
Abstract
Purpose
To characterize the clinical presentation of posterior polymorphous corneal dystrophy (PPCD) in eyes of Indian ethnicity.
Design
Retrospective cohort study from January 1995 to December 2015.
Participants
Patients with the diagnosis of posterior polymorphous corneal dystrophy.
Methods
Medical records of the patients were reviewed for clinical presentation. Histology of corneal specimens of those that underwent keratoplasty was assessed.
Main outcome measures
Descriptive analysis of clinical condition.
Results
Mean age at first evaluation was 32.5 years (range 1–73 years), male:female = 35:18. Majority (44/53; 83 %) of the patients had bilateral involvement. 5/9 (44 %) patients with unilateral presentation were amblyopic in the affected eye. The clinical features documented were vesicles in 94 eyes, band-like pattern in 32 eyes, edema of varying degree in 23 eyes (12 patients, 1 patient was one eyed), and anterior segment changes in 1 eye. 8/45 (17 %) eyes had a regular astigmatism with steep axis >47 D (range 47.2–56.2 D). 16 eyes of 12 patients who had clinically evident corneal edema underwent keratoplasty. Mean age at keratoplasty was 58 years (range 1–73 years). 8 patients had penetrating keratoplasty (PK) and 8 had Descemet stripping endothelial keratoplasty (DSEK). Mean follow-up after keratoplasty was 4.2 years (1 month to 13 years). Except one, all grafts remained clear till the last follow-up. In all specimens, the Descemet membrane was grossly thickened.
Conclusions
In our study, 12/53 (22.6 %) patients required keratoplasty for visually significant corneal edema. Except one, all were older adults. The patients who needed keratoplasty were bilaterally afflicted and had visually significant cornea edema in both eyes. With a mean follow-up duration of 4.2 years after keratoplasty, no recurrences were noted.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Posterior polymorphous corneal dystrophy (PPCD) is a dominantly inherited corneal endothelial dystrophy that is associated with a varied phenotype, ranging from asymptomatic corneal endothelial changes to corneal edema and glaucoma [1]. The genome-wide linkage analyses on different families with PPCD have identified several gene loci responsible for this condition. These include the long arm of chromosome 20 (20p11.2-q11.2) designated as PPCD 1, the short arm of chromosome 1 (1p34.3-p32) designated as PPCD 2, and the short arm of chromosome 10 (10p11.2) now designated as PPCD 3 [2–4].
Exact prevalence of this form of corneal dystrophy is unknown. A high prevalence of this dystrophy is reported in the Czech Republic [5]. It is possible that its prevalence is more than what is expected and reported in literature as a vast majority of the patients are asymptomatic and many cases may fail to be recognized in the clinic. The diagnosis of PPCD is recognized with slit-lamp biomicroscopic findings of corneal endothelial vesicles, bands, and geographic placoid opacities. Even with a considerable amount of clinical experience, PPCD can be confused with Fuchs endothelial dystrophy, Iridocorneal endothelial syndrome, and congenital hereditary endothelial dystrophy (CHED). The clinical findings and pathology of this entity are considerably different, and hence, an attempt must be made to characterize them to the best of the precision. A positive family history, histo-pathological features of multilayered endothelium with cytokeratin positivity, and genetic testing may help in making an accurate diagnosis in confusing case scenarios. Most of the cases are non progressive, asymptomatic, and corneal edema is rare to develop, and hence the condition is often diagnosed on a routine eye exam [1].. Several associations have been described such as corneal steepening, high myopia, and Alport’s syndrome [6–8].
The purpose of this study was to characterize the clinical presentation of PPCD in eyes of Indian ethnicity.
Methods
A retrospective analysis of 98 patients with a diagnosis of PPCD from 1995 to 2015 was done. As the study spanned over a period of 20-year retrospective data analysis, those medical records which were illegible (old scanned records), or where the diagnosis was questionable or documentation of clinical lesions incomplete or had a confounding event such as a prior intraocular surgery were excluded (Total added up to 45) from the analysis. This was to avoid ambiguity and uncertainties in the diagnosis of the condition.
A total of 53 medical charts were identified and reviewed for the clinical presentation, systemic history, familial history, and management plan. Of these 53 patients, 16 eyes of 12 patients underwent keratoplasty for endothelial dysfunction. The histology of the corneal specimens was reviewed.
Results
Demographics
Mean age at first evaluation was 32.5 years (range 1–73 years), male:female ratio was 35:18. 6/12 (50 %) patients that needed keratoplasty gave a definite history of an affected family member (either a parent or sibling) who had undergone keratoplasty in at least one of the eyes for a corneal pathology similar to that in the patient; remaining five patients were either unsure or unaware. 4/12 (44 %) patients that had a keratoplasty done had diabetes. The patients in whom keratoplasty was required had bilateral progression of corneal edema and needed surgery in both eyes for endothelial dysfunction (Table 1).
Clinical features
The clinical features documented were vesicles in 94 eyes, band-like pattern in 32 eyes, edema of varying degree in 23 eyes (12 patients, 1 patient was one-eyed), and anterior segment changes in 1 eye. 2 patients had esotropia, 1 had an absolute eye secondary to lens-induced glaucoma, and 1 patient had extensive myopic chorioretinal degeneration with a neovascular membrane.
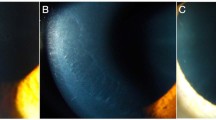
Majority (44/53; 83 %) of the patients had bilateral involvement. 5/9 (44 %) patients with unilateral presentation had meridional amblyopia in the affected eye. Table 2 shows the clinical characterization of the unilateral cases. Figures 1 and 2 illustrate the representative cases that had a unilateral involvement.

(a–e) Representative case (case no 9 in Table 2) that had endothelial abnormalities in one eye (right eye); a slit-lamp photograph in retroillumination showing the band-like pattern at the level of Descemet membrane endothelial complex; b specular microscopy of the involved eye showing the endothelial morphological abnormality and reduced endothelial cell density(=1176 cells/mm2) in the right eye; c specular microscopy of the normal left eye(endothelial cell density = 2641 cells/mm2); (d, e) tomography maps showing K m of 42.3 and 41.3 D in the right and the left eye, respectively

(a–d) Representative case (case no 8 in Table 2) showing a unilateral involvement (right eye); a, b specular microscopy image of the right eye showing reduced endothelial cell density (=1546 cells/mm2) and increased mean cell area compared to the normal left eye (endothelial cell density = 3126 cells/mm2); c, d tomography maps showing the with-the-rule astigmatism in the affected right eye, K m of 47.5 D in the right eye and 44.2 D in the left eye
12 patients had presented to the clinic with complaints of gradually decreasing vision due to corneal edema, 4 patients complained of noticing poor vision in one eye; the remaining visited the clinic either for a routine eye check, consideration of laser refractive surgery, or for diminished vision attributed to causes other than the corneal pathology.
Corneal topography (either Orbscan II/Oculyzer/Galilei) was documented in 45 eyes of 25 patients. 8/45 (17 %) eyes had a symmetric astigmatism with steep axis >47 D (range 47.2–56.2 D). Mean keratometry in patients (n = 12 eyes, 7 patients) that underwent keratoplasty was 43.92 D (range 42.5–46). Mean keratometry in eyes that did not require keratoplasty was 45.75 D (range 41.3–55.15).
Keratoplasty
16 eyes of 12 patients who had clinically evident corneal edema underwent keratoplasty at the institute. Mean age at keratoplasty was 58 years (range 1–73 years). One patient had bilateral symmetrical corneal edema at the time of birth and was initially diagnosed with CHED. However, the corneal specimen had histological features and cytokeratin study consistent with PPCD. 8 patients had PK and 8 had DSEK. Mean follow-up after keratoplasty was 4.2 years (1 month to 13 years). Except one, all grafts remained clear till the last follow-up. One eye developed persistent graft edema when cataract surgery was performed 13 years after keratoplasty. 5/16 eyes had transient episode of raised intraocular pressures after keratoplasty which was controlled with a single anti glaucoma medication. There were no recurrences of the primary pathology in the graft.
Histology
Histology was available for 13 specimens. In all specimens, the Descemet membrane was grossly thickened but without any evidences of guttae as is characteristic of Fuchs Endothelial dystrophy; multilayering of endothelial cells was seen in 9 and pan-Cytokeratin positivity was seen in 8 specimens. Figure 3a and b shows the histology of representative case that underwent DSEK. Figure 3c and d shows the histology of the 1-year-old child who underwent penetrating keratoplasty.

a Histology of the stripped Descemet membrane of 59-year-old male patient who underwent DSEK, 40×; b the same specimen showing positivity for cytokeratin marker; c histology of full-thickness corneal button of 1-year-old child who underwent PK. Note the multilayering of the cells and thickened Descemet membrane, 40×; d cytokeratin positivity seen in the corneal specimen
Discussion
PPCD is a genetically heterogenous condition with extremely variable expression. Lesions generally develop in early childhood and are mostly bilateral but may be asymmetrical or unilateral in some cases. Most patients are asymptomatic with no corneal edema, but progressive visual impairment due to stromal clouding may occur later in life. Rarely, corneal edema may be seen right at the time of birth [9]. In the largest series of PPCD cases reported (120 individuals), thirteen (10.8 %) patients required corneal transplantation [10]. In our study 12/53 (22.6 %), patients required keratoplasty. All were bilaterally afflicted and had visually significant cornea edema in both eyes. All of these were older adults except one case who had bilateral edematous corneas right at birth. This child was referred to us at the age of 1 year for a congenital glaucoma. 50 % of the patients with corneal edema had a definite positive family history of one affected family member. The parents of the child who had congenital corneal edema were, however, normal. These are relevant clinical associations and should be asked for in these subjects to understand the likely course of the disease when seen in the early stages. A positive family history and a corneal edema in one eye at the time of presentation put the patient at a risk of developing corneal edema in future, necessitating a keratoplasty. Keratoplasty in PPCD has a good success rate [1]. There are reports of recurrences of the condition in the graft. In this study, within a mean follow-up duration of 4.2 years, no recurrences were noted.
Aldave et al. have observed corneal steepening in some patients with PPCD and suggested that the eyes with corneal steepening may be at higher risk of endothelial dysfunction. In this study, there was, however, no difference in the mean keratometry of patients that required keratoplasty versus those who did not. More studies with a long-term follow-up are needed to investigate this association. It has been observed that in bilateral cases of PPCD, the more affected eye has a greater degree of astigmatic error, presumably causing amblyopia in that eye [11]. Also, there are a few reports of unilateral PPCD with an astigmatic error in the affected eye and hence leading to anisometric/ meridional amblyopia [12]. We found that 5/9 cases of unilateral PPCD had meridional amblyopia in the affected eye. An important difference in this study population was a relatively low frequency of iris and angle anomalies. The good prognosis following keratoplasty can be attributed to the absence of anterior segment lesions in this study population. Although one patient had unilateral involvement of the anterior segment and underwent keratoplasty (DSEK) in both eyes, there was no evidence of raised intraocular pressure and disc changes in the follow-up period of 8 years.
The literature on PPCD from different geographical areas reports a wide and variable range of phenotypic parameters. Most of the clinical studies are limited to case reports and a few large series. Apart from the endothelial morphological abnormalities, reduced endothelial cell densities have been shown in most studies [1, 10]. The association with steep corneas and glaucoma is variably reported [6, 7, 12–14]. Some studies have also reported an association with extraocular abnormalities [15, 16]. In our study population, all proposed features were noted except for glaucoma and associated extraocular abnormalities.
This study was a retrospective analysis, and hence, the genetic characterization was not ascertained in the patient population. Genetic analysis is in way for the some of the subjects included in this study. Utmost care was taken to include the subjects in the study. Only those records where a proper documentation was available and/or histology was corroborative were included in the analysis.
The variability in the clinical presentation and the course of this condition needs a continued investigation. Considering the variable phenotypes in this relative rare endothelial dystrophy and paucity of literature on its prevalence, it would be useful to have collaborative multicenter studies to ascertain its true prevalence and genotype–phenotype clinical correlation from various parts of the world.
References
Cibis GW, Krachmer JA, Phelps CD, Weingeist TA (1977) The clinical spectrum of posterior polymorphous dystrophy. Arch Ophthalmol 95(9):1529–1537
Heon E et al (1995) Linkage of posterior polymorphous corneal dystrophy to 20q11. Hum Mol Genet 4(3):485–488
Biswas S et al (2001) Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet 10(21):2415–2423
Shimizu S et al (2004) A locus for posterior polymorphous corneal dystrophy (PPCD3) maps to chromosome10. Am J Med Genet A 130A(4):372–377
Liskova P, Gwilliam R, Filipec M, Jirsova K, Reinstein Merjava S, Deloukas P et al (2012) High prevalence of posterior polymorphous corneal dystrophy in the Czech Republic; linkage disequilibrium mapping and dating an ancestral mutation. PLoS One 7(9):e45495
Aldave AJ, Ann LB, Frausto RF et al (2013) Classification of posterior polymorphous corneal dystrophy as a corneal ectatic disorder following confirmation of associated significant corneal steepening. JAMA Ophthalmol 131(12):1583–1590
Shen J, Chixin D, Gu Y (2015) Long-term observation of coexistence of posterior polymorphous corneal dystrophy, resultant high myopia and nonkeratoconic developing corneal astigmatism: a case report of 7-year tracking in a Chinese boy. Medicine (Baltimore) 94(23):e921
Teekhasaenee C, Nimmanit S, Wutthiphan S, Vareesangthip K, Laohapand T, Malasitr P, Ritch R (1991) Posterior polymorphous dystrophy and Alport syndrome. Ophthalmology 98(8):1207–1215
Sella R, Rootman D, Bahar I (2013) Descemet’s stripping automated endothelial keratoplasty for posterior polymorphous corneal dystrophy in an 8-month-old boy. J AAPOS 17(1):94–96
Krachmer JH (1985) Posterior polymorphous corneal dystrophy: a disease characterized by epithelial-like endothelial cells which influence management and prognosis. Trans Am Ophthalmol Soc 83:413–475
DeRespinis PA, Norden RA, Rispoli LC (1996) Posterior polymorphous dystrophy associated with astigmatism and amblyopia in children. J Refract Surg 12(6):709–714
Bozkurt B, Ozkan F, Yilmaz M, Okudan S (2015) Posterior corneal steepening in posterior polymorphous corneal dystrophy. Optom Vis Sci 92(11):414–419
Raber IM, Fintelmann R, Chhabra S, Ribeiro MP, Eagle RC Jr, Orlin SE (2011) Posterior polymorphous dystrophy associated with non-keratoconic steep corneal curvatures. Cornea 30(10):1120–1124
Pang CJ, Jing Y, Li J, Song XH, Wang LY (2011) Clinical observation of posterior polymorphous corneal dystrophy. Zhonghua Yan Ke Za Zhi 47(1):17–21
Jang MS, Roldan AN, Frausto RF, Aldave AJ (2014) Posterior polymorphous corneal dystrophy 3 is associated with agenesis and hypoplasia of the corpus callosum. Vis Res 100:88–92
Nguyen DQ, Hosseini M, Billingsley G, Héon E, Churchill AJ (2010) Clinical phenotype of posterior polymorphous corneal dystrophy in a family with a novel ZEB1 mutation. Acta Ophthalmol 88(6):695–699
Acknowledgments
We would like to thank Dr. Dilip Kumar Mishra, Mr. Sridhar Rao, and Mr. Naidu from the Ocular Pathology Services, LV Prasad Eye Institute for providing histology support.
Author's Contribution
Design and conduct of the study: SCH; collection, management, analysis, and interpretation of the data: SCH, RM, BG; preparation of the manuscript: SCH; review of the manuscript: SCH, MDR; final approval of the manuscript: SCH, MDR, RM, BG, DM, SIM.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
None.
Rights and permissions
About this article

Cite this article
Chaurasia, S., Mittal, R., Bichappa, G. et al. Clinical characterization of posterior polymorphous corneal dystrophy in patients of Indian ethnicity. Int Ophthalmol 37, 945–952 (2017). https://doi.org/10.1007/s10792-016-0360-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10792-016-0360-y