
Abstract
Fibroblast-like synoviocytes (FLSs), the main pathological cells in rheumatoid arthritis (RA), display tumor-like phenotype, including hyper-proliferation, apoptosis resistance, and aggressive phenotype. Excessive proliferation and insufficient apoptosis of RA-FLSs can lead to hyperplastic synovial pannus tissue, excess production of inflammatory mediators, and destruction of joints. In this article, we investigate the effect of PRIMA-1MET on the apoptosis induction and inhibition of pro-inflammatory cytokines in RA-FLSs. Synovial tissue samples were obtained from 10 patients with RA. The FLSs were treated with different concentrations of PRIMA-1MET. The rate of apoptosis and cell survival was assessed by flow cytometry and MTT assay and Real-time quantitative PCR was performed to evaluate the transcription of p53, IL-6, IL-1β, TNF-α, Noxa, p21, PUMA, Bax, Survivin, and XIAP in treated RA-FLSs. The protein level of p53, IκBα, and phospho-IκBα were measured using Western blotting. The results showed that PRIMA-1MET induced apoptosis in RA-FLSs and increased significantly the expression of Noxa, and decreased significantly IL-6, IL-1β, p53, and phospho-IκBα expression. PRIMA-1MET can induce apoptosis in RA-FLSs through induction of Noxa expression while p53 was downregulated. Furthermore, PRIMA-1MET treatment results in the suppression of pro-inflammatory cytokine production and NF-κB inhibition. Given the role of p53 and NF-κB in RA-FLSs, PRIMA-1MET can be considered as a new therapeutic strategy for rheumatoid arthritis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is an inflammatory, chronic, and invasive autoimmune disease, with a prevalence of about 1%. RA is associated with irreversible joint disabilities, systemic complications, premature death, and socioeconomic burden (Hewagama and Richardson 2009). The main pathological features of RA are joint inflammation, proliferative synovitis, and destruction of cartilage and bone. Joint involvement is characterized by the formation of hypertrophied synovium that is called “pannus”. Pannus tissue is composed of aggressive fibroblast- and macrophage-like synoviocytes. The development of the pannus stimulates the production of degrading enzymes such as matrix metalloproteinases (MMPs) and pro-inflammatory cytokines that promote the destruction of cartilage and bone (Firestein 2003).
Intimal lining layer fibroblast-like synoviocytes (FLSs or type B synoviocytes) are among the most critical cells involved in the pathogenesis of RA. In the inflammatory milieu of RA synovium, FLSs display tumor-like phenotype, including hyper-proliferation, apoptosis resistance, and aggressive phenotype. Excessive proliferation and insufficient apoptosis of RA-FLSs can lead to hyperplastic synovial pannus tissue, excess production of inflammatory mediators, and destruction of joints. Therefore, RA-FLSs are directly responsible for inflammation, cartilage destruction, and autoimmunity (Bottini and Firestein 2013).
The p53 tumor suppressor gene plays a pivotal role in cell cycle regulation, particularly apoptosis. P53 protein has a protective role in many autoimmune and inflammatory diseases by inhibiting the production of inflammatory cytokines, extracellular matrix-degrading enzymes, and induction of apoptosis (Yamanishi et al. 2005). Several studies have shown that the function of p53 protein is impaired in malignant neoplasms and non-malignant diseases such as RA (Firestein et al. 1997; Greenblatt et al. 1994). P53 protein dysfunction is the result of somatic mutations caused by genotoxic stress (Yamanishi et al. 2005). It has been reported that more than 40% of p53 cDNAs in RA synovium have mutations, most of which have been found in FLSs (Inazuka et al. 2000). Besides, p53 is overexpressed in synovial samples of arthritis patients compared to healthy synovial samples (Firestein et al. 1996). Despite the increased expression of p53, apoptosis resistance in RA-FLSs has been reported which may be related to the dominant-negative mutations (Han et al. 1999).
P53 reactivation and induction of massive apoptosis 1 (PRIMA-1) is a small non-peptide molecule that was first introduced in the American Cancer Center as a compound with anti-cancer properties (Bykov et al. 2002b). PRIMA-1MET, the methylated form of drug, targets the p53 protein and restores the mutant p53 function. This molecule can induce apoptosis by repairing the mutant p53 protein by alkylating and activating the protein thiol groups (Lambert et al. 2009).
Besides, it has been shown that PRIMA-1-induced apoptosis in NB4 cells, acute promyelocytic leukemia (APL) cell line, was accompanied by repressed nuclear factor κB (NF-κB) activity (Farhadi et al. 2017). NF-κB signaling pathway plays an essential role in the formation of pannus, chronic inflammation, and cartilage destruction by promoting proliferation, inhibiting cell apoptosis, producing inflammatory cytokines, and metalloproteinases (Makarov 2001).
With regards to the PRIMA-1MET effects on apoptosis induction and downregulation of NF-κB activity and given the role of apoptosis and inflammation in RA pathogenesis, in the present study, we evaluated the effect of PRIMA-1MET on RA-FLSs. Our results showed that PRIMA-1MET could induce apoptosis and has an anti-inflammatory effect in RA-FLSs.
Materials and methods
Chemicals and reagents
PRIMA-1MET was purchased from Sigma-Aldrich (Sml1789, USA). Antibodies against CD90 (Thy-1) (ab225), CD13 (ac227663), CD44 (ab6124), CD68 (ab31630), fibroblast surface protein (ab11333), p53 (ab131442), IκBα (ab97783), and β-actin (ab8226) purchased from abcam (USA), and the antibody against phospho-IκBα (#2859) was obtained from cell signaling technology (USA). Anti-rabbit HRP conjugated (PZ5610) was from Kalazist (IRAN).
Study subject
Synovial tissue specimens from 10 inactive patients with rheumatoid arthritis who were candidates for knee replacement surgery were taken from an inflamed joint replacement tissue by an orthopedic surgeon at Shariati Hospital. The mean of patients’ ages and disease durations were 59.12 ± 11.81 and 18.6 ± 2.3 years, respectively. All patients fulfilled the American College of Rheumatology/European league against rheumatism 2010 criteria (ACR/EULAR 2010 criteria) for RA (Aletaha et al. 2010) and provided their written consent to participate in the current study. Prednisolone, Hydroxychloroquine, and Sulfasalazine were the major administrated drugs in patients. This study was approved by the Ethics Committees of Tehran University of Medical Sciences (Approval No: IR.TUMS.VCR.REC.1397.968).
FLS isolation and cell culture
The synovial tissue was first rinsed with 70% ethanol and phosphate-buffered saline (PBS) solution (Gibco) containing 2% penicillin/streptomycin (Biosera) and 2% amphotericin B (Sigma-Aldrich), and then was cut into 5-mm slices. After that, the fragmented tissue was digested and minced with 1 mg/ml collagenase type VIII (Sigma–Aldrich, USA) in high glucose Dulbecco's Modified Eagle’s Medium (DMEM) (Gibco, USA) at 37 °C for 80 min.
After centrifugation (1000 g, 10 min) and washing, the cells were resuspended in DMEM supplemented with 1% penicillin/streptomycin and 10% fetal bovine serum (FBS) (Biosera) and incubated in T25 culture flasks at humidified 37 °C with 5% CO2. The medium was replaced every 48 h and FLSs were sub-cultured when reached 70–80% confluency. FLSs at passage 3 were confirmed and stored in a nitrogen tank. For future experiments, FLSs from passages 4–6 were used.
FLS confirmation: immunofluorescence assay and flow cytometry
Flow cytometry and immunofluorescence assay were used to identify the purity of the isolated cells from synovial tissues. After the third passage, the cells were characterized using immunofluorescence assay for fibroblast surface protein and by four antibodies, CD44, CD13, CD68, and CD90, using flow cytometry. A fluorescence-activated cell sorting (FACS) calibur flow cytometer (BD Biosciences, USA) was used for the analysis.
Cytotoxicity analysis with MTT assay
FLSs were seeded at a density of 7.5 × 103 cells/well into a 48-well culture plate in triplicate and incubated overnight at 37 °C in a CO2 incubator, then treated with various concentrations (25, 30, 35, 40 and 55 μM) of PRIMA-1MET for 12 and 24 h. After that, cells were incubated with 50 μl of MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide] solution (Sigma-Aldrich, USA) for 4 h at 37 °C with 5% CO2. After 4 h incubation, the medium was discarded and 500 μl of dimethyl sulfoxide (DMSO) (Sigma–Aldrich, USA) was added to each well to solve the formazan and the absorbance was measured at 570 nm wavelength by an enzyme-linked immunosorbent assay (ELISA) reader (BioTek-ELx800, USA).
Detection of apoptosis by flow cytometry
FLSs were plated overnight in a 6-well culture plate at a density of 75 × 103 cells/well. The next day, the cells were exposed to different concentrations (25, 30, 35, 40 and 55 μM) of PRIMA-1MET for 24 h. After incubation, cells were harvested and annexin V-FITC and PI solution were added based on the manufacturer’s instruction. The resulting fluorescence was detected by BD FACSCalibur flow cytometer (BD biosciences, USA) and data analysis was performed with the FlowJo software (Tree Star, Ashland, USA) (version 7.6.1).
Quantitative real-time PCR (qRT-PCR)
After 24 h treatment of FLSs with 35 μM of PRIMA-1MET, total RNA was extracted using SinaPure-RNA kit (SinaClonBioScience, Iran). Complementary DNA (cDNA) synthesis was performed with a PrimeScript™ RT reagent kit (Takara BIO INC. Japan), according to the manufacturer's instruction. Quantitative real-time PCR was performed for gene expression analysis using the RealQ Plus Master Mix Green‐high ROX (Ampliqon, Denmark) on a StepOne Real-Time PCR System (Applied Biosystems, USA). Each sample was analyzed in triplicate and data were analyzed based on the comparative Ct (2−ΔΔCt) method. The expression of target genes was normalized to GAPDH expression.
Western blot analysis
After FLSs treatment (35 μM, 24 h), treated and untreated FLSs were lysed in radioimmunoprecipitation assay (RIPA) buffer (Cytomatingene, Iran) containing protease inhibitor cocktail (P8340, Sigma–Aldrich, USA). Equal amounts of cell lysates were used to separate proteins by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membrane (Thermo Scientific, USA). The PVDF was blocked with 5% skim milk (Sigma-Aldrich, USA), followed by incubation with primary antibodies including anti-IκBα, anti-phospho-IκBα, anti-P53 (1:1000), and anti-β-actin as the loading control. After washing three times with Tris Buffered Saline with Tween 20 (TBST), the blots were incubated with goat anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibodies. Finally, the protein expression levels were visualized on X-ray film using an enhanced chemiluminescence (ECL) kit (GE Healthcare, USA).
Statistical analysis
IBM SPSS software version 24 (SPSS Inc., Chicago, IL, USA) was used to perform the analysis. The normal distribution of variants was analyzed using the Kolmogorov–Smirnov test. Mean comparison analysis between two groups (treated and untreated) was performed using Student’s paired t or Wilcoxon signed-rank tests. P values less than 0.05 were considered statistically significant and all data were represented as the mean ± standard error of the mean (SEM). GraphPad Prism software version 8.0.2 (GraphPad Software, La Jolla California USA) was used for drawing the plots.
Results
Identification of FLSs
Flow cytometry and immunofluorescence assay were used to identify isolated cells from synovial tissues. After 3 passages, a typical bipolar configuration was seen under inverted microscopy which confirmed a morphologically homogeneous population of FLSs. Most cells (> 98%) expressed the fibroblast surface protein (Fig. 1a), and were positive for CD90 (99.44% ± 3.7%), CD44 (99.33 ± 2.21%), and CD13 (98.63 ± 2.06%) markers, and were negative for the expression of CD68 (0.16 ± 4.01%) (Fig. 1b). Synoviocytes from passages 4–6 were used for further experiments.

a Confirmation of purity of cultured FLSs by immunofluorescence assay. 1) Specific staining of FLSs using anti-Fibroblast Surface Protein (green). 2) Staining the cell nucleus using DAPI (blue). 3) Merge two pictures indicating the purity of the cultured FLS. (Inverted phase contrast microscope with 200 × magnification). b Characterization of FLSs. After 3 passages, isolated cells were analyzed by flow cytometry for CD90, CD44 and CD13, and CD68. FLSs are positive for CD90, CD44, and CD13 and negative for CD68. FLS, fibroblast-like synoviocytes; FITC, fluorescein isothiocyanate; DAPI, 4′,6-diamidino-2-phenylindole (Color figure online)
PRIMA-1MET reduces RA-FLSs viability
MTT assay was used to determine the effect of different doses of PRIMA-1MET on cell viability. The results showed that PRIMA-1MET treatment leads to decreased cell survival in a dose-dependent manner in RA-FLSs. Using various concentrations (25, 30, 35, 40 and 55 μM) of PRIMA-1MET for 12 and 24 h, we found a significant decrease in cell viability with approximately 65% and 99% at concentrations of 40 and 55 μM, respectively (Fig. 2).

Decreases cell viability and metabolic activity of RA-FLSs. FLSs were cultured in complete medium with different concentrations of PRIMA-1MET in 48-well plates for 12 and 24 h and cell viability was measured using the MTT assay. The metabolic activity of RA-FLSs upon PRIMA-1MET treatment decreased in a dose-dependent manner (n = 2). FLS fibroblast-like synoviocytes, RA rheumatoid arthritis, MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, CO control, DMSO dimethyl sulfoxide
PRIMA-1MET induces apoptosis in RA-FLSs
To determine the cell death induced by PRIMA-1MET through apoptosis, annexin V/PI staining by flow cytometry was used. As shown in Fig. 3, the rate of apoptosis in PRIMA-1MET-treated-RA-FLSs was increased in a dose-dependent manner. The percentage of annexin V positive cells after treatment of RA-FLSs with 25, 30, 35, 40 and 55 μM of PRIMA-1MET was increased by 7.5, 16.11, 35.73, 61.75 and 95.38%, respectively.

Apoptotic effect of PRIMA-1MET on RA-FLSs. Cells were treated with different concentrations (25, 30, 35, 40, and 55 μM) of PRIMA-1MET for 24 h. Then cells were analyzed for annexin-V, PI by flow cytometry (n = 2). FLS fibroblast-like synoviocytes, RA rheumatoid arthritis, PI propidium iodide, CO control, DMSO dimethyl sulfoxide, FITC fluorescein isothiocyanate
PRIMA-1MET treatment downregulates p53 gene and protein expression in RA-FLSs
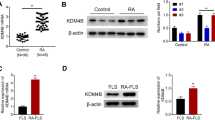
Previous studies have shown that PRIMA-1 treatment in cancer cells has no effect on p53 expression and only affects p53 protein function (Farhadi et al. 2017; Izetti et al. 2014). As described in Fig. 4, using quantitative RT-PCR and Western blotting, there is a significant decrease in p53 expression at the gene (P value = 0.002) and protein levels in the treated group compared to the control group (Supplementary Fig. 1).

Downregulation of p53 in RA-FLSs by PRIMA-1MET. a The effect of PRIMA-1MET on p53 protein expression. After exposure of cells with 35 μM of PRIMA-1MET for 24 h, total cell lysates were prepared and Western blotting was performed (n = 3). Actin serves as a loading control. b The densitometric analysis of Western blots by ImageJ software. c The effect of PRIMA-1MET on p53 mRNA expression. RA-FLSs were treated with 35 μM PRIMA-1MET for 24 h, after which RNA was extracted, and the expression of the p53 gene was measured by quantitative RT-PCR and normalized to the expression of GAPDH (n = 10). FLS fibroblast-like synoviocyte, RA rheumatoid arthritis, mRNA microRNA, GAPDH glyceraldehyde-3-phosphate dehydrogenase, RT-PCR reverse transcription-polymerase chain reaction, UNT untreated
PRIMA-1MET upregulates Noxa as a pro-apoptotic target gene of p53 in RA-FLSs
Previous studies have shown that PRIMA-1MET induces p53 pro-apoptotic target genes such as phorbol-12-myristate-13-acetate-induced protein 1 (Noxa), (cyclin-dependent kinase inhibitor 1A (CDKN1A) or P21, and Bcl-2-associated X protein (BAX) through the restoration of mutant p53 sequence-specific DNA-binding and results in apoptosis in the cancer cells (Liang et al. 2009; Messina et al. 2012; Wang et al. 2007). As indicated in Fig. 5A, unlike a significant decrease in p53 gene and protein expression, mRNA expression of Noxa was significantly increased upon PRIMA-1MET treatment (P value = 0.039). Other pro-apoptotic p53 target genes like P53 upregulated mediator of apoptosis (PUMA), P21, and Bax mRNA expression levels were not altered significantly after PRIMA-1MET treatment (P value = 0.633, 0.109, 0.25 respectively).

RA-FLSs were treated with 35 μM PRIMA-1 for 24 h, then total RNA was extracted, and gene expression was performed using quantitative RT-PCR and normalized to the expression of GAPDH (n = 10). a Modulation of p53 target genes by PRIMA-1MET. b The effect of PRIMA-1MET on the expression of XIAP and Survivin. c PRIMA-1MET decreases NF-κB activity. RA-FLSs were treated with 35 μM PRIMA-1MET for 24 h. Total cell lysates were prepared and Western blot analysis was performed (n = 3). β-actin has been used as a loading control. d The densitometric analysis of P-IκBα and IκBα Western blots by ImageJ software. e Anti-inflammatory effect of PRIM A-1MET in RA-FLSs. FLS fibroblast-like synoviocytes, RA rheumatoid arthritis, GAPDH glyceraldehyde-3-phosphate dehydrogenase, RT-PCR reverse transcription-polymerase chain reaction, PUMA P53 upregulated mediator of apoptosis, BAX Bcl-2-associated X protein, NOXA Phorbol-12-myristate-13-acetate-induced protein 1, XIAP X-linked Inhibitor of Apoptosis, NF-κB nuclear factor-kappaB, P-IκBα phosphorylated IkBα, UNT untreated, Tr treated, IL-6 Interleukin 6, IL-1β interleukin 1 beta, TNF-α tumor necrosis factor-alpha
Effect of PRIMA-1MET on anti-apoptotic genes
It has been shown that PRIMA-1MET can decrease anti-apoptotic genes like Bcl-2 and XIAP expression and has no effect on survivin expression in cancer cells (Farhadi et al. 2017). Paradoxically, it has been reported that a derivative of PRIMA-1 increases XIAP expression (Soans et al. 2014). To investigate the effect of PRIMA-1MET on anti-apoptotic genes, we evaluated XIAP and survivin mRNA expression in PRIMA-1MET –treated RA-FLSs. As described in Fig. 5B, the transcription of XIAP and survivin mRNA were significantly upregulated after 24 h exposure to PRIMA-1MET (P value = 0.012, 0.002, respectively).
PRIMA-1MET treatment inhibits NF-κB through IκBα dephosphorylation
PRIMA-1 treatment in APL cell line (NB4) showed that PRIMA-1 can inhibit NF-κB activity. In the present study, the effect of PRIMA-1MET on NF-κB was evaluated by Western blotting of p-IκBα and IκBα proteins in the treated and control (untreated) groups (Fig. 5C and Supplementary Fig. 1). The results of Western blotting showed that PRIMA-1MET treatment increases IκBα dephosphorylation.
PRIMA-1MET downregulates pro-inflammatory cytokines in RA-FLSs
The effect of PRIMA-1 on inflammation and pro-inflammatory cytokines has not been investigated but it has been reported that a derivative of PRIMA-1 can upregulate tumor necrosis factor-alpha (TNF-α) (Soans et al. 2014). To evaluate whether PRIMA-1MET can reduce inflammatory cytokines in RA-FLSs, PRIMA-1MET-treated FLSs were assessed for pro-inflammatory cytokine genes. As indicated in Fig. 5D, comparison and statistical real-time PCR analysis showed that the exposure to PRIMA-1MET led to a significant decrease in mRNA expression of IL-6 and IL1-β as the most important pro-inflammatory cytokines in RA (P value = 0.016, 0.031 respectively). TNF-α in the treated-FLSs group were not significantly changed compared to the untreated group (P value = 0.055).
Discussion
Transcription factor p53 has multiple functions in the cells, the most important of which is induction of apoptosis (Colman et al. 2000). However, it has been reported that p53 can activate pro-survival pathways, too (Vousden 2006). Reactivation of mutant p53 is a promising and effective therapeutic strategy in cancer cells (Khoo et al. 2014). Therefore, according to the presence of mutant p53 in RA-FLSs, restoration of mutant p53 by recently developed small molecules such as PRIMA-1MET may induce apoptosis in RA-FLSs. PRIMA-1 and its methylated version restore p53 function through modification of thiol group in mutant p53 (Lambert et al. 2009) without any changes in p53 expression (Farhadi et al. 2017; Izetti et al. 2014). However, the results of the present study indicate a significant decrease in p53 expression at both mRNA and protein levels. Since PRIMA-1MET affects multiple targets and pathways, it is possible that downregulation of p53 is not a direct effect of PRIMA-1MET and is related to the activation of other regulatory proteins (Bykov et al. 2018; Rangel et al. 2019).
A number of experimental studies have shown that PRIMA-1 can induce apoptosis and inhibit the growth of tumor cells by induction of pro-apoptotic target genes of p53 (Bykov et al. 2002a, 2003). In this study, the cytotoxic effect of PRIMA-1MET in RA-FLSs was evaluated and we found that PRIMA-1MET induces apoptosis in RA-FLSs in a dose-dependent manner.
It has been reported that PRIMA-1 enhances apoptosis through activation of transcription of pro-apoptotic p53 target genes, including P21, Bax, and Noxa in cancer cells (Farhadi et al. 2017). To determine whether PRIMA-1MET induces apoptosis through induction of pro-apoptotic genes in RA-FLSs, we evaluated the expression of Noxa, Bax, PUMA, and p21. Our results showed that PRIMA-1MET can just induce Noxa expression significantly in RA-FLSs. Noxa is the most important pro-apoptotic target of the p53 gene and can induce apoptosis in FLSs by inactivating Mcl-1, an anti-apoptotic protein of the Bcl-2 family (Cottier et al. 2014). Our results showed that PRIMA-1MET had no effect on the expression of PUMA, Bax, and p21 which is consistent with other studies (Li et al. 2015; Mlakar et al. 2019; Synnott et al. 2017). Concerning these results and due to the pro-survival role of p53(Vousden 2006), it seems that downregulation of p53, which is not reported before, is involved in apoptosis induction in RA-FLSs.
Survivin and XIAP are members of the apoptosis inhibitory protein (IAP) family. Survivin is regulated by p53 and p53 has an inhibitory effect on this protein (Zafari et al. 2019). P53 activation also induces XIAP downregulation (Güllülü et al. 2021). Previous studies have revealed PRIMA-1 has no effect on p53 and survivin expression (Farhadi et al. 2017; Izetti et al. 2014) but in our study PRIMA-1MET treatment led to a significant overexpression of survivin that may be related to decreased expression of p53. Furthermore, PRIMA-1 can downregulate XIAP expression in cancer cells (Farhadi et al. 2017) while our results showed that PRIMA-1MET increases XIAP expression significantly. Our result is consistent with Soans et al. that reported some PRIMA-1 derivatives induced XIAP expression in lung cancer cells (Soans et al. 2014). Besides, given the regulatory role of p53 on XIAP expression, it is not surprising that p53 downregulation upon PRIMA-1MET treatment results in XIAP overexpression.
P53 is required for NF-κB inhibition (Murphy et al. 2011) and both of them inhibit each other’s (Webster and Perkins 1999). Furthermore, some studies have reported p53 activates NF-κB through different factors (Bohuslav et al. 2004; Jänicke et al. 2008). NF-κB activation plays a critical role in RA inflammation (Makarov 2001). Besides, it has been revealed that NF-κB is involved in both the initiation and perpetuation of chronic inflammation in RA. NF-κB contributes to inflammation through the induction of pro-inflammatory cytokines expression like IL-1, IL-6, TNF-α, and IL-17 (Makarov 2001). Given the NF-κB role in RA-FLSs, the inhibition of NF-κB would be a helpful strategy to inhibit pro-inflammatory cytokine production in RA-FLSs. The results of the present study showed that PRIMA-1MET has an inhibitory effect on the NF-κB pathway through decreasing phosphorylation of IκBα protein in RA-FLSs. Our results are consistent with Farhadi et al.’s study which reports that PRIMA-1 can inhibit NF-κB in acute promyelocytic leukemia cell lines (Farhadi et al. 2017). This effect of PRIMA-1MET may be a direct effect or related to the decreased expression of p53 (as an NF-κB activator).
IL-1 is an important player in RA pathogenesis and induces other pro-inflammatory cytokines like IL-6 and TNF-α (Kinne et al. 2007). IL-1 plays a critical role in joint damage through the enhancement of proteoglycan degradation (Ruscitti et al. 2018). Besides, TNF-α and IL-1 act synergistically in the induction of more inflammation and joint destruction (Schett and Gravallese 2012). IL-6 disrupts the balance between Th-17 and regulatory T cells (Tregs) and leads to the differentiation of naive T cells toward Th-17 (Yoshida and Tanaka 2014). Our results showed that PRIMA-1MET can significantly decrease IL-1 and IL-6 and had no effect on TNF-α expression. Given the inhibition of NF-κB upon PRIMA-1MET treatment, it is not surprising to report the downregulation of these key pro-inflammatory cytokines.
Conclusion
In conclusion, our results indicate that PRIMA-1MET can induce apoptosis in RA-FLSs through induction of Noxa expression while p53 was downregulated. Furthermore, PRIMA-1MET treatment results in the suppression of pro-inflammatory cytokine production and NF-κB inhibition. Given the role of p53 and NF-κB in RA-FLSs, PRIMA-1MET can be considered as a new therapeutic strategy for rheumatoid arthritis.
Data availability
The data generated and/or analyzed during the current study are available from the corresponding author on reasonable request. Applicable on requested.
References
Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd et al (2010) 2010 rheumatoid arthritis classification criteria: an American college of Rheumatology/European league against rheumatism collaborative initiative. Arthritis Rheum 62:2569–2581. https://doi.org/10.1002/art.27584
Bohuslav J, Chen LF, Kwon H, Mu Y, Greene WC (2004) p53 induces NF-kappaB activation by an IkappaB kinase-independent mechanism involving phosphorylation of p65 by ribosomal S6 kinase 1. J Biol Chem 279:26115–26125. https://doi.org/10.1074/jbc.M313509200
Bottini N, Firestein GS (2013) Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol 9:24–33. https://doi.org/10.1038/nrrheum.2012.190
Bykov VJ, Issaeva N, Selivanova G, Wiman KG (2002a) Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs: a statistical analysis of information in the National Cancer Institute database. Carcinogenesis 23:2011–2018. https://doi.org/10.1093/carcin/23.12.2011
Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P et al (2002b) Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med 8:282–288. https://doi.org/10.1038/nm0302-282
Bykov VJ, Selivanova G, Wiman KG (2003) Small molecules that reactivate mutant p53. Eur J Cancer 39:1828–1834. https://doi.org/10.1016/s0959-8049(03)00454-4
Bykov VJN, Eriksson SE, Bianchi J, Wiman KG (2018) Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer 18:89–102. https://doi.org/10.1038/nrc.2017.109
Colman MS, Afshari CA, Barrett JC (2000) Regulation of p53 stability and activity in response to genotoxic stress. Mutat Res 462:179–188. https://doi.org/10.1016/s1383-5742(00)00035-1
Cottier KE, Fogle EM, Fox DA, Ahmed S (2014) Noxa in rheumatic diseases: present understanding and future impact. Rheumatol (oxford) 53:1539–1546. https://doi.org/10.1093/rheumatology/ket408
Farhadi E, Safa M, Sharifi AM, Bashash D (2017) PRIMA-1 induces caspase-mediated apoptosis in acute promyelocytic leukemia NB4 cells by inhibition of nuclear factor-κB and downregulation of Bcl-2, XIAP, and c-Myc. Anticancer Drugs 28:51–58. https://doi.org/10.1097/cad.0000000000000426
Firestein GS (2003) Evolving concepts of rheumatoid arthritis. Nature 423:356–361. https://doi.org/10.1038/nature01661
Firestein GS, Nguyen K, Aupperle KR, Yeo M, Boyle DL, Zvaifler NJ (1996) Apoptosis in rheumatoid arthritis: p53 overexpression in rheumatoid arthritis synovium. Am J Pathol 149:2143–2151
Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR (1997) Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci USA 94:10895–10900. https://doi.org/10.1073/pnas.94.20.10895
Greenblatt MS, Bennett WP, Hollstein M, Harris CC (1994) Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 54:4855–4878
Güllülü Ö, Hehlgans S, Rödel C, Fokas E, Rödel F (2021) Tumor suppressor protein p53 and Inhibitor of apoptosis proteins in colorectal cancer—a promising signaling network for therapeutic interventions. Cancers (basel). https://doi.org/10.3390/cancers13040624
Han Z, Boyle DL, Shi Y, Green DR, Firestein GS (1999) Dominant-negative p53 mutations in rheumatoid arthritis. Arthritis Rheum 42:1088–1092. https://doi.org/10.1002/1529-0131(199906)42:6%3c1088::aid-anr4%3e3.0.co;2-e
Hewagama A, Richardson B (2009) The genetics and epigenetics of autoimmune diseases. J Autoimmun 33:3–11. https://doi.org/10.1016/j.jaut.2009.03.007
Inazuka M, Tahira T, Horiuchi T, Harashima S, Sawabe T, Kondo M et al (2000) Analysis of p53 tumour suppressor gene somatic mutations in rheumatoid arthritis synovium. Rheumatol (oxford) 39:262–266. https://doi.org/10.1093/rheumatology/39.3.262
Izetti P, Hautefeuille A, Abujamra AL, de Farias CB, Giacomazzi J, Alemar B et al (2014) PRIMA-1, a mutant p53 reactivator, induces apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic cancer cell lines. Invest New Drugs 32:783–794. https://doi.org/10.1007/s10637-014-0090-9
Jänicke RU, Sohn D, Schulze-Osthoff K (2008) The dark side of a tumor suppressor: anti-apoptotic p53. Cell Death Differ 15:959–976. https://doi.org/10.1038/cdd.2008.33
Khoo KH, Verma CS, Lane DP (2014) Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov 13:217–236. https://doi.org/10.1038/nrd4236
Kinne RW, Stuhlmüller B, Burmester GR (2007) Cells of the synovium in rheumatoid arthritis. Macrophages Arthritis Res Ther 9:224. https://doi.org/10.1186/ar2333
Lambert JM, Gorzov P, Veprintsev DB, Söderqvist M, Segerbäck D, Bergman J et al (2009) PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 15:376–388. https://doi.org/10.1016/j.ccr.2009.03.003
Li XL, Zhou J, Chan ZL, Chooi JY, Chen ZR, Chng WJ (2015) PRIMA-1met (APR-246) inhibits growth of colorectal cancer cells with different p53 status through distinct mechanisms. Oncotarget 6:36689–36699. https://doi.org/10.18632/oncotarget.5385
Liang Y, Besch-Williford C, Hyder SM (2009) PRIMA-1 inhibits growth of breast cancer cells by re-activating mutant p53 protein. Int J Oncol 35:1015–1023. https://doi.org/10.3892/ijo_00000416
Makarov SS (2001) NF-kappa B in rheumatoid arthritis: a pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res 3:200–206. https://doi.org/10.1186/ar300
Messina RL, Sanfilippo M, Vella V, Pandini G, Vigneri P, Nicolosi ML et al (2012) Reactivation of p53 mutants by prima-1 [corrected] in thyroid cancer cells. Int J Cancer 130:2259–2270. https://doi.org/10.1002/ijc.26228
Mlakar V, Jurkovic Mlakar S, Lesne L, Marino D, Rathi KS, Maris JM et al (2019) PRIMA-1(MET)-induced neuroblastoma cell death is modulated by p53 and mycn through glutathione level. J Exp Clin Cancer Res 38:69. https://doi.org/10.1186/s13046-019-1066-6
Murphy SH, Suzuki K, Downes M, Welch GL, De Jesus P, Miraglia LJ et al (2011) Tumor suppressor protein (p)53, is a regulator of NF-kappaB repression by the glucocorticoid receptor. Proc Natl Acad Sci USA 108:17117–17122. https://doi.org/10.1073/pnas.1114420108
Rangel LP, Ferretti GDS, Costa CL, Andrade S, Carvalho RS, Costa DCF et al (2019) p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J Biol Chem 294:3670–3682. https://doi.org/10.1074/jbc.RA118.004671
Ruscitti P, Cipriani P, Liakouli V, Carubbi F, Berardicurti O, Di Benedetto P et al (2018) The emerging role of IL-1 inhibition in patients affected by rheumatoid arthritis and diabetes. Rev Recent Clin Trials 13:210–214. https://doi.org/10.2174/1574887113666180314102651
Schett G, Gravallese E (2012) Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat Rev Rheumatol 8:656–664. https://doi.org/10.1038/nrrheum.2012.153
Soans E, Evans SC, Cipolla C, Fernandes E (2014) Characterizing the sphingomyelinase pathway triggered by PRIMA-1 derivatives in lung cancer cells with differing p53 status. Anticancer Res 34:3271–3283
Synnott NC, Murray A, McGowan PM, Kiely M, Kiely PA, O’Donovan N et al (2017) Mutant p53: a novel target for the treatment of patients with triple-negative breast cancer? Int J Cancer 140:234–246. https://doi.org/10.1002/ijc.30425
Vousden KH (2006) Outcomes of p53 activation–spoilt for choice. J Cell Sci 119:5015–5020. https://doi.org/10.1242/jcs.03293
Wang T, Lee K, Rehman A, Daoud SS (2007) PRIMA-1 induces apoptosis by inhibiting JNK signaling but promoting the activation of Bax. Biochem Biophys Res Commun 352:203–212. https://doi.org/10.1016/j.bbrc.2006.11.006
Webster GA, Perkins ND (1999) Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol 19:3485–3495. https://doi.org/10.1128/mcb.19.5.3485
Yamanishi Y, Boyle DL, Green DR, Keystone EC, Connor A, Zollman S et al (2005) p53 tumor suppressor gene mutations in fibroblast-like synoviocytes from erosion synovium and non-erosion synovium in rheumatoid arthritis. Arthritis Res Ther 7:R12-18. https://doi.org/10.1186/ar1448
Yoshida Y, Tanaka T (2014) Interleukin 6 and rheumatoid arthritis. Biomed Res Int 2014:698313. https://doi.org/10.1155/2014/698313
Zafari P, Rafiei A, Esmaeili SA, Moonesi M, Taghadosi M (2019) Survivin a pivotal antiapoptotic protein in rheumatoid arthritis. J Cell Physiol 234:21575–21587. https://doi.org/10.1002/jcp.28784
Funding
This study was supported by a grant from Tehran University of Medical Sciences (Grant No: 97-03-41-40374), and a grant from Kermanshah University of Medical Sciences (Grant No: 97887).
Author information
Authors and Affiliations
Contributions
MA: acquisition of data, drafting the article, analysis and interpretation of data, final approval of the article. MNT and AS: acquisition of clinical data and patient’s diagnosis and treatment, interpretation of data, drafting the article, and final approval of the article. MT, AJ, MM and EF: the conception and design of the study, revising the article critically, interpretation of data, final approval of the article.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
This study was performed based on the Declaration of Helsinki guidelines and was approved by the Ethics Committee of Tehran University of Medical Sciences (Approval No: IR.TUMS.VCR.REC.1397.968).
Consent to participate
The written informed consent was signed by all participants before enrolling in the study.
Consent to publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Adib, M., Taghadosi, M., Tahmasebi, M.N. et al. Anti-inflammatory effects of PRIMA-1MET (mutant p53 reactivator) induced by inhibition of nuclear factor-κB on rheumatoid arthritis fibroblast-like synoviocytes. Inflammopharmacol 31, 385–394 (2023). https://doi.org/10.1007/s10787-022-01094-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-022-01094-9