
Abstract
Background
Epidemiological evidence suggests that there is an association between rheumatoid arthritis (RA) and Alzheimer’s disease (AD). However, the causal relationship between RA and AD remains unclear. Therefore, this study aimed to investigate the causal relationship between RA and AD.
Methods
Using publicly available genome-wide association study datasets, bidirectional two-sample Mendelian randomization (TSMR) was performed using the inverse-variance weighted (IVW), weighted median, MR‒Egger regression, simple mode, and weighted mode methods.
Results
The results of MR for the causal effect of RA on AD (IVW, odds ratio [OR] = 0.959, 95% confidence interval [CI]: 0.941–0.978, P = 2.752E-05; weighted median, OR = 0.960, 95% CI: 0.937–0.984, P = 0.001) revealed a causal association between genetic susceptibility to RA and an increased risk of AD. The results of MR for the causal effect of AD on RA (IVW, OR = 0.978, 95% CI: 0.906–1.056, P = 0.576; weighted median, OR = 0.966, 95% CI: 0.894–1.043, P = 0.382) indicated that there was no causal association between genetic susceptibility to AD and an increased risk of RA.
Conclusions
The results of this two-way two-sample Mendelian randomization analysis revealed a causal association between genetic susceptibility to RA and a reduced risk of AD but did not reveal a causal association between genetic susceptibility to AD and an increased or reduced risk of RA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a chronic systemic autoimmune disease with a prevalence of 0.5–1% (Smolen et al. 2016). RA affects multiple tissues and organs and causes chronic synovitis, which ultimately leads to joint destruction, chronic disability and shortened life expectancy (Song and Lin 2017). RA occurs due to an interaction between genetic susceptibility, environmental factors and immunological factors, with genetic factors accounting for 50–60% of the risk of RA (Van Der Woude and Van Der Helm-Van Mil 2018; Venetsanopoulou et al. 2022). The disease is characterized by a systemic inflammatory response affecting articular cartilage and bone (Smolen et al. 2016). Alzheimer’s disease is a neurological disorder and neurodegenerative disease (Long and Holtzman 2019). It is the leading cause of cognitive impairment and dementia in people over 65 years of age worldwide (Atri 2019). AD is characterized by a long-term, progressive disease process that begins with pathophysiological changes in the brain years before any clinical manifestations are observed in affected individuals (Jack et al. 2013). It is estimated that by 2050, the global prevalence of AD will exceed 152 million people, and as the population ages, the annual cost of Alzheimer’s disease could exceed $600 billion (Lane et al. 2018; Trzeciak et al. 2021). Its pathophysiological manifestations include accumulation of the toxic substance β-amyloid (amyloid-β, Aβ), formation of neuroprotective fibre tangles by overphosphorylated tau proteins, and neurodegeneration due to secretion of neurotoxins and inflammatory factors (Scheltens et al. 2016; Li et al. 2022; Xu et al. 2022).
Although the relationship between RA and AD is not fully understood, the pathogenesis of both diseases exhibits a chronic inflammatory response and an overreaction of the immune system (Xu et al. 2022). The dysregulation of multiple genetic and environmental factors involved in the inflammatory cascade is a common feature of both RA and AD (Ferraccioli et al. 2012). Existing studies have shown a positive correlation between AD and RA, and observational studies have demonstrated that the incidence of AD is much higher in the population of patients with RA than in healthy individuals (Lin et al. 2018). In another study, people with arthritic diseases, especially RA, had cognitive decline later in life (Wallin et al. 2012). However, some studies have also indicated that there is a negative correlation between RA and AD, that RA may be a protective factor against AD and that the incidence of AD is lower in people with RA (Breitner et al. 1994). These studies have used a variety of methods. However, most of them used small samples of patients with AD and RA; there is a lack of large-sample studies. While researchers have suggested that RA protects against AD, this may be due to the role of nonsteroidal anti-inflammatory drugs (NSAIDs) and methotrexate, which are used to treat RA and may have influenced the observed results (McGeer et al. 1990; Zandi and Breitner 2001). However, in a 7-year randomized controlled trial (RCT), namely, the Alzheimer’s Disease Anti-Inflammatory Prophylaxis Trial (AADPT), the results showed that treatment with naproxen and celecoxib did not reduce the incidence of AD. The results showed that naproxen and celecoxib treatment did not reduce the incidence of AD (Alzheimer’s Disease Anti-inflammatory Prevention Trial Research Group 2013). In addition, the use of etanercept, a TNF-α inhibitor, did not cause significant changes in cognition, behaviour or overall functioning among AD patients (Butchart et al. 2015). These findings suggest that the role of anti-inflammatory drugs in the prevention of AD in patients with RA may have been exaggerated (Price 2003).
Elucidating the causal relationship between rheumatoid arthritis and Alzheimer’s disease is, therefore, crucial for prevention and treatment, but it is unclear whether such a causal relationship exists. Due to various confounding factors in observational clinical studies, observations often do not provide convincing answers regarding the causal relationship between rheumatoid arthritis and Alzheimer’s disease. Causal inferences from observational studies are susceptible to bias due to reverse causation and potential confounders (Evans and Smith 2015), which weakens our understanding of the causal relationship between rheumatoid arthritis and Alzheimer’s disease. Randomized controlled trials (RCTs) are the gold standard for causal inference in epidemiological studies. Some randomized controlled trials are difficult to perform for reasons of medical ethics, subject selection and extrapolation of results. Mendelian randomization (MR) is a technique that uses genetic variation as an instrumental variable (IV) to assess whether observed associations between exposure factors and outcomes are consistent with causal effects (Bae and Lee 2018a). Genetic variation is not influenced by external environmental, social behaviour or other factors and is a stable exposure factor over time. MR can minimize bias by avoiding the influence of confounders and reverse causality on the effects of interest in observational studies. In this study, previously published data were collected and analysed using a bidirectional two-sample Mendelian randomization (TSMR) study to determine whether there was a bidirectional causal relationship between RA and AD.
Methods and materials
Ethics/consent statement
No ethical approval or informed consent was needed, as this study was based on previously published articles and public databases.
Data sources
Relevant genome-wide association study (GWAS) datasets were obtained from the IEU OpenGWAS project (https://gwas.mrcieu.ac.uk). The GWAS dataset for RA was derived from a GWAS that included 3636 cases and 15,554 controls of European ancestry (Okada et al. 2014). The GWAS dataset for AD was derived from another GWAS that included 75,024 cases and 987,844 controls of European ancestry (Schwartzentruber et al. 2021) (Supplementary Table 1).
Screening of IVs
Single-nucleotide polymorphisms (SNPs) were used as IVs, and a P value < 5.0 × 10–8 was set as a threshold. To avoid linkage disequilibrium (LD) bias, LD with significant SNPs associated with exposure factors must meet the following conditions: r2 < 0.001 and a genetic distance of 10,000 kb. SNPs that are significantly associated with exposure factors were extracted from the GWAS dataset of outcome variables, and the resulting IVs were examined to extract the following information: the effect allele, allele effect sizes (beta), standard error, and p value. The F-statistic was used to test the strength of each IV and was calculated using the following formula: F = R2(N − 2)/(1 − R2), where R2 is the proportion of the exposure factor variation explained by each IV (Chen et al. 2022), and N is the sample size of the exposure dataset. When F > 10, there is no weak IV bias (Burgess and Thompson 2011).
Research design
To better estimate the causal effect, three key assumptions should be met when SNPs are used as IVs in the TSMR analysis (Burgess et al. 2013): (1) IVs must be closely related to exposure factors; (2) IVs are independent of confounding factors; and (3) IVs can only influence the outcome through exposure and not through other pathways.
Statistical analysis
Summary statistics for the exposure and outcome datasets were harmonized such that the effect of SNPs on exposure and the effect of SNPs on outcome corresponded to the same alleles. TSMR analyses using inverse-variance weighted (IVW), weighted median, MR‒Egger regression, simple mode, and weighted mode methods were performed to infer causal associations, and weighted mode methods were performed to infer causal associations. We used IVW as the primary method for MR. When each genetic variation met the IVW hypothesis, the IVW method combined with the IVW method was used. hypothesis, the IVW method combined the Wald ratio estimates of the causal effects of different SNPs and provided a consistent estimate of the causal effect of exposure on the outcome effect of exposure on the outcome (Bae and Lee 2018b). The results of the IVW method were most reliable when there was no horizontal pleiotropy of the IVs (Huang et al. 2021). When at least half of the SNPs are effective IVs, the weighted median can provide a consistent estimate of the causal effect (Bowden et al. 2016). MR‒Egger regression is used to confirm whether horizontal pleiotropy of IVs exists, and its intercept represents the effect estimate of horizontal pleiotropy (Burgess and Thompson 2017). When the IVs have horizontal pleiotropy, MR‒Egger regression can still obtain an unbiased estimation of causal association. The weighted median method improves the accuracy of the results compared to the IV method and improves the accuracy of the results compared to the MR‒Egger method (Xiang et al. 2021). Simple mode and weighted mode were used for complementary analyses (Hua et al. 2022). The Mendelian randomization pleiotropy residual sum and outlier (MR-PRESSO) test was used to detect and correct horizontal pleiotropy by removing the outliers (Lin et al. 2022). Statistical analysis was performed using R (version 4.3.1) and R packages (TwoSampleMR and MR-PRESSO). The test level α was 0.05 (P < 0.05), and the difference was statistically significant.
Results
Causal effects of RA on AD
SNPs
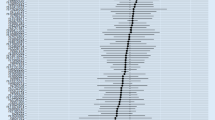
Basic information RA was the exposure factor, and AD was the outcome variable. In total, 56 SNPs were screened and identified as IVs, with F values greater than 10. (Supplementary Table 2). The intercept of the MR‒Egger regression can be used as an indicator to test whether horizontal pleiotropy of the IVs influences the results of TSMR analysis. The intercept was close to 0 (Egger intercept = 0.004, P = 0.074) (Table 1), indicating that there was no horizontal pleiotropy of the IVs, and it was unlikely to influence the results of the TSMR analysis (Fig. 1b).

Forest plot (a), scatter plot (b), sensitivity analysis (c), and funnel plot (d) of the effect of RA on AD
Two-sample Mendelian randomization analysis
The results of MR supported a causal association between genetic susceptibility to RA and reduced risk of AD, i.e., RA may be a protective factor against AD. In the absence of multivariate validity at the level of IVs, IVW was used as the primary method for estimating the causal association between genetic susceptibility to RA and reduced risk of AD (IVW results: OR = 0.959, 95% CI: 0.941–0.978, P = 2.752E-05). The results of other methods were as follows: MR‒Egger, OR = 0.940, 95% CI: 0.914–0.967, P = 0.0001; weighted median, OR = 0.960, 95% CI: 0.937–0.984, P = 0.001; simple mode, OR = 0.964, 95% CI: 0.913–1.017, P = 0.191; weighted mode, OR = 0.951, 95% CI: 0.932–0.971, P = 1.338E-05 (Table 2 and Fig. 1a and b).
Heterogeneity test and sensitivity analysis
Heterogeneity between IVs was detected using IVW and MR‒Egger regression analysis. Heterogeneity was quantified using Cochran’s Q test, and P < 0.05 indicated significant heterogeneity. If heterogeneity existed between IVs, causal effects were estimated using a random effects IVW model. MR‒Egger regression (Cochran’s Q = 83.52, P = 0.006) and IVW (Cochran’s Q = 88.64, P = 0.002) (Table 1 and Fig. 1d) showed heterogeneity among IVs, and therefore, causality was estimated using a random effects IVW model (P = 2.752644e-05). The causal effect of TSMR was estimated using the MR-PRESSO test to remove outlier SNPs (rs5019428) and correct for outliers (P = 0.00003) (Supplementary Table 3).
Sensitivity analysis was performed by removing SNPs one at a time, and the causal effects of the remaining SNPs were compared with the results of the TSMR analysis of all SNPs to determine whether the causal associations were due to a single IV. The sensitivity analysis indicated that the results of the TSMR analysis were robust (Fig. 1c).
Reverse TSMR analysis
In reverse TSMR, MI was the exposure factor, and RA was the outcome variable. In total, 32 SNPs were screened and identified as IVs, with F values greater than 10. The proportion of variance explained by these IVs was 2.6% for MI (Supplementary Table 4). The horizontal pleiotropy test (Egger intercept = − 0.003, P = 0.729) (Table 1) indicated that there was no horizontal pleiotropy for the IVs. The MR results did not support a causal association between the IVs and genetic susceptibility to AD or an increased risk of RA (IVW, OR = 0.978, 95% CI: 0.906–1.056, P = 0.576). The results of other methods were as follows: MR‒Egger, OR = 0.980, 95% CI: 0.870–1.144, P = 0.980; weighted median, OR = 0.966, 95% CI: 0.894–1.043, P = 0.094. − 1.043, P = 0.382; simple mode, OR = 0.892, 95% CI: 0.739–1.077, P = 0.245; and weighted mode, OR = 962, 95% CI: 0.889–1.040, P = 0.340 (Table 3 and Fig. 2a and b). Among the heterogeneity test results, MR‒Egger regression showed relatively small heterogeneity (Cochran’s Q = 63.76, P = 0.0003), IVW regression (Cochran’s Q = 15.18, P = 0.056) showed heterogeneity between IVs (Table 1 and Fig. 2d), and the random effects IVW model was used to estimate the causal relationship (P = 2.752644e-05). The MR-PRESSO test was used to remove the abnormal SNP (rs1859788 rs1878036), and the causal effect of TSMR was estimated after the outliers were corrected (P = 0.448) (Supplementary Table 3). Sensitivity analysis was performed using the leave-one-out method, and the results of TSMR analysis were found to be reliable (Fig. 2c).

Forest plot (a), scatter plot (b), sensitivity analysis (c), and funnel plot (d) of the effect of AD on RA
Discussion
This study analyzed previously published GWAS datasets using a bidirectional TSMR approach to determine whether there is a bidirectional causal association between RA and AD in a European population. Our results indicate that there is a causal association between genetic susceptibility to RA and a reduced risk of AD (IVW, OR = 0.959, 95% CI: 0.941–0.978, P = 2.752E-05). However, our results did not support a causal association between genetic susceptibility to AD and an increased or decreased risk of RA (IVW, OR = 0.978, 95% CI: 0.906–1.056, P = 0.576). Sensitivity analyses indicated that the results of MR were robust and reliable.
Paradoxically, previous observational studies have suggested that RA, an inflammatory disease in its own right, is also associated with risk factors for AD and is often considered a negative risk factor for the development of AD. Recently, a large-sample observational study based on Clinical Practice Research Datalink (CPRD) and a two-sample Mendelian study were performed to investigate the relationship between inflammatory diseases and AD (Huang et al. 2023). The results indicated that the relationship between inflammatory diseases and AD was not as strong as in previous studies (Chou et al. 2016; Yasuoka et al. 2023). The results showed that, consistent with previous observational studies, inflammatory diseases are associated with a higher risk of AD. However, these associations were not supported by MR analyses, and there was no causal relationship, suggesting that the association between inflammatory diseases and AD in observational studies is affected by confounding factors. However, the results of our two-way two-sample Mendelian randomization study suggest that RA may be a protective factor against AD, while the results of the reverse Mendelian randomization showed that AD does not increase the risk of developing RA and that there is no causal relationship. Our results are in agreement with the MR results of Huang et al. (2023) and Policicchio et al. (2017), probably because a different GWAS AD dataset was selected. However, the results of this study are consistent with the previous results of a simple two-sample Mendelian randomization by Bae and Lee (2019). In contrast, we conducted a more comprehensive two-way two-sample Mendelian randomization analysis while applying a newer and larger GWAS database for the study, thus making our results more convincing.
There may be two mechanisms underlying the role of RA as a protective factor against AD. First, this causal effect may be due to the deregulation of cellular and molecular regulatory mechanisms intrinsic to the inflammatory cascade response (Ferraccioli et al. 2012). In an animal study, researchers investigated the effect of RA on β-amyloid (Aβ) disposition in collagen-induced arthritis (CIA) rats and found that microglial hyperplasia and astrocyte hyperplasia were both significantly increased in the brains of CIA rats compared to controls and that these glial cells have a significant effect on neuronal responses (Lai et al. 2021). These glial cells are essential for neuronal and brain health (Castellani and Schwartz 2020; Linnerbauer et al. 2020; Vainchtein and Molofsky 2020). Although the effects of glial cells on Aβ and tau accumulation and clearance are unknown, researchers realize that these are important interactions with potential for therapeutic intervention (Fakhoury 2018; Long and Holtzman 2019). McAlpine et al. (2021) found in mice and humans that astrocyte-derived interleukin-3 (IL-3) reprogrammed microglia to ameliorate Alzheimer’s disease pathology. Upon recognition of Aβ deposition, microglia increase IL-3R (the specific receptor for IL-3), making them responsive to IL-3. Astrocytes constitutively produce IL-3, which triggers transcriptional, morphological and functional reprogramming of microglia, endowing them with an acute immune response program, enhanced motility and the ability to aggregate and clear Aβ and tau aggregates (McAlpine et al. 2021), and astrocytes themselves can take up and degrade Aβ. These changes limit the pathology and cognitive decline in Alzheimer’s disease (McAlpine et al. 2021). In addition, some ADs indicate the potential role of microglia in synaptic remodelling (Wake et al. 2009). Microglia perceive neuronal activity, thereby regulating synaptic plasticity and learning and memory mechanisms. Therefore, they are the main components that determine cognitive function (Morris et al. 2013; Sipe et al. 2016). It has also been found that changes in the expression of microglia-related genes may promote late-onset AD (LOAD) (Hansen et al. 2018).
Second, it has been shown that granulocyte macrophage colony stimulating factor (GM-CSF) is produced in large quantities in RA synovitis and that GM-CSF signalling plays an important role in structural plasticity associated with learning and memory and protects neurons in traumatic brain injury (TBI) mice (Schäbitz et al. 2008; Krieger et al. 2012; Shultz et al. 2014). Additionally, GM-CSF appears to have trophic effects on neuronal cells, such as enhancing brain functions, including short-term memory skills (Boyd et al. 2010; Kiyota et al. 2018). On one hand, GM-CSF induces the differentiation of myeloid cells into specialized cells such as microglia, which transform them into appropriate amyloid scavenging factories that remove amyloid deposits. Amyloid accumulation plays a special role in the pathogenesis of Alzheimer's disease (AD) (Boyd et al. 2010). On the other hand, GM-CSF promotes the recruitment of mononuclear macrophages from the peripheral blood into the brain and affects the clearance of Aβ plaques (Boyd et al. 2010; Darlington et al. 2015; Fu et al. 2016). In addition, GM-CSF attenuates the proinflammatory microglial phenotype in AD (Zuroff et al. 2017).
MR uses genetic variants to estimate the health effects of phenotypes affected by those genetic variants (Riaz et al. 2018). This is a relatively novel epidemiological approach that uses genetic variation to infer causal relationships between exposure factors and outcome variables. MR provides a method of investigating associations that is free of the typical biases inherent in observational epidemiological studies (e.g., reverse causation and potential confounders). Our results differ from the MR results of Cai et al. (2018), possibly because a different GWAS dataset was selected. Therefore, future studies with newer, larger GWAS datasets are needed.
This study has some limitations. First, the MR results are based on a European population, and extrapolation of the results is limited. Further studies are needed to confirm whether causality exists in other populations. Second, the SNPs used for analysis may be correlated with other traits due to genetic polymorphisms, thus leading to confounding bias that could affect causal inference. Third, the strength of IV depends on the sample size of the GWAS, and a larger GWAS is needed to identify more genetic variation in MR.
Conclusions
In conclusion, bidirectional TSMR analyses support a causal association of genetic susceptibility to RA as a protective factor for AD but do not support a causal association between genetic susceptibility to AD and increased or decreased risk of RA. However, due to the limitations of the study, further research is necessary.
Data availability
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Code availability
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
References
Alzheimer’s Disease Anti-inflammatory Prevention Trial Research Group (2013) Results of a follow-up study to the randomized Alzheimer’s disease anti-inflammatory prevention trial (ADAPT). Alzheimers Dement 9:714–723. https://doi.org/10.1016/j.jalz.2012.11.012
Atri A (2019) The Alzheimer’s disease clinical spectrum: diagnosis and management. Med Clin North Am 103:263–293. https://doi.org/10.1016/j.mcna.2018.10.009
Bae SC, Lee YH (2018a) Coffee consumption and the risk of rheumatoid arthritis and systemic lupus erythematosus: a Mendelian randomization study. Clin Rheumatol 37:2875–2879. https://doi.org/10.1007/s10067-018-4278-9
Bae SC, Lee YH (2018b) Vitamin D level and risk of systemic lupus erythematosus and rheumatoid arthritis: a Mendelian randomization. Clin Rheumatol 37:2415–2421. https://doi.org/10.1007/s10067-018-4152-9
Bae SC, Lee YH (2019) Causal association between rheumatoid arthritis and a decreased risk of Alzheimer’s disease: a Mendelian randomization study. Z Rheumatol 78:359–364. https://doi.org/10.1007/s00393-018-0504-8
Bowden J, Smith GD, Haycock PC, Burgess S (2016) Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 40:304–314. https://doi.org/10.1002/gepi.21965
Boyd TD, Bennett SP, Mori T, Governatori N, Runfeldt M, Norden M, Padmanabhan J, Neame P, Wefes I, Sanchez-Ramos J, Arendash GW, Potter H (2010) GM-CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. J Alzheimers Dis 21:507–518. https://doi.org/10.3233/jad-2010-091471
Breitner JC, Gau BA, Welsh KA, Plassman BL, McDonald WM, Helms MJ, Anthony JC (1994) Inverse association of anti-inflammatory treatments and Alzheimer’s disease: initial results of a co-twin control study. Neurology 44:227–232. https://doi.org/10.1212/wnl.44.2.227
Burgess S, Thompson SG (2011) Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol 40:755–764. https://doi.org/10.1093/ije/dyr036
Burgess S, Thompson SG (2017) Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol 32:377–389. https://doi.org/10.1007/s10654-017-0255-x
Burgess S, Butterworth A, Thompson SG (2013) Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 37:658–665. https://doi.org/10.1002/gepi.21758
Butchart J, Brook L, Hopkins V et al (2015) Etanercept in Alzheimer disease: a randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 84:2161–2168. https://doi.org/10.1212/wnl.0000000000001617
Cai Q, Xin Z, Zuo L, Li F, Liu B (2018) Alzheimer’s disease and rheumatoid arthritis: a Mendelian randomization study. Front Neurosci 12:627. https://doi.org/10.3389/fnins.2018.00627
Castellani G, Schwartz M (2020) Immunological features of non-neuronal brain cells: implications for Alzheimer’s disease immunotherapy. Trends Immunol 41:794–804. https://doi.org/10.1016/j.it.2020.07.005
Chen H, Ye R, Guo X (2022) Lack of causal association between heart failure and osteoporosis: a Mendelian randomization study. BMC Med Genomics 15:232. https://doi.org/10.1186/s12920-022-01385-8
Chou RC, Kane M, Ghimire S, Gautam S, Gui J (2016) Treatment for rheumatoid arthritis and risk of Alzheimer’s disease: a nested case-control analysis. CNS Drugs 30:1111–1120. https://doi.org/10.1007/s40263-016-0374-z
Darlington D, Li S, Hou H, Habib A, Tian J, Gao Y, Ehrhart J, Sanberg PR, Sawmiller D, Giunta B, Mori T, Tan J (2015) Human umbilical cord blood-derived monocytes improve cognitive deficits and reduce amyloid-β pathology in PSAPP mice. Cell Transplant 24:2237–2250. https://doi.org/10.3727/096368915x688894
Evans DM, Smith GD (2015) Mendelian randomization: new applications in the coming age of hypothesis-free causality. Annu Rev Genomics Hum Genet 16:327–350. https://doi.org/10.1146/annurev-genom-090314-050016
Fakhoury M (2018) Microglia and astrocytes in Alzheimer’s disease: implications for therapy. Curr Neuropharmacol 16:508–518. https://doi.org/10.2174/1570159x15666170720095240
Ferraccioli G, Carbonella A, Gremese E, Alivernini S (2012) Rheumatoid arthritis and Alzheimer’s disease: genetic and epigenetic links in inflammatory regulation. Discov Med 14:379–388
Fu Y, Hsiao JH, Paxinos G, Halliday GM, Kim WS (2016) ABCA7 mediates phagocytic clearance of amyloid-β in the brain. J Alzheimers Dis 54:569–584. https://doi.org/10.3233/jad-160456
Hansen DV, Hanson JE, Sheng M (2018) Microglia in Alzheimer’s disease. J Cell Biol 217:459–472. https://doi.org/10.1083/jcb.201709069
Hua L, Xiang S, Xu R, Xu X, Liu T, Shi Y, Wu L, Wang R, Sun Q (2022) Causal association between rheumatoid arthritis and celiac disease: a bidirectional two-sample Mendelian randomization study. Front Genet 13:976579. https://doi.org/10.3389/fgene.2022.976579
Huang S, Tian F, Yang X, Fang S, Fan Y, Bao J (2021) Physical activity and systemic lupus erythematosus among European populations: a two-sample Mendelian randomization study. Front Genet 12:784922. https://doi.org/10.3389/fgene.2021.784922
Huang J, Su B, Karhunen V et al (2023) Inflammatory diseases, inflammatory biomarkers, and Alzheimer disease: an observational analysis and Mendelian randomization. Neurology 100:e568–e581. https://doi.org/10.1212/wnl.0000000000201489
Jack CR, Knopman DS, Jagust WJ et al (2013) Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12:207–216. https://doi.org/10.1016/s1474-4422(12)70291-0
Kiyota T, Machhi J, Lu Y, Dyavarshetty B, Nemati M, Yokoyama I, Mosley RL, Gendelman HE (2018) Granulocyte-macrophage colony-stimulating factor neuroprotective activities in Alzheimer’s disease mice. J Neuroimmunol 319:80–92. https://doi.org/10.1016/j.jneuroim.2018.03.009
Krieger M, Both M, Kranig SA, Pitzer C, Klugmann M, Vogt G, Draguhn A, Schneider A (2012) The hematopoietic cytokine granulocyte-macrophage colony stimulating factor is important for cognitive functions. Sci Rep 2:697. https://doi.org/10.1038/srep00697
Lai PH, Wang TH, Zhang NY, Wu KC, Yao CJ, Lin CJ (2021) Changes of blood-brain-barrier function and transfer of amyloid beta in rats with collagen-induced arthritis. J Neuroinflammation 18:35. https://doi.org/10.1186/s12974-021-02086-2
Lane CA, Hardy J, Schott JM (2018) Alzheimer’s disease. Eur J Neurol 25:59–70. https://doi.org/10.1111/ene.13439
Li Q, Wu Y, Chen J, Xuan A, Wang X (2022) Microglia and immunotherapy in Alzheimer’s disease. Acta Neurol Scand 145:273–278. https://doi.org/10.1111/ane.13551
Lin TM, Chen WS, Sheu JJ, Chen YH, Chen JH, Chang CC (2018) Autoimmune rheumatic diseases increase dementia risk in middle-aged patients: a nationwide cohort study. PLoS ONE 13:e0186475. https://doi.org/10.1371/journal.pone.0186475
Lin L, Luo P, Yang M, Wang J, Hou W, Xu P (2022) Causal relationship between osteoporosis and osteoarthritis: a two-sample Mendelian randomized study. Front Endocrinol (lausanne) 13:1011246. https://doi.org/10.3389/fendo.2022.1011246
Linnerbauer M, Wheeler MA, Quintana FJ (2020) Astrocyte crosstalk in CNS inflammation. Neuron 108:608–622. https://doi.org/10.1016/j.neuron.2020.08.012
Long JM, Holtzman DM (2019) Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179:312–339. https://doi.org/10.1016/j.cell.2019.09.001
McAlpine CS, Park J, Griciuc A et al (2021) Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature 595:701–706. https://doi.org/10.1038/s41586-021-03734-6
McGeer PL, McGeer E, Rogers J, Sibley J (1990) Anti-inflammatory drugs and Alzheimer disease. Lancet 335:1037. https://doi.org/10.1016/0140-6736(90)91101-f
Morris GP, Clark IA, Zinn R, Vissel B (2013) Microglia: a new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol Learn Mem 105:40–53. https://doi.org/10.1016/j.nlm.2013.07.002
Okada Y, Wu D, Trynka G et al (2014) Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506:376–381. https://doi.org/10.1038/nature12873
Policicchio S, Ahmad AN, Powell JF, Proitsi P (2017) Rheumatoid arthritis and risk for Alzheimer’s disease: a systematic review and meta-analysis and a Mendelian randomization study. Sci Rep 7:12861. https://doi.org/10.1038/s41598-017-13168-8
Price D (2003) Effect of NSAIDs on risk of Alzheimer’s disease: conclusions on NSAIDs and Alzheimer’s disease were overstated. BMJ 327:752. https://doi.org/10.1136/bmj.327.7417.752
Riaz H, Khan MS, Siddiqi TJ, Usman MS, Shah N, Goyal A, Khan SS, Mookadam F, Krasuski RA, Ahmed H (2018) Association between obesity and cardiovascular outcomes: a systematic review and meta-analysis of Mendelian randomization studies. JAMA Netw Open 1:e183788. https://doi.org/10.1001/jamanetworkopen.2018.3788
Schäbitz WR, Krüger C, Pitzer C et al (2008) A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF). J Cereb Blood Flow Metab 28:29–43. https://doi.org/10.1038/sj.jcbfm.9600496
Scheltens P, Blennow K, Breteler MM, De Strooper B, Frisoni GB, Salloway S, Van Der Flier WM (2016) Alzheimer’s disease. Lancet 388:505–517. https://doi.org/10.1016/s0140-6736(15)01124-1
Schwartzentruber J, Cooper S, Liu JZ et al (2021) Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat Genet 53:392–402. https://doi.org/10.1038/s41588-020-00776-w
Shultz SR, Tan XL, Wright DK, Liu SJ, Semple BD, Johnston L, Jones NC, Cook AD, Hamilton JA, O’Brien TJ (2014) Granulocyte-macrophage colony-stimulating factor is neuroprotective in experimental traumatic brain injury. J Neurotrauma 31:976–983. https://doi.org/10.1089/neu.2013.3106
Sipe GO, Lowery RL, Tremblay M, Kelly EA, Lamantia CE, Majewska AK (2016) Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat Commun 7:10905. https://doi.org/10.1038/ncomms10905
Smolen JS, Aletaha D, McInnes IB (2016) Rheumatoid arthritis. Lancet 388:2023–2038. https://doi.org/10.1016/s0140-6736(16)30173-8
Song X, Lin Q (2017) Genomics, transcriptomics and proteomics to elucidate the pathogenesis of rheumatoid arthritis. Rheumatol Int 37:1257–1265. https://doi.org/10.1007/s00296-017-3732-3
Trzeciak P, Herbet M, Dudka J (2021) Common factors of Alzheimer’s disease and rheumatoid arthritis-pathomechanism and treatment. Molecules 26:6038. https://doi.org/10.3390/molecules26196038
Vainchtein ID, Molofsky AV (2020) Astrocytes and microglia: in sickness and in health. Trends Neurosci 43:144–154. https://doi.org/10.1016/j.tins.2020.01.003
Van Der Woude D, Van Der Helm-Van Mil AHM (2018) Update on the epidemiology, risk factors, and disease outcomes of rheumatoid arthritis. Best Pract Res Clin Rheumatol 32:174–187. https://doi.org/10.1016/j.berh.2018.10.005
Venetsanopoulou AI, Alamanos Y, Voulgari PV, Drosos AA (2022) Epidemiology of rheumatoid arthritis: genetic and environmental influences. Expert Rev Clin Immunol 18:923–931. https://doi.org/10.1080/1744666x.2022.2106970
Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J (2009) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29:3974–3980. https://doi.org/10.1523/jneurosci.4363-08.2009
Wallin K, Solomon A, Kåreholt I, Tuomilehto J, Soininen H, Kivipelto M (2012) Midlife rheumatoid arthritis increases the risk of cognitive impairment two decades later: a population-based study. J Alzheimers Dis 31:669–676. https://doi.org/10.3233/jad-2012-111736
Xiang K, Wang P, Xu Z et al (2021) Causal effects of gut microbiome on systemic lupus erythematosus: a two-sample Mendelian randomization study. Front Immunol 12:667097. https://doi.org/10.3389/fimmu.2021.667097
Xu Y, Yan H, Zhang X, Zhuo J, Han Y, Zhang H, Xie D, Lan X, Cai W, Wang X, Wang S, Li X (2022) Roles of altered macrophages and cytokines: implications for pathological mechanisms of postmenopausal osteoporosis, rheumatoid arthritis, and Alzheimer’s disease. Front Endocrinol (lausanne) 13:876269. https://doi.org/10.3389/fendo.2022.876269
Yasuoka M, Tange C, Nishita Y, Tomida M, Watanabe R, Shimokata H, Otsuka R, Kojima M (2023) Longitudinal changes in physical and cognitive functions among participants with and without rheumatoid arthritis in community-dwelling middle-aged and older adults. Ann Geriatr Med Res 27:58–65. https://doi.org/10.4235/agmr.22.0142
Zandi PP, Breitner JC (2001) Do NSAIDs prevent Alzheimer’s disease? And, if so, why? The epidemiological evidence. Neurobiol Aging 22:811–817. https://doi.org/10.1016/s0197-4580(01)00297-4
Zuroff L, Daley D, Black KL, Koronyo-Hamaoui M (2017) Clearance of cerebral Aβ in Alzheimer’s disease: reassessing the role of microglia and monocytes. Cell Mol Life Sci 74:2167–2201. https://doi.org/10.1007/s00018-017-2463-7
Funding
This study is supported by the Natural Science Foundation of Gansu Province, China. (20JR10RA358).
Author information
Authors and Affiliations
Contributions
GSL, YZY, GRM and PFL: conceptualization, investigation, data curation, writing—original draft. ZZZ, FKZ, QHC and ARZ: conceptualization, investigation, writing—original draft. HF, XY: investigation, writing—review and editing. HZG: conceptualization, writing—review and editing. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval
No ethical approval or informed consent was needed, as this study was based on previously published articles and public databases.
Consent to participate
Not applicable to this article.
Consent to publish
Not applicable to this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, GS., Yang, YZ., Ma, GR. et al. Rheumatoid arthritis is a protective factor against Alzheimer’s disease: a bidirectional two-sample Mendelian randomization study. Inflammopharmacol 32, 863–871 (2024). https://doi.org/10.1007/s10787-023-01397-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-023-01397-5