
Abstract
Purpose
To investigate the potential genetic defects in a five-generation Chinese family with autosomal dominant congenital cataract (ADCC).
Methods
Whole exome sequencing was performed to search the variants in the candidate genes associated with congenital cataract. Sanger sequencing was used to validate the variants and examine their co-segregation in the patients and their relatives. The potential effect of the variants was analyzed using several bioinformatic methods and further examined through Western blotting and co-immunoprecipitation.
Results
A missense variant c. 71 G > T (p. Gly24Val) in the CRYBA4 gene, a known ADCC candidate gene, was identified to be heterozygously present in the patients and co-segregate with cataract in the family. The mutation was absent in all of the searched databases, including our in-house exome sequences of 10,000 Chinese. The alignments of the amino acid sequences of CRYBA4 in a variety of species revealed that the amino acid residue Gly24 was evolutionarily highly conserved, and the in silico analysis predicted that the missense mutation of Gly24Val was damaging for the protein structure and function of CRYBA4. Then, the in vitro expression analysis further revealed that the Gly24Val mutation in CRYBA4 inhibited its binding with CRYBB1. The impaired interaction of β-crystallin proteins may affect their water-solubility and contribute to the formation of precipitates in lens fiber cells.
Conclusion
We identified a novel missense variant in the CRYBA4 gene as a pathogenic mutation of ADCC in a Chinese family. Our finding expanded the CRYBA4 variation spectrum associated with congenital cataracts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital cataract, with a variable prevalence of 0.6–9.3/10,000 live births [1, 2], accounts for 1/10 of the total number of blind eye diseases in children worldwide [3,4,5]. Approximately 1/4 of non-syndromic cataracts are inherited, and they are highly heterogeneous in phenotype and genotype [6, 7]. Of them, autosomal dominant inheritance is most common, autosomal recessive and X-linked inheritance are also reported.
Cataract is caused by the accumulation of insoluble proteins obstructing the light transmission in the lens of eye [1,2,3]. Crystallins, acting as the major structural proteins in the lens, are also the major component of insoluble proteins [3,4,5,6,7,8,9,10]. Crystallins containing 40% α-crystallins, 35% β-crystallins and 25% γ-crystallins [10,11,12] are normally water-soluble proteins. Mutations in crystalline genes would destroy the insolubility and the structural stability of crystallins and result in opacity of the lens [13,14,15].
So far, more than 350 variants located in 13 crystalline genes have been reported to be associated with cataracts (Cat-Map; http://cat-map.wustl.edu/, updated July, 2021) [16]. Among these mutations, the most are reported in the β-crystallin genes including six members of CRYBA1/A3, CRYBA2, CRYBA4, CRYBB1, CRYBB2, and CRYBB3 [11]. The determination of the genetic causes of congenital cataract has significant support for defining clinical diagnoses, performing early treatment, and guiding genetic counseling. In this study, we identified a novel missense mutation in CRYBA4 for a five-generation Chinese family with congenital cataract and showed that the mutation inhibited the formation of heterogeneous oligomers between CRYBA4 and CRYBB1.
Materials and methods
Subjects
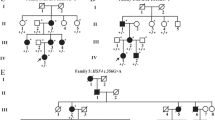
The present study is based on a five-generation Chinese family that consists of 16 individuals, including eight affected individuals (Fig. 1A). The proband (IV:3) was diagnosed with bilateral congenital cataract (Fig. 1B) and his eyes were also amblyopic, strabismal and nystagmic. In addition, his best corrected visual acuity of both eyes was 0.1. His mother (III:3) and son (V:1) were also diagnosed with bilateral congenital cataract at birth, showing nuclear and lamellar opacities in both eyes. All of them underwent surgery shortly after diagnosis. Other systematic abnormality was not found in preoperative examinations. The diagnosis of cataract is based on medical history or the eye examination performed at the department of ophthalmology of West China Hospital, Sichuan University. According to the pedigree, the congenital cataract in this family is suggested to be autosomal dominant inheritance. In order to investigate the genetic variants associated to the congenital cataract in the family, we collected peripheral blood samples for genomic DNA analysis from the proband and his parents, sister and son.

Pedigree and variants in a Chinese family with congenital cataract A The pedigree showing an autosomal dominant inheritance of congenital cataract in the family. Squares and circles indicate males and females, respectively. Black symbols indicate affected individuals and the proband (IV:3) is indicated by an arrow. The diagonal line indicates a deceased family member. B Phenotype of nuclear cataract in the patient (V:1). C Sanger sequencing showing the variants of CRYBA4 in unaffected and affected members in the family
The study followed the tenets of the Declaration of Helsinki and was approved by the Ethics Committee of West China Hospital, Sichuan University [Ethics number: 2019–772]. Written informed consent was obtained from all individuals participating in our study or their guardians.
Whole exome sequencing
Genomic DNA was extracted from peripheral blood of family members using DNeasy Blood & Tissue Kit (No. 69504, Qiagen, Germany). The trio of preceptor-parents underwent targeted next-generation sequencing. The xGen® Exome Research Panel v1.0 (Integrated DNA Technologies, USA) was used to capture the coding regions of disease-associated genes following standard protocols. Enriched pools are added to the flow cell and placed on the Illumina NovaSeq 6000 system for 150 bp double-end sequencing configuration. The patient-parents trio, using the high-throughput sequencing platform Illumina NovaSeq 6000-PE150, had a mean depth of coverage of the targeted regions of 110.5 × across all samples, with an average of 98.77% of targeted regions being covered by ≥ 50 ×.
The sequence reads obtained for each sample were automatically aligned to the human reference sequence (hg19) and variants were called by the software on the MiSeq instrument. Annotation and filtering of variants were done using VariantStudio™ software (Illumina, San Diego, CA). All variants associated with cataract with a population frequency < 0.01 were filtered in a database of 1000 genomes (https://www.internationalgenome.org/), a comprehensive resource of human genetic and mutational information based on a large number of individual genomes. The subsequent variants that were predicted to alter protein function and fit the genetic pattern were retained for further analysis. In all available family members, the candidate pathogenic variants were validated by Sanger sequencing.
In silico analysis
The related amino acid sequences of multiple species were compared using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/) [17]. The evolutionary conservatism of amino acids was analyzed using ConSurf online software (http://consurf.tau.ac.il) [18] to calculate the conserved score by a unique algorithm. Amino acids with scores between 7 and 9 were considered evolutionarily conserved. The secondary structure of proteins was predicted using SOPMA online software (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html) [19] to predict by five independent algorithms. To predict whether pG24V affects the conformation of CRYBA4, homology modeling was performed using Swiss PDB viewer software [20]. To determine the effect of variation on protein stability, we used online software: I-Mutant 2.0 (http://folding.biofold.org/i-mutant/i-mutant2.0) [21], Mupro (http://mupro.proteomics.ics.uci.edu/) [22] and INPS (http://inpsmd.biocomp.unibo.it) [23]. They predicted the change of stability by calculating the change of thermodynamic free energy (ΔΔG) and the direction of change after single point mutation of protein: ΔΔG > 0 indicated stabilization, while a negative value indicated destabilization. The effect of mutation on CRYBA4 solubility was predicted by GETAREA (http://curie.utmb.edu/getarea.html) [24] and ProtScale (https://web.expasy.org/protscale/) [25]. Finally, the pathogenicity of G24V was predicted using Mutpred2 (http://mutpred.mutdb.org/), PolyPhen 2.0 (http://genetics.bwh.harvard.edu/pph2/) [26] and PROVEAN (http://provean.jcvi.org/index.php) [27]. Mutpred2 is a sequence homology-based bioinformatics tool that predicts potential pathogenicity. An amino acid substitution with a score of > 0.5–1 is considered pathogenic. PolyPhen 2.0 is a widely used supervised machine learning Naive Bayes classifier. Mutations with scores above 0.50 are predicted to be pathogenic. PROVEAN is a sequence homology-based tool with independently developed algorithms. Mutations with scores over − 2.5 are predicted to be pathogenic.
Plasmid construction
Two CRYBA4 (NM_001886) expression vectors, pEZ-CRYBA4-M45-eGFP and pEZ-CRYBA4-M46-eGFP, were constructed with HA and Flag tags, respectively, according to our previous report [28]. Also, the CRYBB1 (NM_001887) expression vector pEZ-CRYBB1-M45-eGFP with HA tag was constructed. To generate the mutant c.71G > T in the CRYBA4 gene, targeted mutagenesis was performed using the primers (5′-GGCTTCCAGGTCCGGCGGCACGAGTTCACGGCCGAGTGCC-3′, 5′-GTGCCGCCGGACCTGGAAGCCGTCCTCATCCCACACCACCATCTT-3′) with Fast Mutagenesis Kit V2 (Vazyme Biotech, Nanjing, China). All constructs were sequenced by Sanger sequencing.
Cell culture and transfection
Human embryonic kidney 293 T (HEK293T) cells were grown in DMEM high glucose supplemented with 10% fetal bovine serum (FBS) and maintained at 37 °C with 5% CO2. Cells were plated into 6-well plates one day before transfection and grown until 70–80% confluent. The plasmids were transfected into cells using the ExFect® 2000 Transfection Reagent (Vazyme Biotech). After 48 h of transfection, cultured HEK293T cells were harvested and lysed in RIPAII lysis buffer (Beyotime, Shanghai, China) containing a protease inhibitor mixture (Roche, Basel, Switzerland) at 4 °C.
Western blotting
The supernatant of above cell lysis was subjected in the SDS-PAGE electrophoresis. Then, proteins were transferred from the PAGE gels to nitrocellulose membranes. After the nitrocellulose membranes blocked, they were incubated with rabbit anti-Flag antibody (Proteintech, Wuhan, China) or rabbit anti-HA antibody (Proteintech) overnight at 4 °C. Finally, the membranes were incubated with HRP-labeled goat anti-rabbit antibody (Sangon Biotech) and then developed in TMB substrate and visualized with a gel imager (BioRad, California).
Co-immunoprecipitation (Co-IP)
Considering that the β-crystallins naturally associate into heterogeneous oligomers [29, 30], we examined the interaction between the proteins of mutated-CRYBA4 and CRYBB1 through Co-IP methods. Briefly, the cell lysates were firstly incubated with anti-HA antibodies or IgG. After that, the immune complex was enriched and separated with protein A/G magnetic beads. Finally, the Co-IP proteins were detected by Western blotting with anti-FLAG antibodies.
Results
Identification of a novel missense variant in the CRYBA4 gene
Whole-exome sequencing detected 106,281 variants presented in the proband. Among them, 3896 non-benign variants were predicted according to the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines [31]. Then, the variants located in the 44 candidate genes known to cause ADCC were examined. Finally, a heterozygous missense variant c. 71 G > T (p. Gly24Val) in the CRYBA4 gene and a rare synonymous variant c.183 C > T in the BFSP1 gene were selected to be further investigated.
The BFSP1 c.183 C > T variant was heterogeneously detected in the affected III:3, IV:3 and V:1(Fig. S1). Then, the rare variant was further found to be also presented in the unaffected IV:4 (Fig. S1). Additionally, using the in silico method of Human Splicing Finder (https://www.genomnis.com/access-hsf) [32], we did not find any potential splice sites in the flanking region of the synonymous variant. Therefore, we excluded the correlation between this synonymous variation and congenital cataract in this family.
The G24V mutation may affect the stability and hydrophobicity of CRYBA4 protein
Two online software of Clustal Omega and ConSurf revealed that the amino acid residue Gly24 was evolutionally highly conserved, respectively (Figs. S2A and S2B). The SOPMA software predicted that Gly24 was located at the β-turn angle between a- and b-strand in the Greek Key I Motif of CRYBA4 protein. The present of Gly24 may be critical for CRYBA4 structure since the previous studies have declared that residues between a- and b- strand must contain glycine or serine at key positions to ensure the formation of hairpin loops after folding and that the two hairpin loops are necessary for the stability of tertiary protein structure [33]. As shown in the Swiss homology model of CRYBA4 (Fig. 2A-E), the amino acids of Gln23 and Glu19 flanking Gly24 form only one hydrogen bond in the hairpin loop between a- and b-strand of the Greek Key I motif (Fig. 2B). However, after the change of G24V, the branched chain of valine may clash the interaction of Gln23 or/and Glu19 (Fig. 2C-E) and then potentially destabilize the β-turn. Additionally, the software including I-Mutant 2.0, Mupro and INPS also predicted that G24V may decrease the stability of CRYBA4 structure.

The three-dimensional model and the variation spectrum of CRYBA4 A The modeled structure of CRYBA4 built by Swiss-Pdb Viewer 4.0.1. B The backbone and sidechains of the hairpin loop between a- and b-strand of CRYBA4. C-E Three possible structures of the hairpin loop between a- and b-strand in the mutant G24V protein (backbone and sidechains). The alpha-helix is shown in light red, the beta-chain is shown in light blue, and the other structures are shown in yellow. The gray color is for carbon atoms, the red color is for oxygen atoms, and the blue color is for nitrogen atoms. The green dashed lines are hydrogen bonds and the pink dashed lines represent conflicts between atoms. F Schematic of CRYBA4 mutations. CRYBA4 is folded in four Greek key motifs. The mutation identified in this study was highlighted in red
Since the protein solubility in the lens is directly related to lens transparency, the software of GETAREA and ProtScale were applied to predict the solubility of G24Vmutation. The results showed that G24V may reduce the average solvent-accessible surface area of CRYBA4 (Fig. S2D and E), resulting in the decline of the G24V protein solubility. In addition, the analysis of Mutpred2 (score: 0.958), PolyPhen 2.0(score: 1.0) and PROVEAN (score: -8.54) also predicted that the G24V mutation of CYRBA4 is probably to cause disease. Taken together, the in silico analysis revealed that the missense mutation of Gly24Val was damaging for the protein structure and function of CRYBA4.
The G24Vmutation of CRYBA4 inhibits its interaction with CRYBB1
To further examine the potential deleterious effects of the mutant CRYBA4 proteins of G24V, the recombinant wild-type (WT) and mutant G24V proteins were transiently expressed in the HEK293T cells. The WB analysis showed no significant difference in the expression levels between WT CRYBA4 and G24V (Fig. 3A). Considering that previous studies have showed that the complex were formed between CRYBA4 and CRYBB1 [34], we further investigated the effect of G24V mutation on its interaction with CRYBB1 and observed that the G24V mutation in GRYBA4 destroyed its binding activity with CRYBB1. CRYBB1 has been reported to contribute to the solubility of CRYBA4 [35, 36]. In addition, CRYBB1 is able to interact with CRYBA4 and further forms heterodimers [37]. These results indicated that the missense mutation of G24V inhibited the formation of heterodimers between CRYBA4 and CRYBB1, although the expression level and the stability of the mutant proteins were intact. And this destructed interaction may contribute to the formation of precipitates in lens fiber cells, leading to cataracts.

In vitro expression of the wild–type CRYBA4 and the mutant G24V proteins A Expression of CRYBA4, G24V (CRYBA4*) and CRYBB1 in HEK 293 T cells. The expression of the blank HA vector (NEG-HA) was used as a negative control. B Co-immunoprecipitation analysis showing the interaction between CRYBA4, G24V (CRYBA4*) and CRYBB1. Normal human immunoglobulin G (IgG) was used as a negative control
Discussion
The β-crystallin protein contains two structural domains, each consisting of two highly conserved Greek Key motifs, each of which in turn consists of four reverse parallel β-strands (known as a-, b-, c- and d- strand) [33, 34, 38]. The stable folding of the β chains depends on the hydrogen bonds between the β-strands, and the damage of the hydrogen bonds will affect the structural stability of the protein [39]. Meanwhile, many studies have shown that CRYBA4 has low solubility in water and weak ability to form homodimers [40, 41]. Moreover, CRYBA4 readily oligomerizes with CRYBB1 [34], and the CRYBA4 solubility will be enhanced after combining with CRYBB1 [35, 42]. In this study, we uncovered a missense mutation of G24V in the CRYBA4 protein in an ADCC family. We found that the G24V mutation prevents CRYBA4 from forming a heterodimer with CRYBB1, resulting in the potential insolubility of mutant CRYBA4 protein. According to the ACMG guidelines, we speculated that the G24V mutation is pathogenic.
Most of the pathogenic mutations in the crystalline genes disrupted the Greek key domains of the crystalline proteins [43]. So far, 351 pathogenic mutations have been identified in the crystallin proteins, with the most, 63 in total, in CRYGD. Instead, CRYBA4 is only responsible for approximately 2.56% of congenital cataracts caused by mutations in the crystallin genes, 9 in total [44]. Among the mutations in CRYBA4, six are located in the Greek Key II motif and the other three are each in the Greek Key I motif, Greek Key IV motif and NH2-term (Fig. 3F) [44]. The G24V mutation that we identified in this study is the second one located in Greek Key I motif.
In conclusion, we revealed a novel missense variant c.71G > T (p.G24V) in the CRYBA4 gene in a Chinese ADCC family. The functional analysis confirmed that the mutation could result in the impaired interaction of β-crystallin proteins. Our findings expanded the variation spectrum of CRYBA4 and provided the support for clinical diagnoses and genetic counseling on congenital cataract.
Data availability
The data used to support this study are available from the corresponding author upon request.
References
Robinson GC, Jan JE, Kinnis C (1987) Congenital ocular blindness in children, 1945 to 1984. Am J Dis Child 141(12):1321–1324. https://doi.org/10.1001/archpedi.1987.04460120087041
Apple DJ, Ram J, Foster A, Peng Q (2000) Elimination of cataract blindness: a global perspective entering the new millenium. Surv Ophthalmol 45(Suppl 1):S1–196
Gilbert C, Foster A (2001) Childhood blindness in the context of VISION 2020–the right to sight. Bull World Health Organ 79(3):227–232
Holmes JM, Leske DA, Burke JP, Hodge DO (2003) Birth prevalence of visually significant infantile cataract in a defined U.S. population. Ophthalmic Epidemiol 10(2):67–74. https://doi.org/10.1076/opep.10.2.67.13894
Sheeladevi S, Lawrenson JG, Fielder AR, Suttle CM (2016) Global prevalence of childhood cataract: a systematic review. Eye 30(9):1160–1169. https://doi.org/10.1038/eye.2016.156
Francois J (1982) Genetics of cataract. Ophthalmologica 184(2):61–71. https://doi.org/10.1159/000309186
Haargaard B, Wohlfahrt J, Fledelius HC, Rosenberg T, Melbye M (2004) A nationwide Danish study of 1027 cases of congenital/infantile cataracts: etiological and clinical classifications. Ophthalmology 111(12):2292–2298. https://doi.org/10.1016/j.ophtha.2004.06.024
Hejtmancik JF (2008) Congenital cataracts and their molecular genetics. Semin Cell Dev Biol 19(2):134–149. https://doi.org/10.1016/j.semcdb.2007.10.003
Wirth MG, Russell-Eggitt IM, Craig JE, Elder JE, Mackey DA (2002) Aetiology of congenital and paediatric cataract in an Australian population. Brit J Ophthalmol 86(7):782–786. https://doi.org/10.1136/bjo.86.7.782
Wistow GJ, Piatigorsky J (1988) Lens crystallins - the evolution and expression of proteins for a highly specialized tissue. Annu Rev Biochem 57:479–504. https://doi.org/10.1146/annurev.bi.57.070188.002403
Wistow G (2012) The human crystallin gene families. Hum Genomics 6:26. https://doi.org/10.1186/1479-7364-6-26
Billingsley G, Santhiya ST, Paterson AD, Ogata K, Wodak S, Hosseini SM, Manisastry SM, Vijayalakshmi P, Gopinath PM, Graw GJ, Heon E (2006) CRYBA4, a novel human cataract gene, is also involved in microphthalmia. Am J Hum Genet 79(4):702–709. https://doi.org/10.1086/507712
Xu J, Zhao WJ, Chen XJ, Yao K, Yan YB (2018) Introduction of an extra tryptophan fluorophore by cataract-associating mutations destabilizes betaB2-crystallin and promotes aggregation. Biochem Biophys Res Commun 504(4):851–856. https://doi.org/10.1016/j.bbrc.2018.09.028
Xi YB, Zhang K, Dai AB, Ji SR, Yao K, Yan YB (2014) Cataract-linked mutation RI 88H promotes beta B2-crystallin aggregation and fibrillization during acid denaturation. Biochem Biophys Res Commun 447(2):244–249. https://doi.org/10.1016/j.bbrc.2014.03.119
Zhu S, Xi XB, Duan TL, Zhai Y, Li J, Yan YB, Yao K (2018) The cataract-causing mutation G75V promotes gammaS-crystallin aggregation by modifying and destabilizing the native structure. Int J Biol Macromol 117:807–814. https://doi.org/10.1016/j.ijbiomac.2018.05.220
Shiels A, Bennett TM, Hejtmancik JF (2010) Cat-Map: putting cataract on the map. Mol Vis 16(215–19):2007–2015
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. https://doi.org/10.1038/msb.2011.75
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44(W1):W344–W350. https://doi.org/10.1093/nar/gkw408
Geourjon C, Deleage G (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci 11(6):681–684. https://doi.org/10.1093/bioinformatics/11.6.681
Kaplan W, Littlejohn TG (2001) Swiss-PDB Viewer (Deep View). Brief Bioinform 2(2):195–197. https://doi.org/10.1093/bib/2.2.195
Capriotti E, Fariselli P, Casadio R (2005) I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res 33:306–310. https://doi.org/10.1093/nar/gki375
Cheng J, Randall A, Baldi P (2006) Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 62(4):1125–1132. https://doi.org/10.1002/prot.20810
Savojardo C, Fariselli P, Martelli PL, Casadio R (2016) INPS-MD: a web server to predict stability of protein variants from sequence and structure. Bioinformatics 32(16):2542–2544. https://doi.org/10.1093/bioinformatics/btw192
Fraczkiewicz RW (1998) Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comp Chem 19:319–333
Walker JM (2005) The proteomics protocols handbook. Humana Press, Totowa
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249. https://doi.org/10.1038/nmeth0410-248
Choi Y, Chan AP (2015) PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31(16):2745–2747. https://doi.org/10.1093/bioinformatics/btv195
Liu YQ, Lu YJ, Liu SS, Liao SY (2017) Novel compound heterozygous mutations of ALDH1A3 contribute to anophthalmia in a non-consanguineous Chinese family. Genet Mol Biol 40(2):430–435. https://doi.org/10.1590/1678-4685-Gmb-2016-0120
Yi J, Yun J, Li ZK, Xu CT, Pan BR (2011) Epidemiology and molecular genetics of congenital cataracts. Int J Ophthalmol 4(4):422–432. https://doi.org/10.3980/j.issn.2222-3959.2011.04.20
Li FF, Zhu SQ, Wang SZ, Gao C, Huang SZ, Zhang M, Ma X (2008) Nonsense mutation in the CRYBB2 gene causing autosomal dominant progressive polymorphic congenital coronary cataracts. Mol Vis 14:750–755. http://47mv-v14-750-dimer.pdf
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C (2009) Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37(9):e67. https://doi.org/10.1093/nar/gkp215
Slingsby C, Clout NJ (1999) Structure of the crystallins. Eye 13:395–402. https://doi.org/10.1038/eye.1999.113
Bateman OA, Sarra R, van Genesen ST, Kappe G, Lubsen NH, Slingsby C (2003) The stability of human acidic beta-crystallin oligomers and hetero-oligomers. Exp Eye Res 77(4):409–422. https://doi.org/10.1016/S0014-4835(03)00173-8
Liu BF, Liang JJ (2007) Protein-protein interactions among human lens acidic and basic beta-crystallins. FEBS Lett 581(21):3936–3942. https://doi.org/10.1016/j.febslet.2007.07.022
Marin Vinader L, Onnekink C, Van Genesen ST, Slingsby C, Lubsen NH (2006) In vivo heteromer formation Expression of soluble betaA4-crystallin requires coexpression of a heteromeric partner. FEBS J 273(14):3172. https://doi.org/10.1111/j.1742-4658.2006.05326.x
Bateman JB, Von-Bischhoffshaunsen FR, Richter L, Flodman P, Burch D, Spence MA (2007) Gene conversion mutation in crystallin, beta-B2 (CRYBB2) in a Chilean family with autosomal dominant cataract. Ophthalmology 114(3):425–432. https://doi.org/10.1016/j.ophtha.2006.09.013
Lubsen NH, Aarts HJ, Schoenmakers JG (1988) The evolution of lenticular proteins: the beta- and gamma-crystallin super gene family. Prog Biophys Mol Biol 51(1):47–76. https://doi.org/10.1016/0079-6107(88)90010-7
Rose GD, Fleming PJ, Banavar JR, Maritan A (2006) A backbone-based theory of protein folding. Proc Natl Acad Sci U S A 103(45):16623–16633. https://doi.org/10.1073/pnas.0606843103
Li W, Ji Q, Wei Z, Chen YL, Zhang Z, Yin X, Aghmiuni SK, Liu M, Chen W, Shi L, Chen Q, Du X, Yu L, Cao MJ, Wang Z, Huang S, Jin T, Wang Q (2019) Biochemical characterization of G64W mutant of acidic beta-crystallin 4. Exp Eye Res 186:107712. https://doi.org/10.1016/j.exer.2019.107712
Graw J (2009) Genetics of crystallins: cataract and beyond. Exp Eye Res 88(2):173–189. https://doi.org/10.1016/j.exer.2008.10.011
Marin-Vinader L, Onnekink C, van Genesen ST, Slingsby C, Lubsen NH (2006) In vivo heteromer formation. Expression of soluble beta A4-crystallin requires coexpression of a heteromeric partner. FEBS J 273(14):3172–3182. https://doi.org/10.1111/j.1742-4658.2006.05326.x
Ecroyd H, Carver JA (2009) Crystallin proteins and amyloid fibrils. Cell Mol Life Sci 66(1):62–81. https://doi.org/10.1007/s00018-008-8327-4
Yu Y, Qiao Y, Ye Y, Li J, Yao K (2021) Identification and characterization of six beta-crystallin gene mutations associated with congenital cataract in Chinese families. Mol Genet Genomic Med 9(3):e1617. https://doi.org/10.1002/mgg3.1617
Acknowledgements
This work was supported by Grants from the National Natural Science Foundation of China (No.81773159).
Funding
This work was supported by the National Natural Science Foundation of China (Grant Number: No.81773159).
Author information
Authors and Affiliations
Contributions
XZ: participated in the whole study, drafting of the manuscript, and data analysis; XZ, CL, ML, XL, ZW, SX and XT: carried out the experiments; YY: contributed to data curation; YL: contributed to writing—review, editing, correspondence, and proofs of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare no conflict of interest. The authors have no relevant financial or non-financial interests to disclose.
Consent to publish
The authors affirm that human research participants provided informed consent for publication of the images in Figure 1b.
Ethical approval
The current study was reviewed and approved by the Research Ethics Committee of the West China Hospital, West China Medical School, Sichuan University (2019–772).
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, X., Liang, C., Liu, M. et al. A novel missense variant c.71G > T (p.Gly24Val) of the CRYBA4 gene contributes to autosomal-dominant congenital cataract in a Chinese family. Int Ophthalmol 43, 43–50 (2023). https://doi.org/10.1007/s10792-022-02386-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10792-022-02386-3