
Abstract
Neurodegenerative illness develops as a result of genetic defects that cause changes at numerous levels, including genomic products and biological processes. It entails the degradation of cyclic nucleotides, cyclic adenosine monophosphate (cAMP), and cyclic guanosine monophosphate (cGMP). PDE7 modulates intracellular cAMP signalling, which is involved in numerous essential physiological and pathological processes. For the therapy of neurodegenerative illnesses, the normalization of cyclic nucleotide signalling through PDE inhibition remains intriguing. In this article, we shall examine the role of PDEs in neurodegenerative diseases. Alzheimer’s disease, Multiple sclerosis, Huntington’s disease, Parkinson’s disease, Stroke, and Epilepsy are related to alterations in PDE7 expression in the brain. Earlier, animal models of neurological illnesses including Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis have had significant results to PDE7 inhibitors, i.e., VP3.15; VP1.14. In addition, modulation of CAMP/CREB/GSK/PKA signalling pathways involving PDE7 in neurodegenerative diseases has been addressed. To understand the etiology, treatment options of these disorders mediated by PDE7 and its subtypes can be the focus of future research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Phosphodiesterases (PDEs) are enzymes that regulate cAMP and cGMP levels in the body. Because both cAMP and cGMP are critical in the brain during neurodevelopment, as well as in developing synaptic plasticity and, ultimately, learning and memory, inhibiting PDEs have become a well-studied target for therapy in a wide range of illnesses, including NDDs (Mendiola-Precoma et al. 2016; Sharma et al. 2021a, b). To hydrolyze cAMP, phosphodiesterases (PDEs) show a vital role in neuroinflammation, which is controlled by cAMP levels. PDE superfamily contains 11 subtypes that are encoded by 21 known genes. furthermore, each gene product can have many splice variants (for example, PDE4D1–PDE4D9), resulting in over 100 different PDE proteins (Bollen and Prickaerts 2012). Neurodegenerative diseases, on the other hand, are defined by the progressive loss of particularly sensitive groups of neurons, as opposed to select static neuronal loss induced by metabolic or toxic illnesses. The major NDDs are Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis (MS), Stroke, Epilepsy, and Huntington’s disease (Thapa et al. 2021). Memory loss is followed by the formation of beta-amyloid plaques, hyperphosphorylation of tau protein, and inflammation in Alzheimer’s patients (Mendiola-Precoma et al. 2016; Khan et al. 2022a). PDE7, a cAMP-selective enzyme, is found in the brain and has been investigated as a potential target for treating Alzheimer’s disease, particularly when combined with GSK3 activity. Both cAMP and cGMP have been linked to Alzheimer’s disease. Increasing evidence suggests that up-regulation of the cAMP is implicated in AD and its blockage lowered intellectual impairment and clinical markers (Ribaudo et al. 2020). Dopaminergic neuron loss in the substantia nigra compacta (SNpc) is a hallmark of Parkinson’s disease (Kinoshita et al. 2015). PDE7 inhibitors elevated the number of neural stem cells by enhancing embryonic neural stem cell differentiation into matured dopamine neurons and hastened the differentiation and proliferation of mature progenitor cells in adult rats by raising the levels of pCREB. PDE7 possibly be a promising therapeutic target for PD; blocking PDE7 may have beneficial effects on the disease (Mercier et al. 2017). Multiple pathways, including oxidative stress, excitotoxicity, blood–brain barrier permeability, neuroinflammation, and others, are altered, which explains a portion of the brain’s post-ischemic neurodegeneration (McGeer and McGeer 2007; Khan et al. 2021a; b). Neuroinflammation can be controlled by blocking cAMP-specific phosphodiesterases (PDEs), such as PDE 4, 7, and 8, which raise cAMP levels and, as a result, enhance functional recovery.
Preclinical discoveries, clinical consequences, and upcoming views of cAMP-specific PDE7 inhibition as a new research topic for the treatment of neurodegenerative disease have been shown in recent publications (Ponsaerts et al. 2021). cAMP participates in the transmission of signaling pathways involved in neuroprotection and immune regulation. By modulating the quantities of this nucleotide, inhibition of cAMP-specific PDEs such as PDE7 may modify the pathogenic processes of neuroinflammatory diseases, such as multiple sclerosis (MS) (Mestre et al. 2015; Rehni et al. 2010). PDE7 mRNA and protein are found in many immune cells, and evidence shows that PDE7 may play a role in T-lymphocyte activation, although further research is needed. Consequently, in HD animals, strategies aimed at increasing CREB transcription—and, as a result, the expression of brain-derived neurotrophic factor (BDNF)—have neuroprotective effects (Fusco and Giampa 2015; Gharami et al. 2008). A CREB activation is mediated by a cAMP-dependent protein kinase (PKA), as a result, in the control of neuronal activities, the balance of intracellular cAMP/cGMP levels is critical (Xie et al. 2010; Khan et al. 2022a). As a result, PDEs and inhibitors of PDEs might be useful in the treatment of HD. PDEs are enzymes that catabolize cAMP and/or cGMP in the cell. As a result, inhibiting them may be advantageous in the treatment of CNS neuronal degeneration (Giampà et al. 2013.
Methodology
A systematic literature review of Bentham, Scopus, PubMed, Medline, and EMBASE (Elsevier) Databases was created with the use of keywords, such as Phosphodiesterases; Neurodegenerative disease; PDE 7; cyclic adenosine monophosphate; cGMP. To comprehend the review, the keywords listed above were used in the role of phosphodiesterase-7 in neurodegenerative disorder.
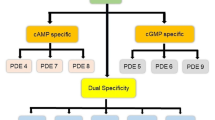
Structure of PDE 7
PDE7 contains two genes that have been discovered in the brain. There are three or four splice variations in PDE7A and PDE7B. PDE7 is a PDE family member that has been discovered as CAMP specialized and was previously known for its role in regulating T-cell activity (Bloom and Beavo 1996. The significance of PDE7 in neurotransmission activity profile in the brain, as well as the drugability of other Phosphodiesterases has encouraged the quest for strong antagonists to cure neurodegenerative disorders (Fig. 1). The PDE7 gene is located on human chromosome 8q13-q22 and is not linked to any known disease factors (Gene Mapping). PDE7A is found in high concentrations in the lungs and hematopoietic cells, while PDE7B activity in the CNS is higher than in other tissues. PDE7B is abundant in the caudate nucleus, the nucleus accumbens, the cortex, and the hippocampus. PDE7B has been found in the pancreas, heart, thyroid, and skeletal muscles (Hetman et al. 2000). PDE7A and PDE7B mRNA are also found in osteoblasts and certain brain regions, such as various cortical regions, the dentate gyrus, most olfactory system elements, the striatum, numerous thalamic nuclei, and hippocampal pyramidal cells. A dual PDE7/PDE4 inhibitor was used to investigate the possible involvement of PDE7 in human T-cell activity (Gil et al. 2008). The development of selective PDE7 inhibitors with attractive ADME qualities for in vivo research would widen the reach of a unique class of medicines with an innovative mode of action that keeps intracellular cAMP levels high. PDE7 was discovered both in neuronal and non-neuronal cell populations. PDE7 regulates inflammatory responses via the cyclic adenosine monophosphate (cAMP) signalling pathway, and so plays a key role in Alzheimer’s disease.

Structure and Illustration of PDE7 mechanism of action via its anti-inflammatory, pro-remyelinating, anti-oxidant, anti-fibrotic, neuroprotective, synaptic plasticity properties in improving the severity of neurodegenerative disease
Expression and localization of PDE’s isoforms
The PDE superfamily contains 11 subtypes, each of which is encoded by one of the 21 known genes (Bender and Beavo 2004). Although the majority of PDE isoforms (PDE1, PDE2, PDE3, PDE4, PDE5A, PDE6, PDE7, PDE8B, PDE9A, PD10A, and PDE11A) are expressed in the brain (Omori and Kotera 2007) (Table 1).
Therapeutic potential of PDE7 inhibitors in several neurodegenerative disorders through multiple downstream signalling pathways
PDE7 and Alzheimer’s disease
Alzheimer’s disease (AD), is marked by gradual memory loss, it is the most prevalent form of senile dementia, and neuronal destruction, as well as cognitive impairment (Sharma et al. 2021a). Neuropathological hallmarks of AD include extracellular amyloid-ß deposition (ß plague), neurofibrillary tan-gel formation (NTFS), and neuroinflammation (Pozueta et al. 2013). Compound 14, a selective PDE7 inhibitor that is orally soluble (5,8-disubstituted pirquinozol) was found to significantly enhance memory in both healthy mice and rats following oral dosage in a variety of behavioral activities (McQuown et al. 2021). The findings show that PDE7 may play a crucial role in the etiology of AD. Recent research has shown that inhibiting PDE7 improves in vitro and in vivo AD-related alterations. At a dose of 3 mg/kg, i.p., VP1.15 (dual inhibitor of PDE7/GSK3), a 5-imino-1,2-4-thiazoles and quinazolines derivatives, was reported to improve episodic, working, and showed fear memory in healthy mice (Lipina et al. 2013). S14 (5 mg/kg, i.p.) is a more effective selective PDE7 inhibitor that improves learning and memory impairment in Aß-amyloid precursor protein/presenilin APP/PS1 twin transgenic mice, a model of Alzheimer’s disease with age-related memory loss. S14 decreased Aß accumulation, Tau protein hyperphosphorylation, and mitochondrial malfunction in SH-SY5Y cells treated in vitro to AB1-42 via elevating CAMP/CREB signalling and lowering GSK3 activity (Perez-Gonzalez et al. 2013). Similarly, PDE7 inhibitors contribute to the treatment of Alzheimer’s disease by controlling early stage mitochondrial dysfunction observed in the condition. Mitochondrial failure contributes to the development of AD by causing the destruction and mutation of mitochondrial DNA, abnormalities in calcium homeostasis, oxidative stress, and synapsis loss and malfunction (Sheng et al. 2012). To ameliorate mitochondrial damage, dosing with the PDE7 inhibitor (S14) increased total mitochondrial mass and the ratio of mitochondrial fission/fusion proteins in the cerebral cortex and hippocampus of APP/PS1 mice while blocking autophagy. According to the study, S14 (10 mg/kg, p.o.) induces neurogenesis in the subventricular zone (SVZ) and subgranular zone (SGZ), hence improving the cognitive function of rats (Fig. 2). In a similar fashion, S14 overexpression of pCREB enhances the multiplication of chief cultured rat SGZ and SVZ neurons, as well as SGZ and olfactory bulb neurogenesis (Morales-Garcia et al. 2017). Anti-inflammatory also anti-excitotoxic mechanisms are involved in the anti-AD actions of PDE7 inhibitors (Blokland et al. 2012). The dual PDE7/GSK3 inhibitor [VP1.14 (14 (5-imino-1, 2, 4-thiadiazole)] decreases inflammation and pyramidal cell loss in the CA1 and CA3 subregions of the hippocampus produced by unilateral hippocampal kainic acid injection (Susín et al. 2012). In glial cells on exposure to lipopolysaccharide (LPS), this PDE7 inhibitor reduces nitrite release and pro-inflammatory cytokines, as well as glutamate-induced excitotoxicity in hippocampal HT22 cells (Susín et al. 2012). Further study suggested that enough data are required regarding current pharmacological, non-pharmacological interventions and other relevant treatments related to AD.

PDE7, a cAMP-selective enzyme, has been revealed to be a promising therapeutic target for neurodegenerative illnesses, such as Alzheimer’s disease, Parkinson’s disease, stroke, epilepsy, Huntington’s disease, and multiple sclerosis, especially in conjunction with GSK3 modulator. Upregulation of cAMP is linked to Alzheimer’s disease and stroke, and its inhibition reduces intellectual impairment and clinical symptoms. CREB activation is mediated by a cAMP-dependent protein kinase (PKA); hence, the balance of intracellular cAMP/cGMP levels is essential for the regulation of neuronal activity. cAMP and cGMP have both been associated with neurodegenerative disorders. PDEs are cellular enzymes that catabolize cAMP and/or cGMP. Therefore, blocking them may be beneficial in the treatment of CNS neuronal degeneration seen in Parkinson’s disease and epilepsy. cAMP levels may have a role in neuroprotection and the neuroinflammatory response, suggesting that managing cAMP levels could cause the pathogenic neuroinflammatory process to be managed and, as a result, delay the development of NDDs, such as multiple sclerosis and Huntington’s disease
PDE7 and Parkinson disease
Parkinson’s disease (PD) is a progressive neurological ailment generated by around 1% of the elderly population is affected. At the molecular level, Parkinson’s disease is characterized by the loss of dopamine-containing neurons in the substantia nigra pars compacta (SNpc), although neuropathology can extend to other areas of the brain (Damier et al. 1999). There is no cure and no effective treatment available at this time (Orr et al. 2005; Tansey et al. 2007). Some studies have suggested that cAMP levels may be significant in neuroprotection and the neuroinflammatory response (Volakakis et al. 2010) implying that regulating cAMP levels might cause the pathogenic neuroinflammatory process to be regulated (McGeer et al. 2003) and, as a result, delay the development of NDDs, such as Parkinson’s disease. Intracellular cAMP levels are influenced by adenylyl cyclase production and breakdown by cyclic nucleotide 3, 5 PDEs (Mehats et al. 2002). With improved brain penetration, the chemical is suited for the MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) treated mice model of Parkinson’s disease. Consequently, the researchers investigated the pharmacokinetic properties of compound 7. Compound 7 concentrations in the brain were tolerated after mice were given 10 mg/kg i.p. of the D2 receptor antagonist haloperidol-induced catalepsy (HIC) (Boulay et al. 2010). Compound 7’s PDE7A IC50 and pharmacokinetic properties were determined. Based on these data, the free brain concentration attained after an i.p. injection of 10 mg/kg was 10 times lower than the mouse PDE7A IC50. In contrast, free brain concentrations at doses of 30 and 100 mg/kg were equivalent to as well as many times greater than the mouse PDE7A IC50, accordingly. The effects of compound 7 at varying doses (1, 3, 10, and 30 mg/kg, i.p.) on the rearing behavior of male C57BL/6 mice with MPTP-induced PD were examined. Compound 7 increased rearing behavior in mice treated with MPTP at suboptimal doses (1, 3, and 10 mg/kg, i.p.) but had no impact at the maximum dose (30 mg/kg, i.p.). The conventional situation treatment of l-dopa/benserazide (BSZ) had a considerable outcome on vertical rearing behavior in these mice. However, by interfering with dopamine D1 receptor-mediated cAMP signaling in the striatal direct route, PDE7 inhibitors may nevertheless boost the effectiveness of l-dopa. Multiple cellular and animal models of Parkinson’s disease have been discovered to exhibit increased PDE7 expression. In treating Parkinson’s disease, neurotoxins such as lipopolysaccharide (LPS) and 6-hydroxydopamine (6-OHDA) are frequently employed. LPS is an endotoxin produced by bacteria that induce neuroinflammation, whereas 6-OHDA, a dopamine analog, can cause rapid and severe dopaminergic cell death in the substantia nigra (SN). After SNpc injection of LPS or 6-OHDA into rats, a rise in PDE7 was detected in dopaminergic neurons and glial cells that had been injured (Morales-Garcia et al. 2020). According to in vitro study, in human neuroblastoma (SH-SY5Y) cells, primary cultures of embryonic ventral mesencephalic (VM) neurons, and glial cells from rats treated with 6-OHDA or LPS, PDE7 expression is enhanced (Morales-Garcia et al. 2020). The overexpression of PDE7 inhibitor seems to be directly persuaded, because expression of a 962 bp in SH-SY5Y cells treated with PDE7, a portion of the Homo sapiens promoter is enhanced with 6-OHDA and glial cells treated with LPS after transfection with a construct containing a 962-bp fragment of the promoter (Morales-Garcia et al. 2020). PDE7 has been demonstrated to be activated in striatal neurons via the cAMP/PKA/CREB pathway and to be related to memory function, suggesting that it might be a potential treatment target for epilepsy, and Huntington’s disease (Peng et al. 2018).
PDE7 and stroke
An ischemic syndrome is characterized by thromboembolic obstruction of a major cerebral artery, which leads to neuroinflammation and neuronal damage as a result of decreased blood supply (Khan et al. 2022b). During the acute phase of an ischemic stroke, Neutrophils, monocytes/macrophages, and T cells are part of the innate and adaptive immune systems, B cells, respectively) contributed to the neuroinflammatory process (Khan et al. 2020; Ponsaerts et al. 2021). T cell proliferation and generation of pro-inflammatory TNF- were unaffected by treatment with the PDE7 inhibitor BRL150481 by LPS-stimulated macrophages. According to these data, PDE7 inhibition by BRL50481 has minimal anti-inflammatory effects. Concurrently, treatment with BRL50481 and rolipram, a pan-PDE4 inhibitor, resulted in significant anti-inflammatory effects (Fig. 2). This opens the prospect of using a combination of PDE4 and PDE7 inhibition to treat neuroinflammation (Smith et al. 2004). T cells have been shown that exhibit a high level of the PDE7 gene family (Xu et al. 2016). Greater levels of PDE7 in T cells have been associated with reducing cAMP and increasing IL-2 expression. According to another study, inhibiting PDE7 decreases T cell proliferation and IL-2 expression (Li et al. 1999). These findings are supported by studies employing the PDE7 inhibitor BC12, which decreased IL-2 expression and T cell proliferation (Xu et al. 2016). PDE7 inhibition promoted neural stem cell differentiation and proliferation in neural stem niches of the rat brain, notably the SGZ of the dentate gyrus and the SVZ, as per a study. PDE7 inhibition was also shown to boost neuronal cell formation in the olfactory bulb and dentate gyrus, leading to better memory and learning in rats (Morales-Garcia et al. 2014). PDE7 inhibition has been known to drastically lower nitrite formation in LPS-stimulated primary astrocytes, microglia, and neurons (Khan et al. 2022c). In addition, PDE7 inhibitors dramatically decreased infarct sizes in mice 48 h after persistent MCAO formation, as well as improved neurological outcomes. These findings supported the idea that inhibiting PDE7 improves the outcome of ischemic stroke (Redondo et al. 2012). According to research, Quinazoline type phosphodiesterase 7 (PDE7) inhibitors based on microwave irradiation have been developed. This approach enhanced yields and reaction times while also allowing for an ascendable technique. These chemicals are pharmacologically intriguing due to their in vivo effectiveness in spinal cord injury and PD models, as demonstrated in prior research from our lab. A study has been showing for the first time that treatment of 3-phenyl-2,4-dithioxo-1,2,3,4-tetrahydroquinazoline (compound 5) alleviated brain injury and improved behavioral outcomes in a permanent middle cerebral artery occlusion (pMCAO) stroke animal (Ponsaerts et al. 2021). Researchers further show that PDE7 inhibitors are effective anti-inflammatory and neuroprotective drugs in primary brain cell cultures. As a result of these findings, studies suggest PDE7 inhibitors as a new class of neuroprotective medicinal medicines.
PDE7 and multiple sclerosis
Multiple sclerosis (MS) is a central nervous system (CNS) inflammatory disease characterized by demyelination and axonal damage, which disrupts normal neurotransmission (Zarei et al. 2015) and causes sensory and motor deficits (Lipina et al. 2013). Using 5-imino-1,2,4-thiadiazoles, PDE7 and GSK-3 inhibitors decreased signs of demyelination in the experimental autoimmune encephalomyelitis (EAE) animal model (as previously described) and improved remyelination in 2 additional demyelinating mice models with low adaptive immune system input (using lysophosphatidylcholine–LPC or cuprizone) (Jankowska et al. 2017). Furthermore, the findings suggested that both PDE7–GSK3 inhibitors, particularly VP3.15, which has a high oral bioavailability and CNS dispersion, might be used as possible anti-inflammation and pro-remyelinating treatments for MS (Medina-Rodríguez et al. 2017). TC3.6 is a small heterocyclic synthetic compound that can control cAMP levels and exhibits the predicted physiological behavior. Research has revealed that the TMEV–IDD model of chronic progressive MS, in which there is a dormancy period after viral injection is before symptoms and motor impairments occur. In the research, TC3.6 given for 12 days (10 mg/kg, i.p.) during the pre-symptomatic period (30 day post-infection) substantially delayed the start of TMEV–IDD was extended to 60 days instead of 45 days in TMEV–vehicle mice (Mestre et al. 2015). Furthermore, it was discovered that the TC3.6 treated mice had improved motor function at 50, 60, and 70 days after infection. At day 60 post-infection, TC3.6 (5 mg/kg) therapy of TMEV-infected mice for 14 days reduced the progression of the clinical score. The administrated of TC3.6 greatly enhanced the performance of spontaneous motor activity around day 75 post-infection, according to the results of the study (McCarthy et al. 2012). Several disease-modified medicines for the treatment of MS are currently being developed, and more are likely to be introduced. It is critical to establish a population-based prevalence of MS in the country, as well as to conduct patient awareness programs and medical training, particularly for internists, to enhance care for MS patients.
PDE7 and epilepsy
Worldwide, 50 million individuals suffer from epilepsy, including an estimated 2–3 million in the United States (Guilbert 2006). A seizure is an uncontrollable shift in behavior that is characterized by abnormalities in sensory feelings or motor activity (movement) as a result of abnormal brain cell firing. Epilepsy is a disorder characterized by repeated seizures, which may include convulsions (tedious twitches of the muscles) (Galanopoulou et al. 2012). In the MES (maximal electroshock seizure) model of epilepsy, SQ22536 in combination with exogenously delivered BRL50481 (1.4 mg/kg, i.p.) and SQ22536 in combination with dipyridamole (1.4 mg/kg, i.p.) resulted in a considerable delay in the commencement of the tonic extensor phase of convulsion. Compared to the DMSO-treated group, the overall convulsion length was significantly longer in the SQ22536 alone-treated and SQ22536 and BRL50481 combination-treated groups. The adenylate cyclase (AC) inhibitor SQ22536 was reported to delay the start of seizures and extend the entire duration of convulsive time in both the PTZ and MES models. In both epilepsy models, SQ22536 significantly increased anticonvulsant activity as well as the percentage of animals protected (Nandhakumar and Tyagi 2010. According to studies, PDE7 is connected to seizures at the molecular level for the following reasons: (a) PDE7B is linked to 6q23-24, one of the most important seizure loci (Torri et al. 2010); (b) PDE7 is widely expressed, particularly in the striatum, cerebellum and cerebral cortex, which are regarded as the brain regions most closely linked to seizures (P’erez-Torres et al. 2003); (c) Based on pharmaco-transcriptomic, pharmacogenomics, and case–control association methods, PDE7B is a promising candidate gene for epilepsy susceptibility (Ikeda et al. 2010; Torri et al. 2010); (d PDE7 inhibiting by VP1.15 (3 mg/kg, i.p.) enhances pre-pulse inhibition (PPI), alters amphetamine-induced PPI deficit, substantially enhanced disturbed latent inhibition (LI) persuaded by the rising prevalence of conditioning trials, noting that it has antipsychotic effects (Lipina et al. 2013). Improved understanding of the processes behind illness development, improved trial designs, medication repurposing tactics, and new cooperation models may all aid in the discovery of viable treatments.
PDE7 and Huntington diseases
Huntington’s disease (HD) is a rare autosomal leading neurological illness that causes physical impairment, cognitive decline, and mental symptoms. Chorea, an involuntary muscle contraction caused by dysfunction of the basal ganglia, which is the major target of HD, is the most common motor symptom (Dayalu and Albin 2015). End-stage HD-like neuropathology and decrease of mitochondrial respiratory chain activity in mice treated with mitochondrial poisons. The brains of HD patients have revealed that energy deficiencies may have an impact on the disease’s progression (Golpich et al. 2017). However, research using HD transgenic systems has found that genes controlled by the cyclic adenosine 3′,5′-monophosphate (cAMP) response element (CRE) binding protein had lower transcription (CREB). BDNF (brain-derived neurotrophic factor) and a host of other genes are among them, with roles ranging from neurotransmission to cholesterol metabolism. Reduced gene transcription has been attributed to sequestration of coactivators that bind Huntington amino terminus, such as CREB binding protein (CBP) or TAFI1130, into inclusions formed by mutant amino-terminal product. CBP, on the other hand, is found in insertions in two polyglutamine neurodegenerative illnesses, DRPLA and SBMA, suggesting that confiscation is a side effect of the disease (Fusco and Paldino 2017). Furthermore, CBP sequestration does not appear to be the cause of the significant decrease in BDNF transcription seen in immortalized STHdhQ111 striatal cells, which properly expressed both normal and mutant huntingtin and do not have amino-terminal product inclusions (Gines et al. 2003). Currently, few high-quality clinical trials are being undertaken, but this is improving because of the creation of networks, such as the Huntington’s Disease Network, which works directly with industry partners to guarantee that all new trial protocols are of high quality and relevant to HD.
Conclusions
Numerous preclinical studies indicate that PDE7 is engaged in a large number of diseases, including Parkinson’s disease, multiple sclerosis, Alzheimer’s disease, and other NDDs disorders, and that PDE7 inhibitors have given way to a new class of drugs for treating these NDDs diseases by regulating signalling pathways differently (Soni 2019). Currently, there are no ongoing clinical trial initiatives, focused on neurodegenerative disease, however, future potential for developing targeted therapeutics for progressive types of NDDs includes lowering disability. Several challenges must be solved in addition to evaluating PDE7 inhibitors in clinical trials to simplify the discovery and the most promising PDE isoforms have been validated for combating cerebral impairment. The specific location of distinct PDEs in the brain, as well as a better knowledge of the localization of PDEs in different sections of the neuron, must be determined. Cyclic nucleotides are key intracellular signalling components and neuroplasticity in the brain. As a result, damaged signalling is likely to cause significant alterations in cell function, potentially leading to pathological results. Indeed, abnormal cyclic nucleotide signalling linked to PDE expression is linked to a variety of neurodegenerative disorders. Normalizing cyclic nucleotide levels might thus be used as a symptomatic treatment for neurodegenerative diseases. This helps to explain why PDE7 inhibitors are such a popular pharmacological drug target right now. However, further study is desired to have an improved comprehension of the connection between PDE7 function and neuropathology.
Availability of data and materials
Not applicable.
Abbreviations
- PDEs:
-
Phosphodiesterases
- cAMP:
-
Cyclic adenosine monophosphate
- cGMP:
-
Cyclic guanosine monophosphate
- CNS:
-
Central nervous system
- mRNA:
-
Messenger ribonucleic acid
- NDDs:
-
Neurodegenerative disease
- AD:
-
Alzheimer’s disease
- MDD:
-
Major depressive disorder
- ALS:
-
Amyotrophic lateral sclerosis
- SNpc:
-
Substantia nigra compacta
- PD:
-
Parkinson disease
- HD:
-
Huntington’s disorder
- GSK3:
-
Glycogen synthase kinase-3
- DNA:
-
Deoxyribonucleic acid
- SVZ:
-
Subventricular zone
- SGZ:
-
Subgranular zone
- LPS:
-
Lipopolysaccharide
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- 6-OHDA:
-
6-Hydroxydopamine
- SN:
-
Substantia nigra
- TNF:
-
Tumour necrosis factor
- IL-2:
-
Interleukin-2
- MCAO:
-
Middle cerebral artery occlusion
- MS:
-
Multiple sclerosis
- EAE:
-
Experimental autoimmune encephalomyelitis
- TMEV-IDD:
-
Theiler’s murine encephalomyelitis virus-induced demyelinating
- MES:
-
Maximal electroshock seizure
- LI:
-
Latent inhibition
- PPI:
-
Pre-pulse inhibition
- CREB:
-
CAMP response element binding protein
References
Amin SA, Bhargava S, Adhikari N, Gayen S, Jha T (2018) Exploring pyrazolo [3, 4-d] pyrimidine phosphodiesterase 1 (PDE1) inhibitors: a predictive approach combining comparative validated multiple molecular modelling techniques. J Biomol Struct Dyn 36(3):590–608. https://doi.org/10.1080/07391102.2017.1288659
Bender AT, Beavo JA (2004) Specific localized expression of cGMP PDEs in Purkinje neurons and macrophages. Neurochem Int 45(6):853–857. https://doi.org/10.1016/j.neuint.2004.03.015
Blokland A, Menniti FS, Prickaerts J (2012) PDE inhibition and cognition enhancement. Expert Opin Ther Pat 22(4):349–354. https://doi.org/10.1517/13543776.2012.674514
Bloom TJ, Beavo JA (1996) Identification and tissue-specific expression of PDE7 phosphodiesterase splice variants. Proc Natl Acad Sci 93(24):14188–14192. https://doi.org/10.1073/pnas.93.24.14188
Bolger GB (2017) The PDE4 cAMP-specific phosphodiesterases: targets for drugs with antidepressant and memory-enhancing action. In: Phosphodiesterases: CNS functions and diseases, pp 63–102. https://doi.org/10.1016/S0065-7743(07)42001-2
Bollen E, Prickaerts J (2012) Phosphodiesterases in neurodegenerative disorders. IUBMB Life 64(12):965–970. https://doi.org/10.1002/iub.1104
Boulay D, Bergis O, Avenet P, Griebel G (2010) The glycine transporter-1 inhibitor SSR103800 displays a selective and specific antipsychotic-like profile in normal and transgenic mice. Neuropsychopharmacology 35(2):416–427. https://doi.org/10.1038/npp.2009.144
Brandon NJ, Rotella DP (2007) Potential CNS applications for phosphodiesterase enzyme inhibitors. Annu Rep Med Chem 42:3–12. https://doi.org/10.1016/S0065-7743(07)42001-2
Chen Y, Wang H, Wang WZ, Wang D, Skaggs K, Zhang HT (2021) Phosphodiesterase 7 (PDE7): a unique drug target for central nervous system diseases. Neuropharmacology 196:108694. https://doi.org/10.1016/j.neuropharm.2021.108694
Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122(Pt 8):1437–1448. https://doi.org/10.1093/brain/122.8.1437
Dayalu P, Albin RL (2015) Huntington disease: pathogenesis and treatment. Neurol Clin 33(1):101–114. https://doi.org/10.1016/j.ncl.2014.09.003
Delhaye S, Bardoni B (2021) Role of phosphodiesterases in the pathophysiology of neurodevelopmental disorders. Mol Psychiatry 26(9):4570–4582. https://doi.org/10.1038/s41380-020-00997-9
Demirbas D, Wyman AR, Shimizu-Albergine M, Cakici O, Beavo JA, Hoffman CS (2013) A yeast-based chemical screen identifies a PDE inhibitor that elevates steroidogenesis in mouse Leydig cells via PDE8 and PDE4 inhibition. PLoS ONE 8(8):e71279. https://doi.org/10.1371/journal.pone.0071279
Dorner-Ciossek C, Kroker KS, Rosenbrock H (2017) Role of PDE9 in cognition. In: Phosphodiesterases: CNS functions and diseases, pp 231–254. https://doi.org/10.1007/978-3-319-58811-7_9
Duarte-Silva E, Chaves Filho AJ, Barichello T, Quevedo J, Macedo D, Peixoto C (2020) Phosphodiesterase-5 inhibitors: shedding new light on the darkness of depression? J Affect Disord 264:138–149. https://doi.org/10.1016/j.jad.2019.11.114
Farmer R, Burbano SD, Patel NS, Sarmiento A, Smith AJ, Kelly MP (2020) Phosphodiesterases PDE2A and PDE10A both change mRNA expression in the human brain with age, but only PDE2A changes in a region-specific manner with psychiatric disease. Cell Signal 70:109592. https://doi.org/10.1016/j.cellsig.2020.109592
Fusco FR, Giampa C (2015) Phosphodiesterases as therapeutic targets for Huntington’s disease. Curr Pharm Des 21(3):365–377. https://doi.org/10.2174/1381612820666140826113957
Fusco FR, Paldino E (2017) Role of phosphodiesterases in huntington’s disease. In: Phosphodiesterases: CNS functions and diseases, pp 285–304. https://doi.org/10.1007/978-3-319-58811-7_11
Galanopoulou AS, Buckmaster PS, Staley KJ, Moshé SL, Perucca E, Engel J Jr, Löscher W, Noebels JL, Pitkänen A, Stables J, White HS, American Epilepsy Society Basic Science Committee and the International League Against Epilepsy Working Group on Recommendations for Preclinical Epilepsy Drug Discovery, M (2012) Identification of new epilepsy treatments: issues in preclinical methodology. Epilepsia 53(3):571–582. https://doi.org/10.1111/j.1528-1167.2011.03391.x
Gharami K, Xie Y, An JJ, Tonegawa S, Xu B (2008) Brain-derived neurotrophic factor over-expression in the forebrain ameliorates Huntington’s disease phenotypes in mice. J Neurochem 105(2):369–379. https://doi.org/10.1111/j.1471-4159.2007.05137.x
Giampà C, Montagna E, Dato C, Melone MA, Bernardi G, Fusco FR (2013) Systemic delivery of recombinant brain derived neurotrophic factor (BDNF) in the R6/2 mouse model of Huntington’s disease. PLoS ONE 8(5):e64037. https://doi.org/10.1371/journal.pone.0064037
Gil C, Campillo NE, Perez DI, Martinez A (2008) PDE7 inhibitors as new drugs for neurological and inflammatory disorders. Expert Opin Ther Pat 18(10):1127–1139. https://doi.org/10.1517/13543776.18.10.1127
Gines S, Seong IS, Fossale E, Ivanova E, Trettel F, Gusella JF, Wheeler VC, Persichetti F, MacDonald ME (2003) Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum Mol Genet 12(5):497–508. https://doi.org/10.1093/hmg/ddg046
Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, Ahmadiani A (2017) Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: pathogenesis and treatment. CNS Neurosci Ther 23(1):5–22. https://doi.org/10.1111/cns.12655
Guilbert JJ (2006) The World Health Report 2006: working together for health. Educ Health (Abingdon) 19(3):385–387. https://doi.org/10.1080/13576280600937911
Gurney ME, D’Amato EC, Burgin AB (2015) Phosphodiesterase-4 (PDE4) molecular pharmacology and Alzheimer’s disease. Neurotherapeutics 12(1):49–56. https://doi.org/10.1007/s13311-014-0309-7
Heckman PR, Blokland A, Bollen EP, Prickaerts J (2018) Phosphodiesterase inhibition and modulation of corticostriatal and hippocampal circuits: clinical overview and translational considerations. Neurosci Biobehav Rev 87:233–254. https://doi.org/10.1016/j.neubiorev.2018.02.007
Hetman JM, Soderling SH, Glavas NA, Beavo JA (2000) Cloning and characterization of PDE7B, a cAMP-specific phosphodiesterase. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.97.1.472
Ikeda M, Tomita Y, Mouri A, Koga M, Okochi T, Yoshimura R, Yamanouchi Y, Kinoshita Y, Hashimoto R, Williams HJ, Takeda M (2010) Identification of novel candidate genes for treatment response to risperidone and susceptibility for schizophrenia: integrated analysis among pharmacogenomics, mouse expression, and genetic case-control association approaches. Biol Psychiatry 67(3):263–269. https://doi.org/10.1016/j.biopsych.2009.08.030
Jankowska A, Swierczek A, Chlon-Rzepa G, Pawlowski M, Wyska E (2017) PDE7-selective and dual inhibitors: advances in chemical and biological research. Curr Med Chem 24(7):673–700. https://doi.org/10.2174/0929867324666170116125159
Johnstone TB, Smith KH, Koziol-White CJ, Li F, Kazarian AG, Corpuz ML, Shumyatcher M, Ehlert FJ, Himes BE, Panettieri RA Jr, Ostrom RS (2018) PDE8 is expressed in human airway smooth muscle and selectively regulates cAMP signaling by β2-adrenergic receptors and adenylyl cyclase 6. Am J Respir Cell Mol Biol 58(4):530–541. https://doi.org/10.1165/rcmb.2017-0294OC
Kelly MP (2018) Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell Signal 42:281–291. https://doi.org/10.1016/j.cellsig.2017.11.004
Khan H, Kashyap A, Kaur A, Singh TG (2020) Pharmacological postconditioning: a molecular aspect in ischemic injury. J Pharm Pharmacol 72(11):1513–1527. https://doi.org/10.1111/jphp.13336
Khan H, Gupta A, Singh TG, Kaur A (2021a) Mechanistic insight on the role of leukotriene receptors in ischemic–reperfusion injury. Pharmacol Rep 73(5):1240–1254. https://doi.org/10.1007/s43440-021-00258-8
Khan H, Tiwari P, Kaur A, Singh TG (2021b) Sirtuin acetylation and deacetylation: a complex paradigm in neurodegenerative disease. Mol Neurobiol 58(8):3903–3917. https://doi.org/10.1007/s12035-021-02387-w
Khan H, Garg N, Singh TG, Kaur A, Thapa K (2022a) Calpain inhibitors as potential therapeutic modulators in neurodegenerative diseases. Neurochem Res. https://doi.org/10.1007/s11064-021-03521-9
Khan H, Grewal AK, Singh TG (2022b) Pharmacological postconditioning by protocatechuic acid attenuates brain injury in ischemia-reperfusion (I/R) mice model: Implications of nuclear factor erythroid-2-related factor pathway. Neuroscience. https://doi.org/10.1016/j.neuroscience.2022.03.016
Khan H, Sharma K, Kumar A, Kaur A, Singh TG (2022c) Therapeutic implications of cyclooxygenase (COX) inhibitors in ischemic injury. Inflamm Res. https://doi.org/10.1007/s00011-022-01546-6
Kinoshita KI, Tada Y, Muroi Y, Unno T, Ishii T (2015) Selective loss of dopaminergic neurons in the substantia nigra pars compacta after systemic administration of MPTP facilitates extinction learning. Life Sci 137:28–36. https://doi.org/10.1016/j.lfs.2015.07.017
Koran MEI (2014) Imaging and genetics of two amyloid related diseases: Alzheimer’s disease and down syndrome. Vanderbilt Univ. https://doi.org/10.1177/0891988710383571
Lee KJ, Chang WC, Chen X, Valiyaveettil J, Ramirez-Alcantara V, Gavin E, Musiyenko A, Madeira da Silva L, Annamdevula NS, Leavesley SJ, Ward A (2021) Suppression of colon tumorigenesis in mutant Apc mice by a novel PDE10 inhibitor that reduces oncogenic β-catenin. Cancer Prev Res 14(11):995–1008. https://doi.org/10.1158/1940-6207.CAPR-21-0208
Li L, Yee C, Beavo JA (1999) CD3- and CD28-dependent induction of PDE7 required for T cell activation. Science 283:848–851. https://doi.org/10.1126/science.283.5403.848
Lipina TV, Palomo V, Gil C, Martinez A, Roder JC (2013) Dual inhibitor of PDE7 and GSK-3–VP1. 15 acts as antipsychotic and cognitive enhancer in C57BL/6J mice. Neuropharmacology 64:205–214. https://doi.org/10.1016/j.neuropharm.2012.06.032
Luo HR, Wu GS, Dong C, Arcos-Burgos M, Ribeiro L, Licinio J, Wong ML (2009) Association of PDE11A global haplotype with major depression and antidepressant drug response. Neuropsychiatr Dis Treat 5:163. https://doi.org/10.2147/ndt.s4771
McCarthy DP, Richards MH, Miller SD (2012) Mouse models of multiple sclerosis: experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. In: Autoimmunity. Humana Press, Totowa, pp 381–401. https://doi.org/10.1007/978-1-60761-720-4_19
McGeer EG, McGeer PL (2007) The role of anti-inflammatory agents in Parkinson’s disease. CNS Drugs 21:789–797. https://doi.org/10.2165/00023210-200721100-00001
McGeer PL, Schwab C, Parent A, Doudet D (2003) Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine administration. Ann Neurol 54:599–604. https://doi.org/10.1002/ana.10728
McQuown S, Paes D, Baumgärtel K, Prickaerts J, Peters M (2021) Pharmacological inhibition of phosphodiesterase 7 enhances consolidation processes of spatial memory. Neurobiol Learn Mem 177:107357. https://doi.org/10.1016/j.nlm.2020.107357
Medina-Rodríguez EM, Bribián A, Boyd A, Palomo V, Pastor J, Lagares A, Gil C, Martínez A, Williams A, de Castro F (2017) Promoting in vivo remyelination with small molecules: a neuroreparative pharmacological treatment for Multiple Sclerosis. Sci Rep 7(1):1–14. https://doi.org/10.1038/srep43545
Mehats C, Andersen CB, Filopanti M, Jin SC, Conti M (2002) Cyclic nucleotide phosphodiesterases and their role in endocrine cell signaling. Trends Endocrinol Metab 13(1):29–35. https://doi.org/10.1016/s1043-2760(01)00523-9
Mendiola-Precoma J, Berumen LC, Padilla K, Garcia-Alcocer G (2016) Therapies for prevention and treatment of Alzheimer’s disease. Biomed Res Int. https://doi.org/10.1155/2016/2589276
Mercier J, Provins L, Hannestad J (2017). Progress and challenges in the development of PET ligands to aid CNS drug discovery. https://doi.org/10.1016/B978-0-12-409547-2.12437-0
Mestre L, Redondo M, Carrillo-Salinas FJ, Morales-García JA, Alonso-Gil S, Pérez-Castillo A, Gil C, Martínez A, Guaza C (2015) PDE 7 inhibitor TC 3.6 ameliorates symptomatology in a model of primary progressive multiple sclerosis. Br J Pharmacol 172(17):4277–4290. https://doi.org/10.1111/bph.13192
Morales-Garcia JA, Palomo V, Redondo M, Alonso-Gil S, Gil C, Martinez A, Perez-Castillo A (2014) Crosstalk between phosphodiesterase 7 and glycogen synthase kinase-3: two relevant therapeutic targets for neurological disorders. ACS Chem Neurosci 5(3):194–204. https://doi.org/10.1021/cn400166d
Morales-Garcia JA, Echeverry-Alzate V, Alonso-Gil S, Sanz-SanCristobal M, Lopez-Moreno JA, Gil C, Martinez A, Santos A, Perez-Castillo A (2017) Phosphodiesterase7 inhibition activates adult neurogenesis in hippocampus and subventricular zone in vitro and in vivo. Stem Cells 35(2):458–472. https://doi.org/10.1002/stem.2480
Morales-Garcia JA, Alonso-Gil S, Santos Á, Perez-Castillo A (2020) Phosphodiesterase 7 regulation in cellular and rodent models of Parkinson’s disease. Mol Neurobiol 57(2):806–822. https://doi.org/10.1007/s12035-019-01745-z
Nakamura T, Zhu G, Ranek MJ, Kokkonen-Simon K, Zhang M, Kim GE, Tsujita K, Kass DA (2018) Prevention of PKG-1α oxidation suppresses antihypertrophic/antifibrotic effects from PDE5 inhibition but not sGC stimulation. Circ Heart Fail 11(3):e004740. https://doi.org/10.1161/CIRCHEARTFAILURE.117.004740
Nandhakumar J, Tyagi MG (2010) Evaluation of seizure activity after phospho-diesterase and adenylate cyclase inhibition (SQ22536) in animal models of epilepsy. Indian J Sci Technol 3(7):710–717. https://doi.org/10.17485/ijst/2010/v3i7.1
Nelson MD, Rader F, Tang X, Tavyev J, Nelson SF, Miceli MC, Elashoff RM, Sweeney HL, Victor RG (2014) PDE5 inhibition alleviates functional muscle ischemia in boys with Duchenne muscular dystrophy. Neurology 82(23):2085–2091. https://doi.org/10.1212/WNL.0000000000000498
Omori K, Kotera J (2007) Overview of PDEs and their regulation. Circ Res 100(3):309–327. https://doi.org/10.1161/01.RES.0000256354.95791.f1
Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM (2005) A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain 128:2665–2674. https://doi.org/10.1093/brain/awh625
Patel NS, Klett J, Pilarzyk K, ik Lee D, Kass D, Menniti FS, Kelly MP (2018) Identification of new phosphodiesterase 9A (PDE9A) isoforms and how their expression and subcellular compartmentalization in brain changes across the lifespan. Neurobiol Aging 65:217. https://doi.org/10.1016/j.neurobiolaging.2018.01.019
Peng T, Gong J, Jin Y, Zhou Y, Tong R, Wei X, Bai L, Shi J (2018) Inhibitors of phosphodiesterase as cancer therapeutics. Eur J Med Chem 150:742–756. https://doi.org/10.1016/j.ejmech.2018.03.046
Perez-Gonzalez R, Pascual C, Antequera D, Bolos M, Redondo M, Perez DI, Pérez-Grijalba V, Krzyzanowska A, Sarasa M, Gil C, Ferrer I (2013) Phosphodiesterase 7 inhibitor reduced cognitive impairment and pathological hallmarks in a mouse model of Alzheimer's disease. Neurobiol Aging 34(9):2133–2145. https://doi.org/10.1016/j.neurobiolaging.2013.03.011
Pérez-Torres S, Cortés R, Tolnay M, Probst A, Palacios JM, Mengod G (2003) Alterations on phosphodiesterase type 7 and 8 isozyme mRNA expression in Alzheimer’s disease brains examined by in situ hybridization. Exp Neurol 182:322–334. https://doi.org/10.1016/S0014-4886(03)00042-6
Ponsaerts L, Alders L, Schepers M, de Oliveira RM, Prickaerts J, Vanmierlo T, Bronckaers A (2021) Neuroinflammation in ischemic stroke: inhibition of cAMP-specific phosphodiesterases (PDEs) to the rescue. Biomedicines 9(7):703. https://doi.org/10.3390/biomedicines9070703
Pozueta J, Lefort R, Shelanski ML (2013) Synaptic changes in Alzheimer's disease and its models. Neuroscience 251:51–65. https://doi.org/10.1016/j.neuroscience.2012.05.050
Redondo M, Zarruk JG, Ceballos P, Pérez DI, Pérez C, Perez-Castillo A, Moro MA, Brea J, Val C, Cadavid MI, Loza MI (2012) Neuroprotective efficacy of quinazoline type phosphodiesterase 7 inhibitors in cellular cultures and experimental stroke model. Eur J Med Chem 47:175–185. https://doi.org/10.1016/j.ejmech.2011.10.040
Rehni AK, Singh TG, Bhateja P, Singh N, Arora S (2010) Involvement of cyclic adenosine diphosphoribose receptor activation in ischemic preconditioning induced protection in mouse brain. Brain Res 1309:75–82. https://doi.org/10.1016/j.brainres.2009.10.071
Reierson GW, Guo S, Mastronardi C, Licinio J, Wong ML (2011) cGMP signaling, phosphodiesterases and major depressive disorder. Curr Neuropharmacol 9(4):715–727. https://doi.org/10.2174/157015911798376271
Ribaudo G, Ongaro A, Zagotto G, Memo M, Gianoncelli A (2020) Therapeutic potential of phosphodiesterase inhibitors against neurodegeneration: the perspective of the medicinal chemist. ACS Chem Neurosci 11(12):1726–1739. https://doi.org/10.1021/acschemneuro.0c00244
Sharma A, Khan H, Singh TG, Grewal AK, Najda A, Kawecka-Radomska M, Kamel M, Altyar AE, Abdel-Daim MM (2021a) Pharmacological modulation of ubiquitin-proteasome pathways in oncogenic signaling. Int J Mol Sci 22(21):11971. https://doi.org/10.3390/ijms222111971
Sheng Z, Lai S, Ma Y, Luo X (2012) The space global weak solutions to the weakly dissipative camassa- holm equation. In: Abstract and applied analysis, vol 2012. Hindawi. https://doi.org/10.1155/2012/693010
Sharma VK, Singh TG, Mehta V (2021b) Stressed mitochondria: a target to intrude Alzheimer’s disease. Mitochondrion 59:48–57. https://doi.org/10.1016/j.mito.2021.04.004
Smith SJ, Cieslinski LB, Newton R, Donnelly LE, Fenwick PS, Nicholson AG, Barnes PJ, Barnette MS, Giembycz MA (2004) Discovery of BRL 50481 [3-(N, N-dimethylsulfonamido)-4-methyl-nitrobenzene], a selective inhibitor of phosphodiesterase 7: in vitro studies in human monocytes, lung macrophages, and CD8+ T-lymphocytes. Mol Pharmacol 66(6):1679–1689. https://doi.org/10.1124/mol.104.002246
Soni NO (2019) Current and future clinical application-phosphodiesterase (PDEs)-inhibitors. https://doi.org/10.20959/wjpps20197-14233
Sun J, Xiao Z, Haider A, Gebhard C, Xu H, Luo HB, Zhang HT, Josephson L, Wang L, Liang SH (2021) Advances in cyclic nucleotide phosphodiesterase-targeted PET imaging and drug discovery. J Med Chem 64(11):7083–7109. https://doi.org/10.1021/acs.jmedchem.1c00115
Susín C, Morales‐Garcia JA, Aguilar‐Morante D, Palomo V, Sanz‐Sancristobal M, Alonso‐Gil S, Gil C, Santos A, Martinez A, Perez‐Castillo A (2012) The new iminothiadiazole derivative VP1. 14 ameliorates hippocampal damage after an excitotoxic injury. J Neurochem 122(6):1193–1202. https://doi.org/10.1111/j.1471-4159.2012.07866.x
Szczypka M (2020) Role of phosphodiesterase 7 (PDE7) in T cell activity. Effects of selective PDE7 inhibitors and dual PDE4/7 inhibitors on T cell functions. Int J Mol Sci 21(17):6118. https://doi.org/10.3390/ijms21176118
Tansey MG, McCoy MK, Frank-Cannon TC (2007) Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol 208:1–25. https://doi.org/10.1016/j.expneurol.2007.07.004
Thapa K, Khan H, Sharma U, Grewal AK, Singh TG (2021) Poly (ADP-ribose) polymerase-1 as a promising drug target for neurodegenerative diseases. Life Sci 267:118975. https://doi.org/10.1016/j.lfs.2020.118975
Torri F, Akelai A, Lupoli S, Sironi M, Amann‐Zalcenstein D, Fumagalli M, Fiume CD, Ben‐Asher E, Kanyas K, Cagliani R, Cozzi P (2010) Fine mapping of AHI1 as a schizophrenia susceptibility gene: from association to evolutionary evidence. FASEB J 24(8):3066–3082. https://doi.org/10.1096/fj.09-152611
Volakakis N, Kadkhodaei B, Joodmardi E, Wallis K, Panman L, Silvaggi J, Spiegelman BM, Perlmann T (2010) NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proc Natl Acad Sci 107(27):12317–12322. https://doi.org/10.1073/pnas.1007088107
Xie Y, Hayden MR, Xu B (2010) BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. J Neurosci 30(44):14708–14718. https://doi.org/10.1523/JNEUROSCI.1637-10.2010
Xu C, Wyman AR, Alaamery MA, Argueta SA, Ivey FD, Meyers JA, Lerner A, Burdo TH, Connolly T, Hoffman CS, Chiles TC (2016) Anti-inflammatory effects of novel barbituric acid derivatives in T lymphocytes. Int Immunopharmacol 38:223–232. https://doi.org/10.1016/j.intimp.2016.06.004
Xu M, Yu X, Meng X, Huang S, Zhang Y, Zhang A, Jia Z (2020) Inhibition of PDE4/PDE4B improves renal function and ameliorates inflammation in cisplatin-induced acute kidney injury. Am J Physiol Renal Physiol 318(3):F576–F588. https://doi.org/10.1152/ajprenal.00477.2019
Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, Chinea A (2015) A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. https://doi.org/10.4103/2152-7806.169561
Zhang H, Yang Z, Lin T, Wu Y, Zou T, Zhang Y (2018) ARL2 regulates trafficking and expression of isoprenylated proteins and is crucial for development of photoreceptor outer segments. Invest Ophthalmol vis Sci 59(9):963–963. https://doi.org/10.4161/org.26710
Zhang P, Xu S, Zhu Z, Xu J (2019) Multi-target design strategies for the improved treatment of Alzheimer’s disease. Eur J Med Chem 176:228–247. https://doi.org/10.1016/j.ejmech.2019.05.020
Acknowledgements
The authors are grateful to the Chitkara College of Pharmacy, Chitkara University, Rajpura, Patiala, Punjab, India for providing the necessary facilities to carry out the research work.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Conceptualization: conceived and designed the experiments: TGS. Analyzed the data: AK. Wrote the manuscript: HK, CT and SC. Visualization: TGS. Editing of the manuscript: HK, AK, TGS. Critically reviewed the article: TGS. Supervision: TGS. Funding: GE–SB. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest.
Consent to participate
Not applicable
Consent to publish
Not applicable
Ethics approval and consent to participate
Not applicable
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Khan, H., Tiwari, C., Grewal, A.K. et al. Pharmacological modulation of phosphodiesterase-7 as a novel strategy for neurodegenerative disorders. Inflammopharmacol 30, 2051–2061 (2022). https://doi.org/10.1007/s10787-022-01072-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-022-01072-1