
Abstract
The initial precipitating injury such as SE progresses to chronic epilepsy through multiple epileptogenic processes. Early epileptogenic events are generally characterized by neuroinflammation, neurodegeneration and abnormal neurogenesis in the hippocampus. Metformin has exhibited anti-inflammatory and neuroprotective properties in numerous studies. The current study attempts to investigate the effect of metformin on seizure-induced inflammation and neuronal degeneration, and the involvement of the mTOR pathway. Status epilepticus (SE) was induced in male Wistar rats with systemic administration of Lithium (127 mg/kg) and Pilocarpine (30 mg/kg). In test rats, Metformin 100 mg/kg or 200 mg/kg was administered orally for 7 days, followed by SE induction. Results indicate that metformin did not alter the SE profile significantly which was evident by the behavioural scoring and electroencephalogram (EEG) recordings. However, metformin 200 mg/kg attenuated the SE-induced glial activation (p < 0.01), up regulated mRNA levels of proinflammatory cytokines (p < 0.001) and chemokines (p < 0.001) and enhanced BBB permeability (p < 0.05). In addition, metformin ameliorated the insult-induced region-specific neuronal damage (p < 0.01) and restored the hippocampal neuronal density. Metformin significantly inhibited phosphorylated S6 ribosomal protein (phospho-S6rp) (p < 0.05), thus demonstrating that the beneficial effects might be partly mediated by the mTOR pathway. The study thus reiterates that mTOR signalling is one of the mechanisms involved in inflammation and neurodegeneration in early epileptogenesis following SE.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epileptogenesis is one among the major grey areas in epilepsy research. Though certain drugs such as rapamycin and natural products have displayed anti-epileptogenic properties in the preclinical studies, they failed to reach the bedside (Clossen and Reddy 2017), due to the lack of understanding of underlying mechanisms. Among the numerous factors driving the epileptogenesis, involvement of immune activation and inflammation is being robustly observed in the preclinical and clinical studies (Vezzani et al. 2013). Persistent seizures such as SE can trigger inflammation, characterized by dramatic up regulation of an array of proinflammatory mediators, activation of astroglia, microglia and endothelial cells, BBB breach, infiltration of plasma proteins and immune cells, which in turn enhance neuronal excitability and neurodegeneration (Vezzani 2016; Tannich et al. 2020). Additionally, several clinical and preclinical studies have shown potential beneficial effects of anti-inflammatory drugs regardless of the pathology and epileptic syndrome (Marchi et al. 2011; Dey et al. 2016). Therefore, targeting inflammation poses a promising therapeutic strategy in the search for new anti-epileptic and anti-epileptogenic drugs.
Metformin, an established, safe anti-diabetic drug surprised the scientific community with its diverse therapeutic responses in various disorders such as polycystic ovarian disease (PCOS) (Lashen et al. 2010), Non-alcoholic fatty liver disease (NAFLD) (Fujito and Inagaki 2017), more recently in cancer (Zi et al. 2018) and ageing (Barzilai et al. 2016). Its pleiotropic effects extend to CNS where it showed promising beneficial effects in several neurological disorders including multiple sclerosis, seizures, Huntington’s and Alzheimer’s diseases and cerebral ischaemia (Arbelaez-Quintero and Palacios 2017; Rotermund et al. 2018; Ursini et al. 2018). Existing studies indicate the beneficial effect of metformin against seizures and epilepsy in PTZ (acute) and PTZ kindling (chronic) models attributed to its anti-oxidant, anti-glycotic and anti-apoptotic actions (Zhao et al. 2014; Mehrabi et al. 2018; Chen et al. 2018). Additionally, metformin was shown to attenuate central as well as peripheral inflammation by attenuating the release of inflammatory mediators such as proinflammatory cytokines, NO, prostaglandins, damage-associated molecular patterns (DAMPs) and reactive oxygen species (ROS) in AMPK/NF-κβ dependent manner in a variety of in vitro studies (Saisho 2015; Jing et al. 2018; Negrotto et al. 2016).
Besides, metformin has been reported to inhibit mTOR (Kalender et al. 2010), and mTOR inhibitors have promising potential as therapeutic strategies for epilepsy (Citraro et al. 2016), underlining metformin as a prospective multifaceted intervention for epilepsy. Many studies have demonstrated the involvement of mTOR in the regulation of neuroinflammatory responses (Wang et al. 2018; Saliba et al. 2017; Yang et al. 2017; Ye et al. 2020) and accordingly, rapamycin has been proposed as treatment for a variety of neurological disorders characterized by chronic inflammation within the CNS (Crino, 2016, Srivastava et al. 2015, Cho et al. 2018). Of note, inhibition of mTOR pathway promoted microglial, M1 to M2 transition, significantly reducing the generation of proinflammatory cytokines and chemokines (IL-1β, TNFα, MCP-1, CCL-22 and iNOS), resulting in improvements in motor deficit in MCAO model of cerebral stroke (Li et al. 2016). Therefore, the present study attempts to investigate the effect of metformin over neuro-inflammation and neurodegeneration in status epilepticus model of rats and the possible involvement of the mTOR pathway.
Materials and methods
Animals
Two-month-old adult male Wistar rats weighing about 180–230 g were obtained from the Advanced facility for small animal research, Post Graduate Institute of Medical Education and Research (PGIMER). Rats were housed in polymer cages at an ambient temperature of 25 ± 2 °C, 60 ± 2% humidity and a 12-h light/dark cycle. Food in form of standard dry pellets and water was available ad libitum. All experiments were carried according to the guidelines of the Committee for the Purpose of Control and Supervision of Experimentation on Animals (CPCSEA) and the experiments were approved by Institutional Animal ethical committee (approval number: 73/IAEC/449) and the Institute Biosafety committee (11/IBC/2014).
Drugs and chemicals
Pilocarpine and lithium (Sigma, USA) were dissolved in saline and administered intraperitoneally. Metformin (Sigma, USA) was dissolved in distilled water and given orally. Rapamycin (Biocon, India), solubilized in 100% ethanol and stored at − 20 °C, was diluted immediately prior administration in a vehicle solution comprising of 5% Tween 80, 5% PEG 400 and 4% ethanol (Chi et al. 2017). Doses of metformin (Ashabi et al. 2015) and rapamycin (Rensing et al. 2015) were selected on the basis of reported literatures. All the solutions were made freshly before the experiment.
Treatment schedule
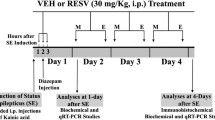
After acclimatization period, rats were randomly divided into five groups: vehicle control (distilled water); Negative control, Lithium- Pilocarpine (Li-Pi); Treatment groups, metformin (100 mg/kg) and metformin (200 mg/kg); Positive control, rapamycin (10 mg/kg). All groups consisted of 16 rats each, whereas metformin (100 mg/kg) group had five rats. SE and metformin groups were treated with distilled water/metformin (100 or 200 mg/kg), respectively, for 7 days, followed by induction of SE, 60 min after the last dose of distilled water/metformin. To investigate whether metformin acts through mTOR pathway, mTOR inhibitor—rapamycin (10 mg/kg)—was administered for 4 subsequent days in rapamycin group followed by SE induction 24 h after the last dose (Rensing et al. 2015). The study design is illustrated in Fig. 1.

Study model for investigating the effect of metformin or rapamycin pretreatment against pilocarpine-induced SE. Subscripts depict the number of days the respective drugs were administered to rats prior to subjecting to SE. DW: distilled water, Met: Metformin, Rap: Rapamycin, Li-Pi-DZP: Lithium-Pilocarpine followed by diazepam, BBB: Blood–brain barrier, H&E: haematoxylin and eosin, FJB: Fluorojade B staining
Electrode implantation and recording
Telemetry device (EPOCH WIRELESS EEG 2-CH transmitters; BIOPAC, CA, USA) was implanted in six rats (250–300 g) for remote EEG recording. Rats were anaesthetised with ketamine (100 mg/kg) and xylazine (10 mg/kg, i.p.). Rat head was shaved and swabbed with 70% ethanol. The head was positioned in stereotactic frame and a midline sagittal incision was made in the scalp. Two holes, just penetrating the skull, were drilled at approximately 4.0 mm posterior and 3.0 mm left and right to the bregma over each hemisphere and bloodswabbed off to ensure dry surface. Electrode tips were trimmed to remove insulation and electrode was positioned in the holes, in contact with the dura mater. Electrodes were adhered with dermabond which assures strong attachment to the skull. The plastic bases were carefully overlaid by dental cement to fix the electrode and scalp was sutured to close the incision. Rats that had undergone surgery were fed with soft diet and monitored for weight to assess recovery. After 7 days’ recovery period, seizures were induced and behavioural analysis was done.
To acquire EEG, rat cage was placed on the receiver plate. Acquiring and analysis were done using Acknowledge software (Biopac developer, CA, USA) with simultaneous video recording for 3 h. Sampling rate of 1000 Hz with filter low-pass filter cut off at 50 Hz and high-pass filter cutoff at 0.5HZ, and a line frequency cut off at 60 Hz (electrical noise) were applied. Automated seizure analysis, EEG frequency analysis and relative power calculation were done on the raw EEG trace. For automated seizure analysis, high-pass filter was applied at 10 Hz to remove artefacts related to movements. Baseline EEG is recorded for each rat prior to seizure induction.
Induction of SE by lithium-pilocarpine (Li-Pi)
All the rats except those in the vehicle group received lithium chloride (127 mg/kg, i.p.). 19–21 h following lithium treatment, pilocarpine (30 mg/kg, i.p.) was administered to induce SE. To limit the peripheral cholinergic side effects, methyl scopolamine bromide (2 mg/kg, i.p) was given 30 min prior to pilocarpine. SE is defined as a condition of continuous grade IV–VII seizures (according to Racine scale) lasting longer than 30 min. To reduce the mortality associated with SE, 120 min after the initiation of SE, diazepam (10 mg/kg, i.p.) was administered to terminate SE. 90% of the rats presented with SE after pilocarpine administration, and rats that did not display SE were not included in the study.
Seizure scoring
The behavioural progression of seizures was assessed using a modified Racine scale (Salem et al. 2018). Seizure activity was recorded as follows:
Stage I: Staring with mouth clonus.
Stage II: Head nodding, automatisms (e.g., scratching, sniffing orientation).
Stage III: Unilateral forelimb clonus.
Stage IV: Forelimb clonus (rearing).
Stage V: Forelimb clonus with rearing and one fall.
Stage VI: Forelimb clonus with rearing and multiple successive falls.
Stage VII: Tonic/clonic seizures (e.g., running and jumping).
Animals were constantly monitored following pilocarpine administration for 2 h. Latency time for the onset of different seizure stages and the maximum stage of seizure activity occurring in each 15-minute interval following SE were recorded as described by Kumar et al. 2014.
RNA isolation and real-time PCR
Rat was killed and brain isolated. Total RNA from rat hippocampi were isolated using Nucleospin RNA extraction kit, Macherey–Nagel, Germany. RNA concentrations in the extracts were measured photometrically and RNA integrity was verified. mRNA (1 µg) was then reverse transcribed into cDNA using oligo (dT) primers and cDNA synthesis kit (Thermo, Rockford, IL). Polymerase chain reaction (PCR) was performed for TNF-α, IL-1β, IL-6, CCL-3, NFκβ, iNOS, COX-2, FoxP3 and IL-10. GAPDH and β-actin were used as housekeeping genes. Real-time PCR was performed with Roche Light Cycler 480 using SYBR Green I (Rani et al. 2015). Primer sequences and annealing temperatures are listed in the following Table 1.
Quantification of BBB permeability
BBB permeability was analysed using brain Evan's Blue (EB) extravasation. Rats were administered a fluorescent tracer that would not penetrate the CNS in physiological conditions. 500 μL of 4% EB in normal saline was administered into the rat tail vein. After 4 h, rats were anaesthetized with ketamine and xylazine, transcardially perfused with normal saline till blue colour disappeared in the effluent. Then, hippocampi were isolated, homogenized in 50% TCA and then centrifuged at 10,000 rcf for 30 min. The absorbance was determined at 620 nm with UV spectrophotometer. The amount of dye in the supernatant was extrapolated from the standard curve and expressed as nanogram of EB per gramme of tissue (van Vliet et al. 2007).
Histopathology and Immunohistochemistry
Rats were perfused transcardially with PBS and 4% paraformaldehyde after 3% pentobarbital anaesthesia. Brains were isolated and stored in paraformaldehyde for 2–3 days. Hippocampus area was grossed. Paraffin blocks were made and 10 μm coronal sections were cut, mounted and used for Haematoxylin and eosin (H&E), Fluorojade B staining and immunohistochemistry (Mehan et al. 2018).
Haematoxylin and eosin staining
Acidophilic neurons are detected by features such as extensive eosinophilic cytoplasm, chromatin disintegration, and absence of an integral nuclear membrane, which serve as a marker for irreversible neuronal damage. Acidophilic neurons were estimated approximately in hippocampus using the following three-point scale: 0 = absence; 0.5—very mild (below 10%); 1—mild (10 to 25%); 1.5—mild-moderate (26 to 45%); 2.0—moderate (46 to 54%); 2.5 = moderate to severe (55 to 75%); 3.0—severe (more than 75%) (Fujikawa et al. 2016; Kumari et al. 2018).
Fluorojade B histochemistry
Fluorojade B (FJB) is a fluoro-Jade derivative, which selectively stains acidophilic cellular components of the dying neurons and appears fluorescein positive under the fluorescent microscope. Paraffin sections were rehydrated in absolute ethanol and then in 70% alcohol. Sections were changed to 0.06% potassium permanganate for 15 min on rotating shaker, subjected to 0.001% FJ staining solution (Millipore, Temecula, CA) and gently shaken for 30 min. Later, the sections were dried, immersed in xylene and coverslipped with mounting media. Slides were then examined under fluorescent microscope with excitation and emission peak of 480 and 525 nm, respectively. Images were captured in three different fields at 40X and the cells were counted using Image J software (Zhu et al. 2019).
Immunohistochemistry analysis
For immunohistochemistry, 10 μM paraffin sections were taken on polylysine-coated slides and deparaffinised, rehydrated and blocked in hydrogen peroxide. Antigen retrieval was performed using citrate buffer pH = 6. Sections were washed in PBS and incubated at 4 °C overnight with the following primary Abs: rabbit polyclonal anti-glial fibrillary acidic protein (DAKO, USA) for astrocytes and rabbit polyclonal to phospho-S6rp-Ser 235/236 (CST, Danvers, MA). Later, sections were incubated in HRP-conjugated IgG anti rabbit secondary antibody prepared in blocking solution for 30 min at RT. Sections were finally washed and developed using the DAB Peroxidase (HRP) substrate kit, 3,3′-diaminobenzidine. Preparations were counterstained with haematoxylin, mounted and observed under light microscope (Singh et al. 2019).
Immunoblotting analysis
Hippocampi were homogenized individually in Tris lysis buffer with protease inhibitor cocktail (Thermo, USA). Homogenate was centrifuged at 13,000 ×g for 10 min at 4 °C and supernatant was isolated for immunoblot analysis. Protein concentration was estimated by Bradford assay (Kumar et al. 2013).
Volume equivalent to 50 μg of the protein was run on 10% SDS polyacrylamide gel and transferred to PVDF membrane. Membrane was blocked with BSA and probed with the primary antibodies against S6 protein (1:1000; CST, Danvers, MA), phospho-S6rp (Ser235/236) protein (1:1000; CST, Danvers, MA) and β-actin (1:1000; Thermo, USA) overnight at 4 °C. Followed by PBST washing, membrane was treated with the horseradish peroxidase (HRP)-labelled anti-rabbit IgG secondary antibody (1: 10,000; Thermo, USA) at RT for 1–2 h. Using enhanced chemiluminescence reagent (Clarity™ ECL reagent, Biorad, Hercules, CA), detection was performed by the Protein Simple (CA, USA). Image J software 1.51j (NIH; Bethesda, MD) was used to measure relative intensity of bands after normalizing with β-actin (Wang et al. 2013).
Statistical analysis
Graph Pad prism was used for applying statistical analyses. Data were represented as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) test followed by Bonferroni post hoc analysis was applied. Probability value (p) less than 0.05 was considered statistically significant.
Results
Effect of metformin or rapamycin pretreatment over pilocarpine induced status epilepticus
Intraperitoneal administration of pilocarpine produced an initial motionless state followed by sequence of olfactory and gustatory automatisms. Over a period of 20–50 min, most of the rats developed sudden clonic jerks, unilateral and bilateral forelimb clonus (motor limbic seizures) with rearing and falling and progressed towards limbic SE.
Latency time to reach different seizure stages in minutes were recorded (Fig. 2a). The cumulative seizure score for every 15 min was noted (Fig. 2b). Pilocarpine administration produced seizures with severe intensity and short latency as indicated by Fig. 2a, b. Pretreatment with both the doses of metformin (100 mg/kg and 200 mg/kg) produced no significant difference on the severity and latency of seizures as compared to pilocarpine group (Fig. 2a, b).

Effect of metformin or rapamycin treatment on the latency to develop various seizure stages (a) and seizure scores at regular time intervals (b) against pilocarpine-induced SE. Data are expressed as mean ± SD (N = 10) except Met 100 (N = 5). ANOVA followed by Bonferroni’s posthoc test. Also, effect of metformin or rapamycin treatment on neuronal morphology (c) and on the number of SE-induced acidophilic neurons (d) at the level of the hippocampus 24 h following SE, stained by haematoxylin and eosin. Control Sect. (4×) showing areas of the hippocampal formation; CA1, CA2, CA3 regions and hilus (H) forming hippocampus proper. Dentate gyrus surrounding hilus with its upper and lower limbs. SE group (4×) showing gross destruction of pyramidal neuronal layer CA3, CA2, CA1, loss of hilar neurons, rarefaction of neuropil and higher magnification (40×) of the same section showing pyramidal neurons of CA3 with eosinophilic cytoplasm (*), few vacuolations (v), shrunken and pyknotic nuclei () and densely stained nuclei indicating chromatin clumping; Metformin (100 mg/kg) treated group with partial restoration of CA3 layer, however, with numerous acidophilic neurons, shrunken cells and sparsely distributed healthy neurons; Metformin (200 mg/kg) treated group showing near to normal morphology with preserved number of neurons compared to control; Rapamycin (10 mg/kg) treated group showed restored morphology relative to the pilocarpine group, however, showing few cells with shrunken and pyknotic nuclei. (D) Data are expressed as mean ± SD (N = 10) except Met 100 (N = 5). ANOVA followed by Bonferroni’s posthoc test
Epidural EEG was recorded to examine the effect of metformin over subclinical seizures. The EEG mean power was significantly increased after SE, which was not altered by the metformin pretreatment. The number of spikes and relatively, the number of seizures were not significantly different among the epileptic and metformin treatment groups (Fig. 3).

Effect of metformin over EEG recording (a) EEG records obtained for 15 min before SE which serve as internal baseline reading, and thereafter for 2 h post SE. The first lane shows the 1–100 Hz filtered signal. For automatic detection of spike events during SE, raw EEG traces were filtered (high-pass at 10 Hz) to eliminate movement-related artifacts. The second lane shows all the signals (indicated as ticks) that crossed the threshold value as detected by the automatic spike detection. Threshold value was calculated as mean ± 3 standard deviations of baseline value. The third lane represents such detected spikes on a time scale. The fourth lane represents seizures detected. The software defines a seizure, when minimum 20 spikes occur in time epoch width of 10seconds. The fifth lane indicates the mean power calculated for 5 min epochs. No. of seizures detected after SE induction (B) and mean power calculated for the 2 h EEG trace in the SE alone and metformin treated groups. Data represent mean ± SD (N = 3)
In order to investigate the involvement of mTOR pathway in SE and associated pathologies, rapamycin (typical mTOR inhibitor) group was included in the study. Pretreatment with rapamycin also did not produce any difference on seizure severity and latency as compared to pilocarpine group (Fig. 2b).
Effect of metformin or rapamycin pretreatment on histological scores in status epilepticus model of rats
Histological analyses revealed significant neuronal damage in the hippocampus of SE undergone rats as compared to control rats. In control rats, hippocampal neurons were arranged in compact fashion with intact morphology as of normal size and shape, prominent dark coloured nucleus. Few hypoxic changes were seen in some sections. Pilocarpine-induced SE revealed gross destruction of pyramidal neurons of CA3 and CA1 with features such as rarefaction of neuropil, eosinophilic cytoplasm, shrunken and pyknotic nuclei in hippocampus as observed by the histopathologist. Pretreatment with metformin (200 mg/kg) restored the CA3, CA2 and CA1 pyramidal neuronal layer as compared to SE rats. Pretreatment with metformin (100 mg/kg) did not produce any notable impact on SE induced histological changes (Fig. 2C).
Particularly in the CA3 region, SE rats exhibited almost complete ablation of healthy neurons with many cells showing shrunken darkened apoptotic nuclei, chromatin clumping, cytoplasmic vacuolations and condensation and hypereosinophilia. However, CA3 region in metformin 200 mg/kg treated rats displayed preserved pyramidal cells, with few apoptotic cells almost near to the control group morphology. Further, rapamycin group revealed moderate improvement with less number of apoptotic cells, angulated in shape and more number of healthy neurons as compared to pilocarpine group (Fig. 2C).
Grading of these sections for acidophilic neurons was done by blinded investigator as per the scale. SE rats displayed significant increase in scores compared to control (p < 0.05) indicating high degree of apoptosis or necrosis undergoing cells. Treatment with metformin 200 mg/kg, but not metformin 100 mg/kg, ameliorated the cell damage with lower number of acidophilic neurons as compared to the pilocarpine group (p < 0.05) indicating the protective effect of metformin over SE-induced neuronal damage (Fig. 2d). Based on the seizure scores and histological grading for neuronal damage, metformin 200 mg/kg was selected as effective dose and used for further molecular studies. Rapamycin pretreatment group did not have significant effect on the number of acidophilic neurons as compared to SE group.
Effect of metformin or rapamycin on SE altered gene expression profile of inflammatory mediators
mRNA levels of proinflammatory cytokines TNFα, IL-6, IL-1β, NF-κβ, CCL-3, COX-2, iNOS and anti-inflammatory cytokine IL-10 were measured in hippocampi at 4 h and 24 h post SE.
Four hours post SE, mRNA expression of TNFα (108-fold change, p < 0.001), IL-1β (23-fold change), NFκβ (60-fold; p < 0.001), CCL-3 (102-fold; p < 0.001), COX-2 (225- fold; p < 0.001) and iNOS (1.6-fold, p < 0.001) were significantly elevated in SE rats as compared to control. Further, this increase in mRNA levels induced by SE was significantly attenuated by metformin pretreatment TNFα (2.8-fold; p < 0.001), IL-1β (25.6-fold; p < 0.001), NFκβ (14-fold; p < 0. 01), CCL-3 (12.9-fold, p < 0.001), COX-2 (7.2-fold; p < 0.001) and iNOS (10.3-fold, p < 0.001) as compared to SE alone rats. Similarly, rapamycin group also displayed significant reduction in TNFα (4.5-fold; p < 0.001), IL-1β (19.2; p < 0.001), NFκβ (tenfold; p < 0.001) and COX-2 (4.8-fold; p < 0.001) gene expression as compared to SE rats (Fig. 4). Post SE 24 h, IL-1β (1.9-fold; p < 0.001), IL-6 (27.4-fold; p < 0.001), NFκβ (110-fold; p < 0.001), COX-2 (173-fold; p < 0.001) and iNOS (1.63-fold change; p < 0.001) were significantly elevated in SE rats and this was reversed by metformin pretreatment, IL-6 (6.68-fold; p < 0.001), IL-1β (9.5-fold; p < 0.001), NFκβ (50-fold; p < 0.001) and iNOS (7.8-fold; p < 0.001) as compared to SE rats. Meanwhile, rapamycin pretreatment also significantly attenuated the SE-elevated gene expression of IL-6 (0.02-fold; p < 0.001) and NFκβ (25-fold; p < 0.001) as compared to SE24 group (Fig. 4).

Effect of metformin or rapamycin on mRNA levels of cytokines TNF α (a), IL-1β (b), IL-6 (c), NFκβ (d), COX-2 (e), iNOS (f) and IL-10 (g) in hippocampus following SE. Data normalized to β-actin and GAPDH and expressed as mean ± SD (N = 5). One-way ANOVA followed by Tukey test. *p ≤ 0.05, ***p ≤ 0.001 as compared to control group. $$p ≤ 0.01 $$$p ≤ 0.001 as compared to pilocarpine 4-h group, #p ≤ 0.01, ##p ≤ 0.01, ###p ≤ 0.001 as compared to pilocarpine 24-h group
IL-10, in contrast to the other cytokines, is an inflammatory cytokine and hence expected to decrease in disease condition. On contrary, IL-10 gene expression was rapidly up regulated (fourfold; p < 0.001) at 4 h post SE, which dramatically reduced to 0.4-fold (p < 0.001) by 24 h as compared to control. However, IL-10 mRNA levels remained at basal levels in metformin (0.65 fold; p < 0.001) and rapamycin (2.9-fold; p < 0.001) groups as compared to SE at 4hours. Rapamycin, but not metformin, restored the IL-10 mRNA levels at 24 h as compared to SE 24 h (1.14-fold, p < 0.001, Fig. 4). This indicates the anti-inflammatory potential of rapamycin in the context of SE induced inflammation. Similar to IL-10, FoxP3 transcription factor was upregulated 4 h post SE (7.8 fold, p < 0.001) as compared to control. However, neither metformin nor rapamycin significantly altered the rise in FoxP3 induced by SE.
Effect of metformin or rapamycin on SE associated changes in BBB permeability
Evans blue dye is commonly used to detect changes in BBB permeability as it binds with serum albumin to form large conjugate which cannot penetrate intact BBB under normal conditions. 24 h post SE, significantly increased dye concentration was found in the brain supernatant of pilocarpine group (p < 0.01) as compared to control rats. This indicates that SE resulted in BBB dysfunction in rats. Pretreatment with either metformin (p < 0.05) or rapamycin (p < 0.01) significantly reduced the brain concentration of dye as compared to pilocarpine group which indicates the protective effect of metformin or rapamycin pretreatment over SE-induced BBB impairment (Fig. 5).

Effect of metformin or rapamycin on the SE-induced BBB dysfunction. Data represented as mean ± S.D (N = 3). One-way ANOVA followed by Tukey test. **p ≤ 0.01 as compared to control group. $p ≤ 0.05, $$p ≤ 0.01 as compared to pilocarpine group
Effect of metformin or rapamycin treatment on SE induced astroglial activation
The activation status of astrocytes was examined in the hippocampal CA1, CA3 and DG areas of hippocampus 24 h following SE through immunostaining against GFAP. Activated astrocytes can be detected by striking alterations such as enhanced number and thickness of GFAP-positive processes. In the control group, resting astroglia appeared as small cell bodies with extensive long and thin processes and exhibited low GFAP-immunoreactivity. 24 h following SE, increased number of reactive astrocytes characterized by hypertrophic cell bodies with short and thick processes, filled with intermediate filaments, were robustly present demonstrating typical astroglial activation. These reactive astrocytes were found to be most prominent in hilar region and molecular layer above CA1 region of hippocampus. Both metformin (p < 0.01) and rapamycin (p < 0.001) pretreatment individually attenuated the astroglial activation induced by SE as evident by the decreased number of reactive astrocytes in the hilar as well as CA1 regions as compared to pilocarpine (Fig. 6).

Immunohistochemical analysis of GFAP-immunopositive astrocytes in the hippocampal hilar region 24 h following SE in different groups at 40x. Quantitation of GFAP positive cells in (B) hilar and (C) CA1 regions of hippocampus. Data represented as mean ± S.D (N = 5). One-way ANOVA followed by Tukey test. ***p ≤ 0.001 as compared to control group, #p ≤ 0.05 as compared to SE group
Effect of metformin pretreatment on SE-induced neuronal damage assessed by FJB histochemistry
Using FIJI software, numbers of fluorescent cells were estimated in a particular region keeping threshold and area constant for each rat section. As compared to the control, post SE 24 h, rats exhibited significantly high number of FJB positive neurons in CA3 (p < 0.001) and CA1 (p < 0.05) areas. Metformin-treated group revealed significantly reduced number of FJB positive neurons more particularly in CA3 region (p < 0.01) of hippocampus as compared to pilocarpine group indicating protective effect of metformin over SE-induced neurodegeneration. These results also indicate the region-specific neuroprotective effect of metformin. In addition, rapamycin group also exhibited similar reduction in CA3 region (p < 0.05), whereas CA1 region remains unaltered as compared to the pilocarpine group suggesting that both metformin and rapamycin attenuated the SE-induced neurodegeneration (Fig. 7).

Effect of metformin pretreatment on SE-induced neuronal damage in hippocampus assessed by FJB staining. (A) Photomicrographs of rat coronal brain section, at the level of the hippocampus CA1 and CA3 showing FJB positive degenerating neurons post SE (20 ×). (B) Quantitation of FJB positive degenerating neurons in hippocampus Data represented as mean ± S.D (N = 5). One-way ANOVA followed by Tukey test. ***p ≤ 0.001 as compared to control group, #p ≤ 0.05 as compared to SE group
Effect of metformin or rapamycin pretreatment over SE-induced mTOR hyperactivation
To study alterations in the mTOR signaling pathway after SE, protein lysis samples from the hippocampus were separated by electrophoresis and analysed for S6, downstream regulator of mTOR pathway, by western blotting. Both total S6rp and phosphorylated (active) form of S6rp, p-S6rp, were assessed. As shown in Fig. 8, the level of phospho-S6rp was raised 24 h following SE as compared to control (p < 0.001). Metformin-treated group significantly (p < 0.05) suppressed the SE-increased p-S6 levels as compared to the pilocarpine group. As expected, rapamycin, typical mTOR inhibitor, also significantly (p < 0.01) blocked the S6rp phosphorylation induced by SE, whereas total S6 levels remained unaltered in all the groups (Fig. 8).

Effect of metformin or rapamycin treatment on hippocampal phosphorylated S6 expression following SE. (A) Immunoblotting analysis of phospho-S6 expression in rat hippocampus in different groups following SE. (B) Quantitative representation of the effect of metformin or rapamycin on expressions of phospho-S6. Data were expressed as mean ± S.D. of five independent animals. One-way ANOVA followed by Tukey test. **p ≤ 0.01 as compared to control group. $p ≤ 0.05 and $$p ≤ 0.01 as compared to SE group. c Immunohistochemical staining of phospho S6-immunopositive cells in the dentate gyrus (DG) and CA3 regions of hippocampus 24 h post SE at 20×
The above results were also confirmed using immunohistochemistry for p-S6 at the level of hippocampus. In control hippocampi, immunostaining with pS6 was predominantly observed in neurons in the CA3 region. There was no immunoreactivity in the granule neurons and pyramidal neurons in CA2 and CA1 regions. However, 24 h following SE, granular and CA3 pyramidal neurons revealed significantly increased immunoreactivity for p-S6 as compared to control hippocampi (Fig. 8). Treatment with rapamycin totally abolished the phosphorylation of S6rp, thereby reducing p-S6 expression in all the regions of hippocampus as expected with a mTOR inhibitor. Further, metformin treatment also reduced the p-S6 expression in the both the DG and CA3 regions as comparable to the control group (Fig. 8).
Discussion
In Li-Pi model of SE, we demonstrated that metformin by alleviating inflammatory responses and neurodegeneration showed a protective effect on SE-induced early epileptogenic events. The Li-Pi model was selected for the study as the central and peripheral inflammation are well studied and characterized (Tannich et al. 2020). Recent in-vitro and in-vivo studies showed metformin to have anti-epileptic and anti-convulsant potential attributed to its anti-glycotic, anti-apoptotic and anti-oxidant mechanisms (Zhang et al. 2014, Yang et al. 2017). Though metformin has proven immune-modulatory features in various diseases (Matsushita and Kawaguchi 2018), its role in seizure-induced inflammation remains unexplored. Our study analysis too showed that the metformin pretreatment attenuated the up regulation of proinflammatory cytokines, chemokines and enzymes, BBB breach, astroglial activation and thereby ameliorated the neurodegeneration. We further indicated the possible mTOR involvement in the protective responses produced by metformin in the pilocarpine-induced seizures.
In the current study, the behavioural seizure analysis suggested that metformin lacks a direct anti-convulsant effect against SE induced by Li-Pi model, which was also confirmed with the EEG recordings. However, Mehrabi et al. demonstrated the anti-convulsant activity of metformin against spontaneous recurrent seizures (SRS) in post SE model of TLE (Mehrabi et al. 2017). Metformin by reducing the insult induced secondary damage, as observed in our study, could have inhibited the generation of chronic SRS (Mehrabi et al. 2017). Based on the behavioural (seizure scoring and seizure latency times) and H&E data, we found that metformin 100 mg/kg showed no effect on SE; therefore, metformin 200 mg/kg was chosen for further studies.
It is well known that gene and protein expression of proinflammatory pathways is rapidly elevated after seizures in animal models as well as patients (Plata et al. 2000). Accordingly, in the present study, mRNA levels of TNFα, IL-1β, IL-6, NFκβ and CCL-3 were dramatically elevated. These cytokines are primarily released from activated microglia and astrocytes and maintain excitatory inputs by enhancing glutamate, whereby mediating excitotoxicity contributing to epileptogenesis (Alyu and Dikmen 2016). Pretreatment with metformin reduced the up regulation of IL-1β, IL-6, NFκβ, CCL-3 and TNF-α genes indicating metformin could attenuate the cytokine-mediated neuronal hyper-excitability. Previous studies also reported the inhibitory effect of metformin over the rise in proinflammatory cytokines and chemokines in LPS treated macrophages via pathways involving AMPK or NF-κβ or STAT-3 mechanisms (Hyun et al. 2013; Kim and Choi 2012).
Overexpression of COX-2 during seizures, particularly SE, is implicated to play critical role in epileptogenic events including BBB breakdown, leukocyte infiltration, astroglial and microglial activation (Dudek 2012). In the present study, COX-2 was found to be upregulated post SE. COX-2 is also associated with seizure-induced neurodegeneration; moreover, COX-2 inhibitors reduced neuronal death and provided neuroprotection in number of studies (Lalitha et al. 2017; Dudek 2012). Meanwhile, metformin pretreatment attenuated the rise in SE-induced COX-2 mRNA levels, thereby preventing the COX-2 mediated epileptogenesis. To our knowledge, this is the first study which shows that metformin pretreatment can cause reduction in COX-2 mRNA levels.
Generally, iNOS is not expressed in nervous cells. However, hours after SE, enhanced iNOS levels overproduce NO mainly in astrocytes and microglial cells which participate in oxidative stress and inflammation-mediated neuronal death (Liu et al. 2010). Our study showed such increase in iNOS mRNA levels post SE, which were dramatically inhibited by metformin pretreatment to levels lower than controls, indicating a strong iNOS inhibitory potential of metformin, concurrent with other studies (Kim and Choi 2012; Nakatake et al. 2018).
IL-10 is a key anti-inflammatory cytokine that can combat the pro-inflammatory responses. In the present study, surge in IL-10 mRNA levels was observed immediately 4 h after SE reflecting a possible negative feedback regulation over the ongoing inflammation. Similarly, rapid increase in serum IL-10 levels was found in neonatal seizure patients (Youn et al. 2013; Kim et al. 2017). Spike in IL-10 transcripts was not observed in metformin-treated rats as the degree of inflammation was less in them as compared to SE group. Together these findings highlight the anti-inflammatory potential of metformin in seizures which is in agreement with the existing literature in various other diseases (Saisho 2015; Kelly et al. 2015). FoxP3 is a transcription factor needed for T cell differentiation and is essential for maintaining immune tolerance. In the present study, FoxP3 mRNA levels rapidly increased after SE which is in agreement with a recent study showing neuroprotective role of elevated glial FoxP3 in the pathogenesis of epilepsy (Wang et al. 2017). There was, however, no influence of metformin pretreatment on FoxP3 expression.
Rapamycin pretreatment for 4 days prior to SE induction significantly inhibited the SE-induced upregulation of inflammatory cytokine genes suggesting anti-inflammatory action of rapamycin. For the first time in our study, we report that rapamycin treatment increased IL-10 levels significantly 24 h following SE, to curtail the seizure-induced inflammation. This is supported by the studies where rapamycin attenuated the inflammatory cytokine, chemokine expression in microglia and macrophages and increased the anti-inflammatory activities of regulatory T cells in focal ischemic model of rats (Xie et al. 2014; Zhong et al. 2012).
Intense seizure activity such as SE results in BBB dysfunction through mechanisms such as inflammation, metabolic failure, angiogenesis, etc., leading to brain albumin extravasation and evidence suggests immediate restoration of BBB integrity as an effective therapeutic strategy to abate epileptogenesis (Bankstahl et al. 2018). We observed SE-induced BBB disruption which was restored by metformin treatment in accordance with in-vitro and in-vivo studies in ischemia (Takata et al. 2013; Liu et al. 2014). Rapamycin pretreatment also prevented the SE enhanced permeability of BBB in accordance with a previous study (van Vliet et al. 2012). This protective effect exerted by metformin and rapamycin can be due to a direct protective effect on endothelial cells of BBB or indirectly by decreasing inflammation, as inflammation is a major contributor to BBB damage (Vezzani et al. 2015).
Present study showed persistent astroglial activation 24 h post SE in hippocampus particularly in hilar and CA1 region. Previous studies suggest causative role of astrocytes in epileptogenesis following brain insults (Wetherington et al. 2008; Robel 2016) and thereby contributing to neuronal hyperexcitability and seizures (Robel 2016). Metformin clearly decreased the reactive astrocytes as suggested by decreased number of GFAP positive cells and hypertrophy. Similarly, metformin attenuated glial activation and improved memory in diabetic mice (Oliveira et al. 2016). Further, rapamycin also attenuated the astroglial activation indicating the possible involvement of mTOR pathway in astrocytic activation. In line with the current findings, rapamycin inhibited the enhanced mTOR signaling in reactive astrocytes and improved the motor deficit in a mice model of spinal cord injury (Codeluppi et al. 2009).
In the current study, significantly high numbers of FJB positive neurons were observed in CA3 and CA1 areas of hippocampus as a result of SE, indicating SE-induced neurodegeneration. Research suggests that patients and animal models of MTLE exhibit neurodegeneration in different brain areas particularly hilar interneurons and CA3 and CA1 pyramidal neurons (Peixoto-Santos et al. 2015). Pretreatment with metformin for 7 days demonstrated neuroprotective activity by preventing the hippocampal neurons from dying as indicated by low FJB positivity in CA3 region. These results are in consensus with studies demonstrating the anti-apoptotic effect of metformin in in-vitro primary cortical neurons, (El-Mir et al. 2008) as well as in in-vivo studies in stroke, ICH and epilepsy (Li et al. 2010; Qi et al. 2017; Chen et al. 2018; Ge et al. 2017). As it is proven that inflammation leads to neurodegeneration, metformin reduced the degree of seizure-induced neuronal damage by abating neuroinflammation, It was also demonstrated that rapamycin treatment also moderately protected the neurodegeneration.
In the present study, increased hippocampal S6rp phosphorylation was observed 24 h post SE which is in line with the previous findings where abnormal activation of mTOR signalling was observed after seizures (Citraro et al. 2016). Meanwhile, metformin attenuated the SE upregulated mTOR pathway comparable to rapamycin (Shi et al. 2012; Kickstein et al. 2010). Previous research shows the involvement of mTOR in the regulation of CNS inflammatory responses and neuronal death (Wang et al. 2018; Saliba et al. 2017; Ye et al. 2020) and accordingly, mTOR inhibition has been a promising intervention for numerous neuroinflammatory and neurodegenerative disorders (Srivastava et al. 2015, Crino, 2016; Cho et al. 2018). Besides, a direct link between mTOR pathway and microglial pro-inflammatory activation has also been well established where mTOR pathway disruption promoted microglial, M1 (pro) to M2 (anti-inflammatory) type transition and ameliorated brain inflammation (Li et al. 2016). Therefore, metformin might have mediated its beneficial effect against seizure-induced inflammation and neuronal death, partly via mTOR modulation. On the other hand, in the present study, metformin also inhibited seizure-induced upregulation of NF-κβ gene expression which might also have rendered the anti-inflammatory and anti-apoptotic effects. The mTOR inhibition can also indirectly control proinflammatory immune responses by modulating NF-κB activity (Dello Russo et al. 2009; Sahan-Firat et al. 2018) (Fig. 9).

Metformin as an anti-inflammatory target to curtail epileptic pathogenesis. Systemic administration of Li-Pi, muscarinic agonist, causes direct neuronal hyperexcitability inducing seizures. Prolonged excitation of neurons results in damage of cells. Damaged cells then release DAMPs such as HMGB1and ATP that spread into the parenchyma and are promptly detected by microglia. Microglia polarize toward DAMP signals activated by signaling pathways such as IL-1R/TLR4- NFκβ axis with morphological and functional changes and overproduce proinflammatory cytokines, chemokines, prostaglandins, ROS and complement proteins. Also neurons express TLRs, thereby activating inflammatory cascade, increased iNOS and COX-2 and also enhanced glutamate release causing excitotoxicity. Extravasated albumin activates TGFβ signaling in astrocytes, thereby inducing production of proinflammatory mediators (TNFα, IL-6, NO, ATP, TGFβ, GDNF, BDNF) and altered glutamate homeostasis leading to increased neuronal hyperexcitability. Inflamed neurons, reactive astrocytes and microglial cells along with infiltrated immune cells produce a flared immune and inflammatory mileu which aggravate seizures to further ensue. Conversely, seizures also promote persistent and unresolved inflammation. Reactive astrocytes then group the lesion site to form the glial scar, suppressing synaptic plasticity, remyelination and axonal repair contributing to scar and lesion formation. All these maladaptations contribute to neuronal death progressing to chronic epilepsy Simultaneously, on the other hand, peripheral inflammation ensues as Li-Pi simultaneously activate the peripheral immune system and stimulates cytokine production by leukocytes. Both central and peripheral rise in proinflammatory mediators induce upregulation of integrins and cell adhesion molecules, increase BBB permeability-enables serum albumin, leukocyte and monocyte extravasation. Infiltrated immune cells further contribute to central inflammation
Conclusion
Altogether, these findings demonstrate that metformin inhibition exhibited neuroprotective and disease-modifying actions by restraining SE-induced neuroinflammation and neurodegeneration, both of which have been recognized as epileptogenic events or precursors for chronic epilepsy development. The study also showed the possible implication of mTOR pathway in the inflammatory mechanisms of epilepsy and, therefore, targeting mTOR pathway might be a novel therapeutic target for epilepsy particularly those induced by inflammatory triggers.
Abbreviations
- mTOR:
-
Mammalian target of rapamycin
- SE:
-
Status epilepticus
- BBB:
-
Blood–brain barrier
- FJB:
-
Fluorojade B
- Li-Pi:
-
Lithium pilocarpine
References
Alyu F, Dikmen M (2016) Inflammatory aspects of epileptogenesis: contribution of molecular inflammatory mechanisms. Acta Neuropsychiatr 29:1–16
Arbelaez-Quintero I, Palacios M (2017) To use or not to use metformin in cerebral ischemia: a review of the application of metformin in stroke rodents. Stroke Res Treat 2017:9756429
Bankstahl M, Breuer H, Leiter I, Märkel M, Bascuñana P, Michalski D et al (2018) Blood–brain barrier leakage during early epileptogenesis is associated with rapid remodeling of the neurovascular unit. ENEURO 5:0123–0218
Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA (2016) Metformin as a tool to target aging. Cell Metab 23:1060–1065
Chen J, Zheng G, Guo H, Shi ZN, Jiang J, Wang XY et al (2018) The effect of metformin treatment on endoplasmic reticulum stress induced by status epilepticus (SE) via the PERK-eIF2α-CHOP pathway. Bosn J Basic Med Sci 18:49–54
Chi X, Huang C, Li R, Wang W, Wu M, Li J, Zhou D (2017) Inhibition of mTOR pathway by rapamycin decreases P-glycoprotein expression and spontaneous seizures in pharmacoresistant epilepsy. J Mol Neurosci 61:553–562
Cho C, Michailidis V, Martin LJ (2018) Revealing brain mechanisms of mTOR-mediated translational regulation: implications for chronic pain. Neurobiol Pain 4:27–34
Citraro R, Leo A, Constanti A, Russo E, De Sarro G (2016) mTOR pathway inhibition as a new therapeutic strategy in epilepsy and epileptogenesis. Pharmacol Res 107:333–343
Clossen BL, Reddy DS (2017) Novel therapeutic approaches for disease-modification of epileptogenesis for curing epilepsy. Biochim Biophys Acta Mol Basis Dis 1863:1519–1538
Codeluppi S, Svensson CI, Hefferan MP, Valencia F, Silldorff MD, Marsala M et al (2009) The Rheb-mTOR pathway is upregulated in reactive astrocytes of the injured spinal cord. J Neurosci 29:1093–1104
Crino P (2016) The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol 12:379–392
Dello Russo C, Lisi L, Tringali G, Navarra P (2009) Involvement of mTOR kinase in cytokine-dependent microglial activation and cell proliferation. Biochem Pharmacol 78:1242–1251
Dey A, Kang X, Qiu J, Du Y, Jiang J (2016) Anti-Inflammatory small molecules to treat seizures and epilepsy: from bench to bedside. Trends Pharmacol Sci 37:463–484
Drion CM, Borm LE, Kooijman L, Aronica E, Wadman WJ, Hartog AF et al (2016) Effects of rapamycin and curcumin treatment on the development of epilepsy after electrically induced status epilepticus in rats. Epilepsia 57:688–697
Dudek FE (2012) Mechanisms of seizure-induced inflammation of the brain: many possible roles for neuronal COX-2. Epilepsy Curr 12:115–117
El-Mir MY, Detaille D, R-Villanueva G, Delgado-Esteban M, Guigas B, Attia S et al (2008) Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J Mol Neurosci 34:77–87
Fujikawa DG, Pais ES, Aviles ER, Hsieh KC, Bashir MT (2016) Methamphetamine-induced neuronal necrosis: the role of electrographic seizure discharges. Neurotoxicology 52:84–88
Ge XH, Zhu GJ, Geng DQ, Zhang HZ, He JM, Guo AZ et al (2017) Metformin protects the brain against ischemia/reperfusion injury through PI3K/Akt1/JNK3 signaling pathways in rats. Physiol Behav 170:115–123
Guo D, Zou J, Wong M (2017) Rapamycin attenuates acute seizure-induced astrocyte injury in mice in vivo. Sci Rep 7:2867
Hyun B, Shin S, Lee A, Lee S, Song Y, Ha NJ et al (2013) Metformin down-regulates TNF-α secretion via suppression of scavenger receptors in macrophages. Immune Netw 13:123–132
Jing Y, Wu F, Li D, Yang L, Li Q, Li R (2018) Metformin improves obesity-associated inflammation by altering macrophages polarization. Mol Cell Endocrinol 461:256–264
Kalender A, Selvaraj A, Kim SY, Gulati P, Brûlé S, Viollet B et al (2010) Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab 11:390–401
Kelly B, Tannahill GM, Murphy MP, O’Neill LA (2015) Metformin inhibits the production of reactive oxygen species from NADH: Ubiquinone Oxidoreductase to limit induction of Interleukin-1β (IL-1β) and boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-activated macrophages. J Biol Chem 290:20348–20359
Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J et al (2010) Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A 107:21830–21835
Kim K, Kwak BO, Kwon A, Ha J, Kim SJ, Bae SW (2017) Analysis of plasma multiplex cytokines and increased level of IL-10 and IL-1Ra cytokines in febrile seizures. J Neuroinflamm 14:200
Kim SA, Choi HC (2012) Metformin inhibits inflammatory response via AMPK-PTEN pathway in vascular smooth muscle cells. Biochem Biophys Res Commun 425:866–872
Kumar A, Lalitha S, Mishra J (2013) Possible nitric oxide mechanism in the protective effect of hesperidin against pentylenetetrazole (PTZ)-induced kindling and associated cognitive dysfunction in mice. Epilepsy Behav 29:103–111
Kumar A, Lalitha S, Mishra J (2014) Hesperidin potentiates the neuroprotective effects of diazepam and gabapentin against pentylenetetrazole-induced convulsions in mice: Possible behavioral, biochemical and mitochondrial alterations. Indian journal of pharmacology 46:309–315
Kumari P, Singh N, Saha L (2018) Potentiation of pentylenetetrazole-induced neuronal damage by dimethyl sulfoxide in chemical kindling model in rats. Indian J Pharmacol 50:84–87
Lalitha S, Minz RW, Medhi B (2017) Understanding the controversial drug targets in epilepsy and pharmacoresistant epilepsy. Rev Neurosci 29:333–345
Lashen H (2010) Role of metformin in the management of polycystic ovary syndrome. Ther Adv Endocrinol Metab 1:117–128
Li D, Wang C, Yao Y, Chen L, Liu G, Zhang R et al (2016) mTORC1 pathway disruption ameliorates brain inflammation following stroke via a shift in microglia phenotype from M1 type to M2 type. Faseb 30:3388–3399
Liu J, Wang A, Li L, Huang Y, Xue P, Hao A (2010) Oxidative stress mediates hippocampal neuron death in rats after lithium-pilocarpine-induced status epilepticus. Seizure 19:165–172
Liu Y, Tang G, Li Y, Wang Y, Chen X, Gu X et al (2014) Metformin attenuates blood-brain barrier disruption in mice following middle cerebral artery occlusion. J Neuroinflamm 11:177
Marchi N, Granata T, Freri E, Ciusani E, Ragona F, Puvenna V et al (2011) Efficacy of anti-inflammatory therapy in a model of acute seizures and in a population of pediatric drug resistant epileptics. PLoS ONE 6:e18200
Matsushita M, Kawaguchi M (2018) Immunomodulatory Effects of Drugs for Effective Cancer Immunotherapy. J Oncol 8653489.
Mehan S, Monga V, Rani M, Dudi R, Ghimire K (2018) Neuroprotective effect of solanesol against 3-nitropropionic acid-induced Huntington’s disease-like behavioral, biochemical, and cellular alterations: Restoration of coenzyme-Q10-mediated mitochondrial dysfunction. Indian J Pharmacol 50:309–319
Mehrabi S, Sanadgol N, Barati M, Shahbazi A, Vahabzadeh G, Barzroudi M (2018) Evaluation of metformin effects in the chronic phase of spontaneous seizures in pilocarpine model of temporal lobe epilepsy. Metab Brain Dis 33:107–114
Nakatake R, Iida H, Ishizaki M, Matsui K, Nakamura Y, Kaibori M et al (2018) Metformin inhibits expression of the proinflammatory biomarker inducible nitric oxide synthase in hepatocytes. Funct food health dis 8:175–192
Negrotto L, Farez MF, Correale J (2016) Immunologic Effects of Metformin and Pioglitazone Treatment on Metabolic Syndrome and Multiple Sclerosis. JAMA Neurol 73:520–528
Oliveira WH, Nunes AK, França ME, Santos LA, Lós DB, Rocha SW et al (2016) Effects of metformin on inflammation and short-term memory in streptozotocin-induced diabetic mice. Brain Res 1644:149–160
Peixoto-Santos JE, Velasco TR, Galvis-Alonso O, Araujo D, Kandratavicius L, Assirati JA et al (2015) Temporal lobe epilepsy patients with severe hippocampal neuron loss but normal hippocampal volume: extracellular matrix molecules are important for the maintenance of hippocampal volume. Epilepsia 56:1562–1570
Plata-Salamán CR, Ilyin SE, Turrin NP, Gayle D, Flynn MC, Romanovitch AE et al (2000) Kindling modulates the IL-1beta system, TNF-alpha, TGF-beta1, and neuropeptide mRNAs in specific brain regions. Brain Res Mol Brain Res 75:248–258
Qi B, Hu L, Zhu L, Shang L, Wang X, Liu N et al (2017) Metformin attenuates neurological deficit after intracerebral hemorrhage by inhibiting apoptosis, oxidative stress and neuroinflammation in rats. Neurochem Res 42:2912–2920
Rani L, Minz RW, Sharma A, Anand S, Gupta D, Panda NK et al (2015) Predominance of PR3 specific immune response and skewed TH17 vs. T-regulatory milieu in active granulomatosis with polyangiitis. Cytokine 71:261–267
Rensing N, Han L, Wong M (2015) Intermittent dosing of rapamycin maintains antiepileptogenic effects in a mouse model of tuberous sclerosis complex. Epilepsia 56:1088–1097
Robel S (2016) Astroglial scarring and seizures: a cell biological perspective on epilepsy. Neuroscientist 2016:1073858416645498
Rotermund C, Machetanz G, Fitzgerald JC (2018) The therapeutic potential of metformin in neurodegenerative diseases. Front Endocrinol (Lausanne) 9:400. https://doi.org/10.3389/fendo.2018.00400
Sahan-Firat S, Temiz-Resitoglu M, Guden DS, Kucukkavruk SP, Tunctan B, Sari AN et al (2018) Protection by mTOR inhibition on zymosan-induced systemic inflammatory response and oxidative/nitrosative stress: contribution of mTOR/MEK1/ERK1/2/IKKβ/IκB-α/NF-κB signalling pathway. Inflammation 41:276–298
Saisho Y (2015) Metformin and Inflammation: Its Potential Beyond Glucose-lowering Effect. Endocr Metab Immune Disord Drug Targets 15:196–205
Salem NA, El-Shamarka M, Khadrawy Y, El-Shebiney S (2018) New prospects of mesenchymal stem cells for ameliorating temporal lobe epilepsy. Inflammopharmacology 26:963–972
Saliba SW, Vieira EL, Santos RP, Candelario-Jalil E, Fiebich BL, Vieira LB et al (2017) Neuroprotective effects of intrastriatal injection of rapamycin in a mouse model of excitotoxicity induced by quinolinic acid. J Neuroinflamm 14:25
Shi WY, Xiao D, Wang L, Dong LH, Yan ZX, Shen ZX (2012) Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis 3:e275
Srivastava IN, Shperdheja J, Baybis M, Ferguson T, Crino PB (2016) mTOR pathway inhibition prevents neuroinflammation and neuronal death in a mouse model of cerebral palsy. Neurobiol Dis 85:144–154
Singh N, Saha L, Kumari P, Singh J, Bhatia A, Banerjee D et al (2019) Effect of dimethyl fumarate on neuroinflammation and apoptosis in pentylenetetrazol kindling model in rats. Brain Res Bull 144:233–245
Takata F, Dohgu S, Matsumoto J, Machida T, Kaneshima S, Matsuo M et al (2013) Metformin induces up-regulation of blood-brain barrier functions by activating AMP-activated protein kinase in rat brain microvascular endothelial cells. Biochem Biophys Res Commun 433:586–590
Tannich F, Tlili A, Pintard C, Chniguir A, Eto B, Dang PM et al (2020) Activation of the phagocyte NADPH oxidase/NOX2 and myeloperoxidase in the mouse brain during pilocarpine-induced temporal lobe epilepsy and inhibition by ketamine. Inflammopharmacology 28:487–497
Ursini F, Russo E, Pellino G, D’Angelo S, Chiaravalloti A, De Sarro G et al (2018) Metformin and autoimmunity: a “new deal” of an old drug. Front Immunol 9:1236
van Vliet EA, da Costa AS, Redeker S, van Schaik R, Aronica E, Gorter JA (2007) Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 130:521–534
van Vliet EA, Forte G, Holtman L, Den Burger JC, Sinjewel A, De Vries HE et al (2012) Inhibition of mammalian target of rapamycin reduces epileptogenesis and blood–brain barrier leakage but not microglia activation. Epilepsia 53:1254–1263
Vezzani A, Friedman A, Dingledine RJ (2013) The role of inflammation in epileptogenesis. Neuropharmacology 69:16–24
Vezzani A, Dingledine R, Rossetti AO (2015) Immunity and inflammation in status epilepticus and its sequelae: possibilities for therapeutic application. Expert Rev Neurother 15:1081–1092
Vezzani A, Lang B, Aronica E (2016) Immunity and inflammation in epilepsy. Cold Spring Harb Perspect Med 6:a022699
Wang FX, Xiong XY, Zhong Q, Meng ZY, Yang H, Yang QW (2017) Foxp3 exhibits antiepileptic effects in ictogenesis involved in TLR4 signaling. FASEB J 31:2948–2962
Wang SJ, Bo QY, Zhao XH, Yang X, Chi ZF, Liu XW (2013) Resveratrol pre-treatment reduces early inflammatory responses induced by status epilepticus via mTOR signaling. Brain Res 1492:122–129
Wang C, Wang Q, Lou Y, Xu J, Feng Z, Chen Y et al (2018) Salidroside attenuates neuroinflammation and improves functional recovery after spinal cord injury through microglia polarization regulation. J Cell Mol Med 22:1148–1166
Wetherington J, Serrano G, Dingledine R (2008) Astrocytes in the epileptic brain. Neuron 58:168–178
Xie L, Sun F, Wang J, Mao X, Xie L, Yang SH et al (2014) mTOR signaling inhibition modulates macrophage/microglia-mediated neuroinflammation and secondary injury via regulatory T cells after focal ischemia. J Immunol 192:6009–6019
Ye X, Zhu M, Che X, Wang H, Liang XJ, Wu C (2020) Lipopolysaccharide induces neuroinflammation in microglia by activating the mTOR pathway and downregulating Vps34 to inhibit autophagosome formation. J Neuroinflamm 17:18
Yang MT, Lin YC, Ho WH, Liu CL, Lee WT (2017) Everolimus is better than rapamycin in attenuating neuroinflammation in kainic acid-induced seizures. J Neuroinflamm 14:15
Youn Y, Sung IK, Lee IG (2013) The role of cytokines in seizures: interleukin (IL)-1β, IL-1Ra, IL-8, and IL-10. Korean J Pediat 56:271–274
Zeng LH, Rensing NR, Wong M (2009) The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci 29:6964–6972
Zhao RR, Xu XC, Xu F, Zhang WL, Zhang WL, Liu LM et al (2014) Metformin protects against seizures, learning and memory impairments and oxidative damage induced by pentylenetetrazole-induced kindling in mice. Biochem Biophys Res Commun 448:414–417
Zhong LM, Zong Y, Sun L, Guo JZ, Zhang W, He YD (2012) Resveratrol inhibits inflammatory responses via the mammalian target of rapamycin signaling pathway in cultured LPS-stimulated microglial cells. PLoS ONE 7:e32195
Zhu X, Liu J, Chen O, Xue J, Huang S, Zhu W et al (2019) Neuroprotective and anti-inflammatory effects of isoliquiritigenin in kainic acid-induced epileptic rats via the TLR4/MYD88 signaling pathway. Inflammopharmacology 27:1143–1153. https://doi.org/10.1007/s10787-019-00592-7
Zi F, Zi H, Li Y, He J, Shi Q, Cai Z (2018) Metformin and cancer: An existing drug for cancer prevention and therapy. Oncol Lett 15:683–690
Acknowledgements
This work was supported by the Department of Biotechnology (DBT), India. Research fellowship was provided by the Council of Scientific & Industrial Research (CSIR), India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interest
No competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bojja, S.L., Medhi, B., Anand, S. et al. Metformin ameliorates the status epilepticus- induced hippocampal pathology through possible mTOR modulation. Inflammopharmacol 29, 137–151 (2021). https://doi.org/10.1007/s10787-020-00782-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-020-00782-8