRO with Cu Kα radiation) between 20° and 80° at room temperature. Raman spectra were obtained using a RENISHAW inVia Reflex MicroRaman spectroscopy system with a 514 nm laser to study the structure of the carbon material. High-resolution transmission electron microscopy (HRTEM, FEI Tecnai F20) was used to observe the morphology of the prepared samples. The BET surface area and pore volume were obtained from the nitrogen adsorption–desorption isotherm measured with a Quantachrome Nova 2000. X-ray photoelectron spectroscopy (XPS) was performed on a VersaProbe II by Ulvac-Phi, using monochromatic Al Kα light at 1486.6 eV and an emission angle of 45°. The silicon, carbon, and A-CNT contents were determined by thermogravimetric analysis (NETZSCH STA 449F1 Jupiter, TGA). Energy-dispersive X-ray spectroscopy (EDX) was measured using an EDX detector equipped with the JEOL JSM-7500F.
2.3 Electrochemical performance
The electrochemical performance was evaluated using three-electrode Swagelok® half-cells. Three kinds of Si-based anode materials (Si, Si/CNT/C, and Si/A-CNT/C) have been investigated. All electrodes were prepared by mixing 85 wt % active material, 10 wt % PAA (polyacrylic acid) as a binder, and 5 wt % conductive carbon black additive (SuperC65, TIMCAL, Switzerland). Prior to slurry preparation, PAA (Alfa Aesar, USA) was dissolved in an ethanol/water solution, 70:30, v:v % before mixing it with carbon black and active material using ultrasonic and high-energy dispersing techniques. The slurry was cast on an untreated Cu foil (Schlenk Metallfolien GmbH & Co. KG, Germany) by a wet-chemical doctor-blade process. The electrode was pre-dried (90 °C, 8 h under vacuum), cut into circular 12-mm-diameter disks, dried (90 °C, 24 h under vacuum), and weighed before assembly. The typical mass load of the active material was about 0.7 mg cm−2. The three-electrode Swagelok® cells using Li (0.38 mm thick, Sigma Aldrich Co., USA) as both counter and reference electrodes and an Si-based working electrode were assembled in an argon-filled glove box using a 1 M solution of LiPF6 in EC/DMC (1/1, m/m) as an electrolyte (LP30, BASF SE, Germany) with 5 wt % FEC (Sigma Aldrich Co., USA). The thickness of the electrode film was about 7 µm. Galvanostatic cycling experiments were performed at a constant temperature of 25 °C using a Maccor series 4000 battery tester, and the reversible capacity of the active materials was measured based on mass of the entire composite. After SEI formation, the cells were cycled in the potential range of 0.01–1.2 V versus Li/Li+ in a full potential range. Cyclic voltammetry measurements were carried out with a BioLogic VMP3 potentiostat in the aforementioned potential range with a scan rate of 10 µV s−1. Electrochemical impedance spectroscopy (frequency range 10 mHz to 100 kHz with an amplitude of 5 mV) was performed using a coaxial measurement setup as suggested by Klink et al. [13]. All measurements were taken after formation at a potential of 1.2 V versus Li/Li+ and after full relaxation for 48 h.
3 Results and discussion
3.1 Preparation of the Si/A-CNT/C composites
In this study, we focused on two challenges of Si-based anodes: the severe volume expansion and contraction of Si during lithiation/delithiation and the contact and conductivity loss between the active anode material and the current collector.
To account for the volume expansion, a monolithic polymerization process with subsequent pyrolysis based on styrene/divinylbenzene (Sty/DVB) was applied to provide a porous, electrically conductive backbone for the Si nanoparticles. The monolithic process technology has several advantages compared to commonly used emulsion polymerization or suspension polymerization: Because only additive-free constituent components become part of the polymer composite, the resulting material is pure and free of any surfactant or water. The method therefore prevents the need for purification. Secondly, the porosity of the produced polymer can be tailored by adding a suitable solvent acting as a porogen during polymerization. We chose acetonitrile and toluene as co-solvents for polymerization. Each component has a specific role: Acetonitrile is a good dispersing media for both Si nanoparticles and CNTs, while toluene ensures good dispersion of the monomers prior to polymerization. Acetonitrile and toluene are fully miscible and all components including, monomer, initiator, nano Si, and CNT form a homogeneous mixture without phase separation. As a result, a polymer composite with a high surface area and porosity can be obtained. Howdle et al. report that a polymer synthesized with only acetonitrile as a solvent shows a negligible porosity and a surface area of around 9 m2 g−1 [14]. The addition of toluene as a co-solvent however increases the surface area to 110–250 m2 g−1.
CNTs were added to enhance the conductivity between the silicon nanoparticles and the conductive backbone formed after pyrolysis and to strengthen the mechanical integrity. We used the as-received CNTs as well as CNTs functionalized with a polar silane coupling agent (SCA) because the dispersibility of CNTs and the stability of the respective dispersions were limited. APTS (3-amino-propyl-triethoxysilane) was chosen as an SCA as it enables the introduction of nitrogen into the carbon network thus facilitating an enhanced conductivity due to nitrogen doping. The addition of amine-terminated groups on CNT also increases the electronegativity which facilitates the formation of permanent dipoles at neutral graphitic surfaces, enhancing the surface energy [15]. The nitrogen in amine groups can also be protonated and deprotonated when changing pH [16] which is crucial for achieving complete encapsulation of Si particles in this work. Similarly, Lee et al. reported an improved encapsulation of Si particles with N-doped graphitic graphene [16].
Si nanoparticles and 5 wt % (based on Si) of CNTs ((both, as-received CNT (further abbreviated as CNT) and functionalized with APTS (further abbreviated as A-CNT)) were well dispersed in the co-solvent of acetonitrile and toluene, mixed with a monomer solution of styrene and divinylbenzene, and polymerized using AIBN as a starter. The resulting Si/A-CNT/polymer composite was subsequently pyrolyzed in an oven at 850 °C for 3 h to convert the polymer into pyrolytic conductive carbon. The performance of the composite material was improved when using surface-modified CNT (A-CNT). TEM images in Fig. 2 show the difference in morphology of Si/CNT and Si/A-CNT. The latter shows excellent wrapping of the Si nanoparticles, while the composite with CNT does not show uniform encapsulation of the Si particles.

HR-TEM images of a pristine Si nanoparticles. b Si/CNT. c Si/A-CNT/C composite materials
Figure 3 shows TEM images of A-CNTs and Si/A-CNT/C composite material at several levels of magnification. From the images, a very unique bamboo-like morphology which can accommodate enough space for volume expansion of silicon nanoparticles during the cycling can be observed. TEM images of Si/A-CNT/C composite material also show that silicon nanoparticles are well wrapped by A-CNTs and carbon layers (Fig. 3b).

HR-TEM images of a A-CNTs and b Si/A-CNT/C composite materials measured with different magnifications
SEM images of the resulting Si/A-CNT/C composite materials are displayed in Fig. 4 and show the surface morphology and distribution of CNTs of the Si/A-CNT/C composite material. The SEM images indicate that the silicon nanoparticles are well incorporated by A-CNTs which are well distributed in the entire composite material.

SEM images of a pristine Si nanoparticles and b Si/A-CNT/C composite materials
The preparation of a monomer dispersion with the as-received CNT suffered from poor dispersibility in the porogenic solvent due to the hydrophobic nature of CNT and revealed a far inferior dispersibility of Si particles and CNT (see Fig. 5).

Dispersions of pristine CNT, A-CNT, and Si/A-CNT in porogenic solvent
3.2 Characterization of the Si/A-CNT/C composites
Figure 6 shows the XRD patterns and Raman spectra of pristine Si, CNT, Si/CNT/C, and Si/A-CNT/C composite. The diffraction peaks can be well indexed to peaks at 2θ of 28.4, 47.4, 56.2, 69.2, and 76.5°, which are assigned to the (111), (220), (311), (400), and (331) planes of crystalline Si, respectively. A Rietveld refinement of the diffraction pattern indicates a mean crystallite size of 21 nm (see Figure S1 in the supplementary material). After pyrolysis, the Si nanoparticles are still nanocrystalline and did not change into silicon carbide since no apparent changes can be observed for the Si peaks. One broad peak in the diffraction pattern of Si/CNT/C at around 24° indicates that the carbon is amorphous, as also observed by Noh et al. [17]. Otherwise, there is no apparent difference between the XRD patterns of Si/CNT/C and Si/A-CNT/C composite materials.

a XRD and b Raman spectra of pristine Si, pristine CNT, Si/CNT/C, and Si/A-CNT/C
The Raman spectra (see Fig. 6b) clearly indicate the existence of Si, carbon, and CNTs in the composite. The signal at 520 cm−1 represents the Si band. The signals in the range of 1200–1460 cm−1 and 1470–1730 cm−1 can be attributed to the D-band (K-point phonons of A1g symmetry) and G-band (E2g phonons of the C sp2 atoms) of the carbon and/or CNTs, which confirms the existence of CNTs and carbon in the composite. The peak intensity ratio between the peaks at 1333 and 1592 cm−1 (ID/IG) generally provides a useful index for the degree of crystallinity of various carbon materials: the smaller the ID/IG ratio, the higher the degree of ordering in the carbon material [18]. The ID/IG values of the Si/CNT/C and Si/A-CNT/C composites are 1.4 and 0.9, respectively, indicating an enhancement of the degree of graphitization using A-CNT.
We suggest that one reason for the enhanced graphitization is attributed to the nitrogen content in the composite originating from APTS. This assumption is supported by the fact that XPS analyses reveal two types of N sites. Besides quaternary N (NQ), pyridinic N (NP) is observed, which might force the formation of graphitic structures [19] (see Fig. 7).

XPS spectra of pristine CNT, CNT-COOH (intermediate CNT for producing the A-CNT), and Si/A-CNT/C composite
The quantitative mass loading of silicon, carbon, and CNT contents of Si/CNT/C and Si/A-CNT/C was determined by thermogravimetric analysis (TGA) following a method proposed in a report from Guo et al. [20]. The weight loss, after correcting for oxidation of Si, was used to calculate the carbon fraction. The calculation of weight percent of silicon, A-CNT, and carbon is described in the supplementary material. TGA analysis performed on the Si/CNT/C and Si/A-CNT/C composite indicates the amount of Si to be close to 70 wt % (Figure S2 in the supplementary material). Furthermore, the EDX analyses (Figure S3 in the supplementary material) of the Si/A-CNT/C clearly shows the existence of silicon. Low oxygen signal was also detected, which could be resulted from the surface oxidation of Si NPs in the air. Aluminum signal could be also detected from the EDX sample holder. N2 gas adsorption isotherms measured at 77 K were used to derive the specific surface area as well as the pore volume of the pyrolized composite. Table 1 lists the respective values for the Barrett–Joyner–Halenda (BJH) pore volume and the surface area. It is obvious that our approach toward a porous support for the silicon nanoparticles was quite successful.
3.3 Battery performance
Besides capacity and cycle stability which will be discussed later, both composite materials show very similar electrochemical properties. Therefore, results will primarily be discussed for Si/A-CNT/C. The electrochemical performance of anode materials was characterized in Swagelok® half-cells (see experimental section). A galvanostatic mode was employed in the potential range of 0.01–1.2 V (vs. Li/Li+). Lithium foil was used for both reference and counter electrodes. The working electrode consists of 85 wt % active material (Si-based materials), 5 wt % of carbon black, and 10 wt % of poly(acrylic acid) (PAA) binder. The mass loading of active material was 0.7 mg cm−2 that had a relatively high tap density (0.61 g cm−3), which is much higher than that of pristine Si nanoparticles (0.16 g cm−3).
Figure 8 shows the result of the first three cycles of the Si/A-CNT/C electrode during cyclic voltammetry (CV) measurement. The first cathodic peaks starting at around 2.25 V vs. Li/Li+ during the initial lithiation can be explained by a reduction of the electrolyte components including fluoroethylene carbonate (FEC) (used as a stabilizer) and contaminations including trace water present in the electrolyte and on the surface of the Si/A-CNT/C electrode, resulting in the formation of a solid electrolyte interface (SEI) [21]. Further cathodic peaks starting at around 0.3 V versus Li/Li+ can be attributed to the lithiation of the Si/A-CNT/C electrode and the formation of LixSi [22]. During delithiation, anodic peaks starting at around 0.2 V versus Li/Li+ are observed and can be related to the delithiation of carbon and silicon resulting in the formation of an amorphous Si phase [23]. In the second and third cycles, the cathodic lithiation peaks display much higher potentials starting at around 0.4 V versus Li/Li+ as a result of the amorphization reaction during initial lithiation of Si [21].

Electrochemical characteristics of Si/A-CNT/C composite. a, b Cyclic voltammetry curves of Si/A-CNT/C anode for the first three cycles (from 3.0 to 0.01 V vs. Li/Li+)
Figure 9 exhibits voltage profiles of Si/A-CNT/C composite electrodes for the initial electrode activation cycle and subsequent cycles during the galvanostatic cycling. The first lithiation curve indicates a long flat plateau below 0.12 V versus Li/Li+ which is related to the conversion reaction of Si to LixSi [22]. Afterwards, the lithiation/delithiation curves show the characteristics of an amorphous Si phase [24]. With an increasing number of cycles, the mean voltage during lithiation/delithiation moves to slightly lower/higher potentials, respectively, suggesting a gradual decrease in the electrochemical reaction kinetics [25]. The voltage profiles exhibit a stabilization of the electrode capacity after 200 cycles indicating a very good reversible electrode performance.

Electrochemical characteristics of the investigated materials: a voltage profiles of Si/A-CNT/C anode; b galvanostatic cycling performance of Si only, Si/CNT/C, and Si/A-CNT/C at 0.5 C rate; c coulombic efficiencies of Si/CNT/C and Si/A-CNT/C. The first two cycles for all electrodes shown in (b, c) were carried out at 0.05 and 0.1 C rates, respectively; and d rate capability tests (0.2–10 C rates, 1 C: 2500 mA g−1) for Si/A-CNT/C
The cycling performances and Coulombic efficiencies (CE) of Si, Si/CNT/C, and Si/A-CNT/C composite electrodes are compared in Fig. 9b and c. Apparently, the pure Si electrode exhibits an abrupt capacity decay within 50 cycles, presumably due to the well-known effects of contact loss with conductive carbon black and formation of an unstable solid–electrolyte interface (SEI) [26, 27]. Due to the large internal resistance caused by contact losses between active material and current collector, dealloying of Li from Si is not completed, leading to high irreversibility and poor cyclability [28]. It is however worth to note that our Si electrode shows slower capacity decay than commonly cited in the literature (fast decay within 20 cycles, e.g., Ref. [26]). This is attributed to the size of our Si particles significantly below the critical crystallite size of around 150 nm [29].
The delithiation capacity of several anode materials and coulomb efficiencies (CEs) at the first and after several cycles are presented in Table 2. It is well known that micron-sized commercial graphite shows a relatively good CE value of around 87 % in the first cycle [30, 33, 34]. However, as we mentioned above, it has a very low delithiation capacity (~330 mAh g−1) and thus it cannot be used as a high-capacity anode material. Compared to graphite anodes, anode materials with nanostructured materials such as nanoparticles or nanowires have the advantage that they can readily release the mechanical strain during charging, which reduces the potential for pulverization during the first cycle [35]. However, the highly irreversible capacity of the first cycle in these nanostructured electrodes is largely attributed to side reactions and unstable SEI formation due to their high surface area [35]. These reactions occur at the anode–electrolyte interface and lead to a poor CE value. Since the Si/A-CNT/C composite anode material in this study is nanostructured and has a relatively high surface area to provide buffer space for silicon volume expansion during cycling, it shows a relatively low efficiency during the first 50 cycles compared to commercial graphite. However, it should be noted that the CE value becomes more stable continuously (Fig. 9c) and levels off at 99.9 %.
High capacity as well as good cyclability was observed for both Si/CNT/C and Si/A-CNT/C electrodes (see Fig. 9b). We attributed this to two reasons: The porous pyrolytic carbon provides a continuous electrically conductive network that is well connected to the Si particles. It acts as a porous host matrix that accommodates the volume change of Si. In addition, CNTs support the contact to the porous conductive network leading to an initial discharge capacity of 2050 mAh g−1 for the Si/CNT/C composite. Taking into account the silicon content of 70 wt %, 78 % of the silicon could be activated (neglecting the contribution of CNTs and the porous pyrolytic carbon), and even after 200 cycles measured at 0.5 C (1.25 A g−1), the capacity retention was about 71 % (1460 mAh g−1).
A further remarkable improvement of the Si/CNT/C system was achieved when CNT was substituted by A-CNT. As described above, A-CNT could be much better dispersed than the as-received CNTs. This supports the formation of a well-interconnected conductive framework and provides a complete integration of the Si particles as can be calculated from the initial discharge capacity of 2704 mAh g−1. In addition, the obviously homogeneous distribution of A-CNT causes a significantly improved cyclability (see Fig. 5). The capacity fading is low resulting in 2293 and more than 2100 mAh g−1 after 200 and 500 cycles, respectively. It should be noted that this correlates to a capacity retention of 78 % after 500 cycles, which is almost sufficient for applications in consumer electronics.
The cycling efficiency of Si/A-CNT/C is represented in Fig. 9c. The initial lithiation and delithiation capacities of Si/A-CNT/C are 3601 and 2704 mAh g−1, respectively, which corresponds to a CE value of 75 %. This value is significantly higher than the CE values of around 50 % for other active materials with Si/CNTs/C composite reported in literature [10]. It should be noted that superior CE values (75, 94, and 99.8 % after initial, second, and after 500 cycles, respectively) are observed for the Si/A-CNT/C composite. Furthermore, the Si/A-CNT/C demonstrates an excellent rate capability. When the C-rate is sequentially increased from 1 C (2.5 A g−1) to 2 C (5 A g−1) and 5 C (12.5 A g−1), a good capacity retention is maintained. The specific capacity drops from 2500–1950 to 1000 mAh g−1. Note that the applied specific current of lithiation is equivalent to delithiation at the corresponding C-rates (see Fig. 9d), and the capacity is still more than 1000 mAh g−1 in case of measurements at 5 C (12.5 A g−1).
The excellent interconnection between silicon nanoparticles and the A-CNTs is also obvious from EIS measurements. Similar to the results reported by Jiang et al. [36], the Nyquist plot shown in Fig. 10 is dominated by a depressed semicircle and a straight line. The latter one corresponds to the capacitive effect of the charged cell. The experimental data were fitted to an equivalent circuit consisting of a resistor for the ohmic portion of the electrode impedance (REl) in series with an R/CPE element representing the intrinsic electronic resistance and the contact resistance of the electrode (Rint), while the straight low-frequency part is represented by a Warburg impedance.

Nyquist plot of EIS measurements (dots) and fitted data (lines) taken at 1.2 V versus Li/Li+. The inset shows the ohmic portion of the electrode impedance and the small semicircle obtained for the Si/A-CNT/C composite in detail
The ohmic portion of the electrode impedance (the intercept with the real axis of the impedance, see inset in Fig. 10) decreases from 19.7 Ohm (Si) to 9.6 Ohm (Si/A-CNT/C), indicating an enhanced DC-conductivity with addition of CNTs and A-CNT, respectively (see also Table 3). More importantly, the diameter of the semicircle and therefore the intrinsic electronic and contact resistance are slightly affected by the addition of CNT but almost vanish in case of the Si/A-CNT/C composite.
4 Conclusions
We present a high-performance anode material using surface-modified A-CNT that greatly enhances the performance of Li-ion battery anode materials in contrast to high-cost conductive carbonaceous materials such as conducting polymer and graphene, and our approach utilizes low-cost materials and is easy to scale up. The unique structure of the Si/A-CNT/C was designed in order to buffer the severe volume change and maintain a stable electric conductive network for active Si. Furthermore, a full encapsulation of Si particles by tangled A-CNT can help prevent cracking and pulverization. It maintains the integrity of electrodes and stabilizes the electronic conductive network even for several hundred cycles. Si/A-CNT/C exhibits remarkable battery performance, especially in terms of cycle life and rate capability. It shows excellent cycling performance with an exceptionally high capacity of over 2100 mAh g−1 with a capacity retention of 78 % for 500 cycles. The novel porous Si/A-CNT/C material is a promising candidate for high-performance anode materials for Li-ion batteries.
References
Armand M, Tarascon JM (2008) Building better batteries. Nature 451(7179):652–657
Kasavajjula U, Wang C, Appleby AJ (2007) Nano- and bulk-silicon-based insertion anodes for lithium-ion secondary cells. J Power Sour 163:1003–1039. doi:10.1016/j.jpowsour.2006.09.084
Park MH, Kim MG, Joo J, Kim K, Kim J, Ahn S, Cui Y, Cho J (2009) Silicon nanotube battery anodes. Nano Lett 9:3844–3847. doi:10.1021/nl902058c
Kummer M, Badillo JP, Schmitz A, Bremes HG, Winter M, Schulz C, Wiggers H (2014) Silicon/polyaniline nanocomposites as anode material for lithium ion batteries. J Electrochem Soc 161:A40–A45. doi:10.1149/2.020401jes
Zhou Z, Xu Y, Liu W, Niu L (2010) High capacity Si/DC/MWCNTs nanocomposite anode materials for lithium ion batteries. J Alloy Comp 493:636–639. doi:10.1016/j.jallcom.2009.12.171
Wang L, Ding CX, Zhang LC, Xu HW, Zhang DW, Cheng T, Chen CH (2010) A novel carbon–silicon composite nanofiber prepared via electrospinning as anode material for high energy-density lithium ion batteries. J Power Sour 195:5052–5056. doi:10.1016/j.jpowsour.2010.01.088
Cui LF, Ruffo R, Chan CK, Peng H, Cui Y (2009) Crystalline-amorphous core−shell silicon nanowires for high capacity and high current battery electrodes. Nano Lett 9:491–495. doi:10.1021/nl8036323
Wiggers H (2009) Novel material properties based on flame-synthesized nanomaterials. KONA Powder Part J 27:186–194. doi:10.14356/kona.2009017
Magasinski A, Dixon P, Hertzberg B, Kvit A, Ayala J, Yushin G (2010) High-performance lithium-ion anodes using a hierarchical bottom-up approach. Nat Mater 9:353–358. doi:10.1038/nmat2725
Xue L, Xu G, Li Y, Li S, Fu K, Shi Q, Zhang X (2013) Carbon-coated Si nanoparticles dispersed in carbon nanotube networks as anode material for lithium-ion batteries. ACS Appl Mater Inter 5:21–25. doi:10.1021/am3027597
Forney MW, DiLeo RA, Raisanen A, Ganter MJ, Staub JW, Rogers RE, Ridgley RD, Landi BJ (2013) High performance silicon free-standing anodes fabricated by low-pressure and plasma-enhanced chemical vapor deposition onto carbon nanotube electrodes. J Power Sour 228:270–280. doi:10.1016/j.jpowsour.2012.11.109
Yuen SM, Ma CCM, Chiang CL, Teng CC (2008) Morphology and properties of aminosilane grafted MWCNT/polyimide nanocomposites. J Nanomat 2008:1–15. doi:10.1155/2008/786405
Klink S, Madej E, Ventosa E, Lindner A, Schuhmann W, La Mantia F (2012) The importance of cell geometry for electrochemical impedance spectroscopy in three-electrode lithium ion battery test cells. Electrochem Comm 22:120–123. doi:10.1016/j.elecom.2012.06.010
Howdle SM, Jerábek K, Leocorbo V, Marr PC, Sherrington DC (2000) Reversibly collapsible macroporous poly(styrene-divinylbenzene) resins. Polymer 41:7273–7277. doi:10.1016/S0032-3861(00)00018-5
Lee WJ, Lee DH, Han TH, Lee SH, Moon HS, Lee JA, Kim SO (2011) Biomimetic mineralization of vertical N-doped carbon nanotubes. Chem Comm 47:535–537. doi:10.1039/c0cc04237d
Lee WJ, Hwang TH, Hwang JO, Kim HW, Lim J, Jeong HY, Shim J, Han TH, Kim JY, Choi JW, Kim SO (2014) N-doped graphitic self-encapsulation for high performance silicon anodes in lithium-ion batteries. Energy Environ Sci 7:621–626. doi:10.1039/c3ee43322f
Noh M, Kwon Y, Lee H, Cho J, Kim Y, Kim MG (2005) Amorphous carbon-coated tin anode material for lithium secondary battery. Chem Mater 17:1926–1929. doi:10.1021/cm0481372
Lee JI, Choi NS, Park S (2012) Highly stable Si-based multicomponent anodes for practical use in lithium-ion batteries. Energy Environ Sci 5:7878–7882. doi:10.1039/c2ee21380j
Bulusheva LG, Okotrub AV, Kurenya AG, Zhang H, Zhang H, Chen X, Song H (2011) Electrochemical properties of nitrogen-doped carbon nanotube anode in Li-ion batteries. Carbon 49:4013–4023. doi:10.1016/j.carbon.2011.05.043
Guo ZP, Milin E, Wang JZ, Chen J, Liu HK (2005) Silicon/disordered carbon nanocomposites for lithium-ion battery anodes. J Electrochem Soc 152:A2211–A2216. doi:10.1149/1.2051847
Pereira-Nabais C, Światowska J, Chagnes A, Ozanam F, Gohier A, Tran-Van P, Cojocaru CS, Cassir M, Marcus P (2013) Interphase chemistry of Si electrodes used as anodes in Li-ion batteries. Appl Surf Sci 266:5–16. doi:10.1016/j.apsusc.2012.10.165
Obrovac MN, Christensen L (2004) Structural changes in silicon anodes during lithium insertion/extraction. Electrochem Solid-State Lett 7:A93–A96. doi:10.1149/1.1652421
Fuchsbichler B, Stangl C, Kren H, Uhlig F, Koller S (2011) High capacity graphite–silicon composite anode material for lithium-ion batteries. J Power Sour 196:2889–2892. doi:10.1016/j.jpowsour.2010.10.081
Li J, Dahn JR (2007) An In situ X-Ray diffraction study of the reaction of Li with crystalline Si. J Electrochem Soc 154:A156–A161. doi:10.1149/1.2409862
Ren JG, Wu QH, Hong G, Zhang WJ, Wu H, Amine K, Yang J, Lee ST (2013) Silicon–graphene composite anodes for high-energy lithium batteries. Energy Technol 1:77–84. doi:10.1002/ente.201200038
Zhang Y, Zhang XG, Zhang HL, Zhao ZG, Li F, Liu C, Cheng HM (2006) Composite anode material of silicon/graphite/carbon nanotubes for Li-ion batteries. Electrochim Acta 51:4994–5000. doi:10.1016/j.electacta.2006.01.043
Xu M, Li W, Lucht BL (2009) Effect of propane sultone on elevated temperature performance of anode and cathode materials in lithium-ion batteries. J Power Sour 193:804–809. doi:10.1016/j.jpowsour.2009.03.067
Park CM, Kim JH, Kim H, Sohn HJ (2010) Li-alloy based anode materials for Li secondary batteries. Chem Soc Rev 39:3115–3141. doi:10.1039/b919877f
Kim H, Lee EJ, Sun YK (2014) Recent advances in the Si-based nanocomposite materials as high capacity anode materials for lithium ion batteries. Mater Today 17:285–297. doi:10.1016/j.mattod.2014.05.003
Han YS, Jung JH, Leec JY (2004) Investigation on the first-cycle charge loss of graphite anodes by coating of the pyrolytic carbon using tumbling CVD. J Electrochem Soc 151:A291–A295. doi:10.1149/1.1639156
Huang R, Fan X, Shen W, Zhu J (2009) Carbon-coated silicon nanowire array films for high-performance lithium-ion battery anodes. Appl Phys Lett 95:133119. doi:10.1063/1.3238572
Esmanski A, Ozin GA (2009) Silicon inverse-opal-based macroporous materials as negative electrodes for lithium ion batteries. Adv Func Mat 19:1999–2010. doi:10.1002/adfm.200900306
Yen YC, Chao SC, Wu HC, Wu NL (2009) Study on solid-electrolyte-interphase of Si and C-coated Si electrodes in lithium cells. J Electrochem Soc 156:A95–A102. doi:10.1149/1.3032230
Eker Y, Kierzek K, Raymundo-Piñero E, Machnikowski J, Béguin F (2010) Effect of electrochemical conditions on the performance worsening of Si/C composite anodes for lithium batteries. Electrochim Acta 55:729–736. doi:10.1016/j.electacta.2009.09.011
Szczech JR, Jin S (2011) Nanostructured silicon for high capacity lithium battery anodes. Energy Environ Sci 4:56–72. doi:10.1039/c0ee00281j
Jiang T, Zhang S, Qiu X, Zhu W, Chen L (2007) Preparation and characterization of silicon-based three-dimensional cellular anode for lithium ion battery. Electrochem Comm 9:930–934. doi:10.1016/j.elecom.2006.11.031
Acknowledgments
This work has been funded by the German Federal Ministry of Education and Research through the NaKoLiA (Nanocomposites for Lithium-Ion Anodes) project. We thank Beate Endres, Jasmina Kovacevic, and Ulrich Hagemann (University of Duisburg-Essen) for BET, TGA, and XPS measurements, respectively.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sehlleier, Y.H., Dobrowolny, S., Plümel, I. et al. High-yield and scalable synthesis of a Silicon/Aminosilane-functionalized Carbon NanoTubes/Carbon (Si/A-CNT/C) composite as a high-capacity anode for lithium-ion batteries. J Appl Electrochem 46, 229–239 (2016). https://doi.org/10.1007/s10800-015-0897-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-015-0897-x