
Abstract
Rheumatoid arthritis (RA) is an incurable, systemic autoimmune disease that decreases quality of life and can lead to severe disability. While there are many medications available to treat RA, the first-line of therapy is low-dose methotrexate (MTX), a small molecule disease-modifying anti-rheumatic drug (DMARD). MTX is the recommended therapy due to its affordability and efficacy in reducing symptoms in most RA patients. Unfortunately, there is great person-to-person variability in the physiological response to MTX, with up to 50% of patients showing little response to the medication. Thus, many RA patients initially placed on MTX do not experience an adequate reduction of symptoms, and could have benefited more in both the short and long terms if initially prescribed a different drug that was more effective for them. To combat this problem and better guide treatment decisions, many research groups have attempted to develop predictive tools for MTX response. Currently, there is no reliable, clinical-grade method to predict an individual’s response to MTX treatment. In this review, we describe progress made in the area of MTX non-response/resistance in RA patients. We specifically focus on application of the following elements as predictive markers: proteins related to MTX transport and function, intracellular MTX concentration, immune cell frequencies, cytokines, and clinical factors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
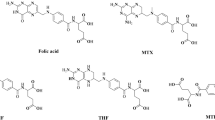
Rheumatoid arthritis (RA) is a chronic disease that results in joint inflammation, pain, and swelling, and can often lead to irreversible deformity (McInnes and Schett 2011). Most RA patients become unable to work full-time within 10 years of symptom onset (Woolf and Pfleger 2003). Both the American College of Rheumatology (ACR) (Singh et al. 2016) and the European League Against Rheumatism (EULAR) (Smolen et al. 2017) currently recommend low-dose methotrexate (MTX; Fig. 1) monotherapy as the initial treatment for patients newly diagnosed with RA. The low-dose distinction is used to draw a contrast with the high doses of MTX used in cancer treatment, which are 100–1000 times larger (Cronstein 2005; Smolen et al. 2017).The ACR’s and the EULAR’s reasons for choosing low-dose MTX are that MTX is inexpensive, and there is a lack of evidence that more expensive treatments have better long-term efficacy (Singh et al. 2016; Smolen et al. 2017).

Structures of MTX and folic acid. MTX is a folate derivative
However, there is a significant variation in MTX efficacy between individual RA patients. Prevalence estimates of non-response/resistance to MTX therapy in RA patients range widely from 30 to 50% (Braun et al. 2008; Klareskog et al. 2004; Lima et al. 2015; Mori et al. 2010; Weinblatt et al. 1994). The mechanisms for this variable response are complex and unclear. Despite the obvious need, there is no validated clinical method available to predict MTX response in RA patients. In this review, non-response and resistance will be used interchangeably.
Methotrexate mechanism
MTX (Fig. 1) is classified as a small molecule disease-modifying anti-rheumatic drug (DMARD). In broad terms, MTX is a folate derivative which acts as a competitive inhibitor for multiple folate-dependent enzymes, leading ultimately to the inhibition of DNA synthesis and elevation of extracellular adenosine. The result is a decrease in both cell proliferation and secretion of pro-inflammatory cytokines (Cronstein 2005; Inoue and Yuasa 2014; Kremer 2004; Tian and Cronstein 2007). In the immune system, MTX most dramatically affects T cells (Gerards et al. 2003; Herman et al. 2005), but is also reported to have anti-proliferative or anti-inflammatory effects on B cells (Witte 2015), monocytes (Herman et al. 2005; Wijngaarden et al. 2005; Witte 2015), and dendritic cells (DCs) (Yu et al. 2013).
MTX is believed to prevent DNA synthesis and, therefore, normal cell division via inhibition of enzymes involved in de novo nucleotide synthesis (Fig. 2). Specifically, MTX inhibits the enzymes such as thymidylate synthetase (TYMS), aminoimidazole carboxamide ribonucleotide transformylase (ATIC), and dihydrofolate reductase (DHFR). TYMS is involved in thymidine synthesis. ATIC is involved in de novo purine synthesis. DHFR reduces dihydrofolate (DHF) to tetrahydrofolate (THF), whose derivatives are cofactors for TYMS and ATIC. Thus, MTX decreases the activity of TYMS and ATIC by directly inhibiting these enzymes and by decreasing access to THF cofactors (Inoue and Yuasa 2014; Kremer 2004; Tian and Cronstein 2007). Inhibition of TYMS and ATIC decreases the amount of nucleotides available for DNA synthesis, particularly thymine (Budzik et al. 2000; Kremer 2004) and guanine (Budzik et al. 2000; Hryniuk et al. 1975), and thus reduces DNA synthesis (Budzik et al. 2000; Hryniuk et al. 1975; Kremer 2004; Tian and Cronstein 2007). This molecular mechanism is particularly important for preventing T-cell proliferation because activated T cells almost exclusively rely on de novo nucleotide synthesis as opposed to the nucleotide salvage pathway, and DNA replication is required prior to cell division (Budzik et al. 2000; Herman et al. 2005; Tian and Cronstein 2007; Witte 2015).

Mechanism of MTX. Boxes indicate proteins. Red, italicized type indicates those enzymes whose activity is reduced by MTX, or compounds whose levels are reduced by MTX. Transport of MTX into the cell occurs by RFC1, while transport out of the cell occurs by ABCB1, ABCC1, or ABCG2. MTX is polyglutamated by FPGS to become MTX-PG, and this process is reversed by GGH. MTX-PG inhibits TYMS, DHFR, and ATIC. Inhibition of DHFR decreases THF levels, leading to a decrease in the THF derivatives which are cofactors for TYMS and ATIC. This lack of access to cofactors contributes to the MTX-PG-induced decrease in TYMS and ATIC activity. Decreases in TYMS and ATIC activity result in reduced synthesis of thymidine and purines, respectively. As a result, there are decreased amounts of thymine and purine nucleotides available for DNA synthesis, thus reducing DNA synthesis. The decrease in ATIC activity also leads to a build-up of AICAR. The increase in AICAR inhibits ADA and AMPDA, resulting in a build-up of adenosine and AMP. Adenosine, AMP, ADP, and ATP can be interconverted. The excess adenosine, AMP, ADP, and ATP are secreted into the extracellular space. Outside the cell, ATP, ADP, and AMP are hydrolyzed into adenosine by CD39 and CD73. Extracellular adenosine binds to A2A receptors, resulting in anti-inflammatory signaling
MTX promotes the increase of extracellular adenosine (Fig. 2) by decreasing the activity of ATIC, resulting in an excess of ATIC’s substrate aminoimidazole carboxamide ribonucleotide (AICAR). AICAR in turn inhibits adenosine deaminase (ADA) and AMP deaminase (AMPDA), resulting in a build-up of their respective substrates, adenosine and AMP. Adenosine, AMP, ADP, and ATP can interconvert into each other by phosphorylation and hydrolysis (Cronstein 2005; Inoue and Yuasa 2014; Kremer 2004; Tian and Cronstein 2007). Excess adenosine, AMP, ADP, and ATP are released into the extracellular space. The mechanism of this release is unclear, but the proposed mechanisms include vesicle exocytosis and facilitated diffusion (Antonioli et al. 2013). Extracellular AMP, ADP, and ATP are converted into adenosine by cell surface enzymes CD39 and CD73. Then, adenosine binds to adenosine receptors (Antonioli et al. 2013; Kremer 2004; Tian and Cronstein 2007).
Adenosine receptors are expressed on immune cells including neutrophils, macrophages, DCs, T cells, and B cells. These receptors are G protein coupled receptors. The adenosine receptor subtypes A2A and A2B activate Gs proteins, which activate the adenylate cyclase–cAMP–protein kinase A (PKA) pathway. In contrast, adenosine receptor subtypes A1 and A3 activate Gi proteins to inhibit the adenylate cyclase–cAMP–PKA pathway (Hasko et al. 2008). The receptor of greatest relevance to MTX-induced effects is believed to be the A2A receptor (Tian and Cronstein 2007).
Through the A2A receptors, extracellular adenosine induces anti-inflammatory effects in several types of immune cells. In neutrophils, A2A receptor signaling decreases expression of pro-inflammatory cytokines and prevents apoptosis. In macrophages and mature DCs, A2A receptor signaling decreases the expression of pro-inflammatory cytokines and increases expression of anti-inflammatory cytokines. In CD4 + T cells, A2A receptor signaling reduces both proliferation and expression of pro-inflammatory cytokines (Hasko et al. 2008). In activated B cells, A2A receptor signaling decreases antibody production, but this is usually balanced by A1 and A3 receptor signaling which promotes antibody production (Sakowicz-Burkiewicz et al. 2012).
Alterations in gene sequences
Variations in gene sequences are attractive as potential predictive markers for MTX response because DNA sequences are static over the course of a lifetime. As a result, it does not matter if the patient samples were acquired before or after beginning MTX treatment. Much work has been done evaluating correlations between MTX efficacy and variations in the DNA sequences of genes involved in MTX pharmacokinetics and pharmacodynamics. Progress in the pharmacogenetics of MTX response in RA has been recently reviewed elsewhere (Zhu et al. 2014), and will not be extensively discussed here. Several studies report associations between MTX response and single gene polymorphisms. However, most of these findings have either been contradicted by other studies, or have not yet been independently confirmed (Zhu et al. 2014).
For example, ATP-binding cassette B1 (ABCB1) codes for a protein that removes MTX from cells, and one of its polymorphisms is rs1045642, 3435C > T. The TT genotype was reported to be associated with increased MTX efficacy in a Polish (Drozdzik et al. 2006) and a Japanese (Kato et al. 2012) cohort of RA patients. However, this same genotype was associated with decreased MTX efficacy in another Japanese cohort (Takatori et al. 2006), and it was the CC genotype that was associated with increased efficacy in an Indian cohort (Sharma et al. 2008). No association between this SNP and MTX efficacy was found in studies with other populations of RA patients (Bohanec Grabar et al. 2008; Lima et al. 2015; Muralidharan et al. 2015; Prasad et al. 2014; Stamp et al. 2010). Unfortunately, in the field of the pharmacogenetics of MTX, such direct contradictions are common (Zhu et al. 2014).
Many reasons have been suggested for the lack of consistency. Many of these studies could be considered small for genetics studies (well under 1000 people), and thus the results may not be replicable (Zhu et al. 2014). It is possible that some of the positive findings may be false positives, with the positive result due to random chance (Dervieux 2009). MTX response is affected by many genes, and the effect of any one gene is likely to be small. Small effects are difficult to consistently detect, but more importantly, a gene with a small effect is unlikely to be a helpful biomarker in personalized medicine. Additionally, the genes may interact with each other, and linkage disequilibrium may play a role (Zhu et al. 2014). So, models utilizing multiple polymorphisms and haplotype blocks have also been developed (Fransen et al. 2012; Wessels et al. 2007; Zhu et al. 2014).
Alterations in protein expression and activity
Expression levels and activity of some MTX-associated proteins have been evaluated as potential predictors of the MTX response. These proteins described below are grouped by function.
Absorption: transport into the bloodstream
After oral delivery, MTX is absorbed into enterocytes of the small intestine by proton-coupled folate transporter (PCFT). PCFT transports physiological folates with Michaelis constant KM 1–3 uM, and it can also transport MTX with KM 3–6 uM because MTX is a folate derivative. It is conceivable that differences in the expression level or activity of PCFT could contribute to differences in response to MTX (Inoue and Yuasa 2014).
A study in RA patients indicated there is no association between PCFT expression and MTX efficacy (Tazoe et al. 2015). But PCFT has been shown to be inhibited in vitro by various compounds such as the DMARD sulfasalazine, nonsteroidal anti-inflammatory drugs (NSAIDs) diclofenac and indomethacin, tea flavonoid epigallocatechin-3-gallate, and wine flavonoid myricetin (Furumiya et al. 2014; Inoue and Yuasa 2014; Yuasa et al. 2009). The inhibitory effects of sulfasalazine on PCFT may explain why the combined therapeutic effects of MTX and sulfasalazine are less than additive (Inoue and Yuasa 2014; Yuasa et al. 2009).
Distribution: transport into cells
Figure 2 illustrates the pathway of MTX transport into the cell.
MTX is transported into cells by reduced folate carrier 1 (RFC1)/solute carrier 19A1 (SLC19A1), which physiologically transports folates (Inoue and Yuasa 2014; Ranganathan et al. 2008; Tian and Cronstein 2007). RFC1 is expressed in a variety of tissues, with the highest expression in the placenta, liver, and leukocytes (Inoue and Yuasa 2014). The high expression of RFC1 in the placenta may contribute to the efficacy of high-dose MTX to induce abortion (Kulier et al. 2011). Additionally, while it has not been shown that low-dose MTX leads to miscarriage, the RFC1 expression in the placenta could be used as a biochemical rationale to support the current recommendation that low-dose MTX not be used to treat RA during pregnancy (Visser et al. 2009).
One study in RA patients indicated that there is a positive correlation between RFC1 expression in peripheral blood mononuclear cells (PBMCs) and MTX efficacy. Subjects with higher expression of RFC1 were more likely to experience lower disease activity while on MTX, which is consistent with the concept that MTX enters target cells through RFC1 (Tazoe et al. 2015). However, another study found that low RFC1 expression in PBMCs is associated with MTX efficacy in RA patients with low activity from ABCB1, a transporter that removes MTX from cells (Wolf et al. 2005). These studies only measured RFC1 expression after MTX treatment and not before (Tazoe et al. 2015; Wolf et al. 2005). For RFC1 expression to be of benefit to patients and truly predictive, the markers must be observed before the patient is on MTX, as MTX treatment has been shown to increase RFC1 expression (Stamp et al. 2013). It is unknown if this difference in RFC1 expression was present prior to MTX administration. The observed differences in RFC1 expression in MTX response compared to MTX-resistant patients could be a result of MTX’s effects, and it is possible that the non-response could be due to the lack of MTX’s effect to increase RFC1.
Distribution: transport out of cells
Figure 2 illustrates the pathway of MTX transport out of the cell.
There are several ABC transporters involved in the efflux of MTX from cells. These include ABCB1/multidrug-resistant protein 1 (MDR1)/P-glycoprotein (P-gp), ABCC1/multidrug resistance-associated protein 1 (MRP1), and ABCG2/breast cancer resistance protein (BCRP) (Inoue and Yuasa 2014; Ranganathan et al. 2008). Theoretically, increased expression or function of these transporters should decrease MTX concentrations in target cells, resulting in lack of therapeutic response to MTX.
ABCB1/MDR1/P-gp
ABCB1 is present on the surface of peripheral blood leukocytes in RA patients and healthy controls (Micsik et al. 2015; Prasad et al. 2014; Wolf et al. 2005), but absent in RA synovial tissue (van der Heijden et al. 2009). Studies have been performed to assess the association between ABCB1 expression and function with MTX resistance.
Prior to treatment, there is no difference in lymphocyte ABCB1 expression between RA patients who will respond to MTX and those will not. After 4 months of MTX treatment, lymphocyte ABCB1 expression decreased in RA patients whose symptoms had also significantly improved, while ABCB1 expression levels were unchanged for those whose symptoms had not improved with MTX (Prasad et al. 2014). Although changes in ABCB1 expression may be indicative of MTX response, these findings are not practical for prediction as the patient has already been exposed to MTX.
It has been proposed that the functional activity of ABCB1 may be more meaningful than its expression. The functional activity is measured by fluorescence of cells incubated with calcein, a fluorescent substrate for ABCB1, with and without an inhibitor of ABCB1 (Micsik et al. 2015). There are conflicting results concerning associations between ABCB1 activity and response to MTX. One study found that lymphocyte ABCB1 activity between responders and non-responders is the same at baseline. After MTX treatment, lymphocyte ABCB1 activity decreased relative to baseline in RA patients whose symptoms had also significantly improved, while ABCB1 activity was unchanged for those whose symptoms resisted MTX (Prasad et al. 2014). The results of this study are in agreement with the accepted role of ABCB1 in MTX pharmacokinetics.
However, in a study utilizing blood samples after MTX treatment, it was found that there was no association between ABCB1 activity in lymphocytes and MTX resistance, and that high ABCB1 activity in granulocytes is associated with MTX efficacy (Micsik et al. 2015). More specifically, in RA patients whose PBMCs express high levels of RFC1, higher ABCB1 activity in PBMCs is associated with increased MTX efficacy (Wolf et al. 2005). The results from these two latter studies conflict with the accepted role of ABCB1 in MTX pharmacokinetics since high ABCB1 should remove MTX and thus decreases the ability of MTX to influence target cells. It is clear that the role of ABCB1 in the overall action of MTX needs additional clarification.
ABCC1/MRP1
ABCC1 is expressed on the surface of peripheral blood leukocytes in RA patients and healthy controls (Micsik et al. 2015; Wolf et al. 2005). Additionally, this protein is present in low levels on synovial T cells and synovial macrophages in a subset of RA patients, and completely absent from synovial tissue in other RA patients (van der Heijden et al. 2009). An ovarian cancer cell line (Hooijberg et al. 2014) study suggests that increased ABCC1 leads to MTX resistance. However, one study utilizing blood samples from RA patients after MTX treatment found no association between ABCC1 activity and MTX resistance. The functional activity was measured by fluorescence of cells incubated with calcein, a fluorescent substrate for ABCC1, with and without an inhibitor of ABCC1 (Micsik et al. 2015).
ABCG2/BCRP
ABCG2 is present in synovial macrophages, T cells, and endothelial cells in RA patients, but present in very few synovial cells in patients without RA (van der Heijden et al. 2009). A study has been performed to assess the association between ABCG2 expression and DMARD resistance. In this study, the RA patients were treated with either MTX or leflunomide, another small molecule that also inhibits nucleotide synthesis. High expression of ABCG2 in synovial tissue after DMARD treatment is associated with DMARD resistance. Correlation between ABCG2 expression before and after treatment suggested that differences in ABCG2 expression between responders and non-responders may have been present before treatment, and thus could be used to predict response to MTX (van der Heijden et al. 2009).
Adenosine pathway
Figure 2 illustrates how MTX increases extracellular adenosine.
One of the downstream effects of MTX is release of ATP and ADP into the extracellular environment. Extracellular ATP and ADP are hydrolyzed into AMP by cell surface-bound CD39, also known as ectonucleoside triphosphate diphosphohydrolase-1. AMP is then hydrolyzed into adenosine by cell surface-bound CD73, also known as ecto-5′-nucleotidase. As described earlier, this extracellular adenosine then causes anti-inflammatory signaling. Both CD39 and CD73 are highly expressed on Tregs, and the CD39 and CD73-mediated production of adenosine contributes to the anti-inflammatory effect of this cell type (Antonioli et al. 2013). A recent study found that low expression of CD39 in Tregs prior to MTX treatment is associated with lack of response to MTX (Peres et al. 2015). CD39 expression in Tregs could potentially be used as a powerful biomarker to predict response to MTX.
Polyglutamated methotrexate as a marker
The MTX concentration in blood or in cells could be used as a pharmacokinetic marker. However, plasma MTX is not used because plasma MTX concentrations are insignificant 24 h after a dose (Kremer et al. 1986). Since plasma MTX is transported into a cell, polyglutamated, and then able to act on its target enzymes, intracellular polyglutamated MTX (MTX-PG) is a better choice. Even though MTX’s therapeutic effects are thought to be due to its interactions with enzymes in leukocytes and synovial cells, the MTX-PG level in the RBCs is more frequently measured. This is due to the greater number of RBCs in the blood and due to the longer lifetimes of RBCs relative to some of the leukocytes, such as neutrophils. It was reported that there is an association between MTX efficacy and higher MTX-PG in RBCs (Angelis-Stoforidis et al. 1999). However, in a recent paper utilizing lower doses of MTX and shorter time between MTX initiation and sample collection, no association was found with RBC MTX-PG and disease activity (Kato et al. 2012).
MTX-PG is a promising tool to study the pharmacokinetics of MTX but is limited as a predictor for MTX response, since the patient must take MTX for several weeks before RBC MTX-PG becomes stabilized.
Immune cell frequencies as markers
Given the importance of immune cells in RA, the relative number of immune cell subtypes has been examined as possible markers of MTX response. One study found that a higher naïve T-cell frequency before treatment is associated with remission induced by MTX monotherapy. This observation was found in both a pilot cohort and a validation cohort, though the pilot cohort was frozen blood samples from RA patients on either MTX monotherapy or MTX and TNF inhibitors. The association between higher naïve T-cell frequency and remission on MTX is consistent with the idea that the T-cell balance may be dysregulated in MTX-resistant RA (Ponchel et al. 2014).
Another study found that a lower Th17 cell frequency before MTX treatment is associated with greater decrease in inflammation induced by MTX monotherapy (Yue et al. 2010). This was a very small study and their observations have yet to be repeated by other larger studies, but this observed association between Th17 cells and MTX response is consistent with the purported importance of Th17 in sustaining RA-associated inflammation.
A recent study found that higher number of circulating monocytes is associated with a lack of response to MTX (Chara et al. 2015). This is consistent with the finding that MTX can activate DCs, which are closely related to and can be derived from monocytes (Shurin et al. 2009). However, there was another study that found no significant differences in circulating monocyte counts between MTX responsive and non-responsive RA patients (Seitz et al. 2003).
Additionally, in a study utilizing blood taken after at least 2 months of MTX monotherapy, it was found that RA patients who are responsive to MTX have lower number of circulating polymorphonuclear leukocytes (PMN) than those who have a partial or no response to MTX (Angelis-Stoforidis et al. 1999). However, this information cannot be used to predict MTX response as this difference in PMN numbers was not determined prior to MTX treatment.
Cytokines and chemokines as markers
Given the importance of cytokines and chemokines in RA pathogenesis, their baseline concentrations have been explored as possible biomarkers to predict MTX response. Favorable MTX response is mildly associated with increased baseline serum concentration of soluble TNF receptor (TNFR), which decreases TNF-α signaling by competing with membrane-bound TNFR for binding to TNF-α (Seitz et al. 2003). Conversely, decreased baseline serum concentration of TNF-α has been reported as predictive of MTX response (Maillefert et al. 2010). However, in another study focusing on early RA patients, serum TNF-α was not predictive of MTX response (Ally et al. 2015). Additionally, it was reported that there is an association between MTX resistance and increased baseline serum concentration of MPIF-1, a chemokine which attracts monocytes and naïve T cells (Dhir et al. 2013). Multiple studies found that serum concentrations of the following cytokines are not correlated with MTX efficacy: IL-1 receptor antagonist (Ally et al. 2015; Seitz et al. 2003), IL-1β, IL-6, IL-8, IL-10, and IL-12 (Ally et al. 2015; Maillefert et al. 2010). These studies indicate that baseline levels of cytokines are not consistently predictive of a clinical response to MTX treatment.
A similar approach is to culture PBMCs and then measure the concentration of secreted cytokines. Through this approach, it was found that a favorable MTX response was associated with increased IL-1β and soluble TNFR secretion from untreated PBMC, prior to in vivo MTX treatment (Seitz et al. 2003). Another group cultured PBMCs in the presence of varying concentrations of MTX, and then calculated half maximal inhibitory concentration (IC50) values for the concentration of MTX required to suppress cytokine production. This group found a negative correlation between the clinical response and IC50 values for TNF-α and IFN-γ. Patients who experienced a larger clinical response to MTX tended to require a lower concentration of in vitro MTX to decrease the secretion of TNF-α and IFN-γ (Haroon et al. 2008).
Other proteins as markers
Autoantibodies are a common occurrence in RA. Two of the best-studied, common RA autoantibody types are anti-citrullinated protein antibody (ACPA), against citrullinated peptides, and rheumatoid factor (RF), against the constant region of IgG antibodies. In most studies exclusively with patients on MTX monotherapy, it has been found that ACPA status is not predictive of response to MTX (Ally et al. 2015; Ponchel et al. 2014; Saevarsdottir et al. 2011a). However, one study found that ACPA-positive RA patients were more likely than ACPA-negative RA patients to achieve remission with MTX and prednisone (Wevers-de Boer et al. 2012). Furthermore, RF status was not shown to be predictive of response to MTX (Ally et al. 2015; Ma et al. 2012; Maillefert et al. 2010; Ponchel et al. 2014; Saevarsdottir et al. 2011a; Wevers-de Boer et al. 2012).
Myeloid-related proteins 8 and 14 are endogenous proteins that activate TLR4 signaling and are found in increased levels in the serum of RA patients. They are secreted by monocytes, macrophages, and neutrophils in pro-inflammatory environments. Recently, it was found that a higher concentration of myeloid-related proteins 8 and 14 in the serum of RA patients before MTX monotherapy is associated with larger therapeutic response to MTX. Thus, serum myeloid-related proteins 8 and 14 are promising biomarkers that could be used to predict MTX response. (Patro et al. 2016)
Clinical predictors of methotrexate resistance
Disease-related
Disease duration has also been examined as a possible factor for MTX resistance. Some (Maillefert et al. 2010; Saevarsdottir et al. 2011a) but not all studies found that MTX is slightly less efficacious if the disease symptoms were of longer duration before treatment. For example, in a cohort with a wide range of symptom durations, MTX responders had symptoms for 3.1 ± 5.4 years prior to treatment while MTX non-responders had symptoms for 10.2 ± 10 years prior to treatment, and this was statistically significant (Maillefert et al. 2010). In cohorts with symptom onset less than 1 year prior to MTX treatment initiation, Saevarsdottir et al. (Saevarsdottir et al. 2011a) reported that the odds ratio for response was 0.88–0.99 per month increase in disease duration, but Ponchel et al. (Ponchel et al. 2014) reported this odds ratio to be 0.89–1.2, which is not significant. Additionally, in a cohort with symptom onset less than 2 years prior to MTX treatment initiation, the median symptom duration was 24 weeks for responders and 25 weeks for non-responders, and this was not statistically significant (Wessels et al. 2007).
Patient-related
A number of studies have examined the impact of patient-related clinical factors on MTX efficacy. Most studies found that MTX is more likely to be effective in male RA patients compared to female RA patients. The odds of effective response to MTX are approximately double for male compared to female RA patients (Saevarsdottir et al. 2011a; Seitz et al. 2003; Wessels et al. 2007; Wevers-de Boer et al. 2012).
Age of the patient has not shown to be consistently predictive. Some studies indicate increased MTX efficacy with increasing age (Saevarsdottir et al. 2011a; Sharma et al. 2009). For example, Saevarsdottir et al. (Saevarsdottir et al. 2011a) reported that the odds ratio for response was 1.3 per 10 years increase in age, and Sharma et al. (Sharma et al. 2009) reported that the average age was 45 years old for MTX responders and 41 years old for MTX non-responders. However, other studies indicate that age is not associated with significant changes in likelihood of response to MTX (Seitz et al. 2003; Wessels et al. 2007; Wevers-de Boer et al. 2012).
People with the shared epitope (SE), HLA-DR alleles sharing a specific motif, are more susceptible to RA. According to a study focusing on RA patients taking MTX and sulfasalazine, SE+ RA patients are more likely than SE− RA patients to be resistant to these DMARDs (Mori et al. 2010).
Lifestyle factors have also been examined, as they can be altered in addition to being theoretically used in the prediction of MTX efficacy. Smoking is the best-established environmental risk factor for RA onset (Boissier et al. 2012; McInnes and Schett 2011). There have been studies indicating that smoking does not predict MTX response (Mori et al. 2010; Wessels et al. 2007). However, in a recent, large study on patients with early RA, it was found that current smoking may predict MTX resistance, but history of smoking is not associated with MTX resistance (Saevarsdottir et al. 2011b). While smoking cessation is advisable to increase the probability of MTX efficacy, it is apparently not a large enough effect by itself to reliably predict who will respond to MTX and who will not.
Since caffeine is an adenosine receptor antagonist and increasing adenosine receptor signaling is part of MTX’s mechanism, there has been some concern that caffeine could reduce MTX’s efficacy. While a small study initially indicated that high caffeine intake decreases the efficacy of MTX (Nesher et al. 2003), a larger study later found that caffeine did not significantly impact MTX efficacy (Benito-Garcia et al. 2006).
MTX is a folate derivative, and it interferes with folate metabolism (Cronstein 2005; Inoue and Yuasa 2014; Kremer 2004; Shea et al. 2013; Tian and Cronstein 2007). Folic acid supplementation prevents MTX-induced liver toxicity and gastrointestinal side effects. While there are concerns that this folic acid supplementation might reduce the efficacy of MTX, meta-analysis indicates that folic acid supplementation does not significantly change the efficacy of MTX (Shea et al. 2013). As a result, EULAR recommends folic acid supplementation in conjunction with MTX (Smolen et al. 2017). In the United States, roughly half of all MTX-treated patients have also been prescribed folic acid (Al-Dabagh et al. 2013).
There is a lack of studies examining association of other dietary components with MTX efficacy in RA patients.
Conclusion
MTX is an effective, well-tolerated drug for most RA patients. However, there remains a lack of efficacy in up to 50% of patients (Braun et al. 2008; Klareskog et al. 2004; Lima et al. 2015; Mori et al. 2010; Weinblatt et al. 1994). In addition, this drug can cause significant side effects in a small minority of RA patients (Weinblatt et al. 1994). A reliable clinical test to determine MTX response in RA patients would be a major clinical advance. Such a test would promote earlier use of non-MTX-based therapies and avoid the possibility of MTX-induced adverse effects in patients who would have a poor response to MTX. Unfortunately, despite many studies in this research area, there is still no reliable test that can be applied clinically to predict MTX response. Given the complexity of MTX transport, cellular delivery, and action, it is likely that MTX response is dependent on multiple variables, including clinical characteristics. There is clearly an unmet need to develop a clinical predictive method for determining MTX response in this devastating disease.
Abbreviations
- ABC:
-
ATP binding cassette
- ACPA:
-
Anti-citrullinated protein antibody
- ACR:
-
American College of Rheumatology
- ADA:
-
Adenosine deaminase
- AICAR:
-
Aminoimidazole carboxamide ribonucleotide
- AMPDA:
-
AMP deaminase
- ATIC:
-
Aminoimidazole carboxamide ribonucleotide transformylase
- BCRP:
-
Breast cancer resistance protein
- DC:
-
Dendritic cell
- DHF:
-
Dihydrofolate
- DHFR:
-
Dihydrofolate reductase
- DMARD:
-
Disease-modifying anti-rheumatic drug
- EULAR:
-
European League Against Rheumatism
- IC50:
-
Half maximal inhibitory concentration
- KM :
-
Michaelis constant
- MDR:
-
Multidrug Resistance Protein
- MRP:
-
Multidrug resistance-associated protein
- MTX:
-
Methotrexate
- MTX-PG:
-
Polyglutamated methotrexate
- NSAID:
-
Nonsteroidal anti-inflammatory drug
- PBMC:
-
Peripheral blood mononuclear cells
- PCFT:
-
Proton-coupled folate transporter
- PKA:
-
Protein kinase A
- PMN:
-
Polymorphonuclear leukocytes
- RA:
-
Rheumatoid arthritis
- RF:
-
Rheumatoid factor
- RFC:
-
Reduced folate carrier
- SE:
-
Shared epitope
- SLC:
-
Solute carrier
- THF:
-
Tetrahydrofolate
- TNFR:
-
TNF receptor
- TYMS:
-
Thymidylate synthetase
References
Al-Dabagh A, Davis SA, Kinney MA, Huang K, Feldman SR (2013) The effect of folate supplementation on methotrexate efficacy and toxicity in psoriasis patients and folic acid use by dermatologists in the USA. Am J Clin Dermatol 14:155–161. https://doi.org/10.1007/s40257-013-0017-9
Ally MM, Hodkinson B, Meyer PW, Musenge E, Tintinger GR, Tikly M, Anderson R (2015) Circulating anti-citrullinated peptide antibodies, cytokines and genotype as biomarkers of response to disease-modifying antirheumatic drug therapy in early rheumatoid arthritis. BMC Musculoskelet Disord 16:130. https://doi.org/10.1186/s12891-015-0587-1
Angelis-Stoforidis P, Vajda FJ, Christophidis N (1999) Methotrexate polyglutamate levels in circulating erythrocytes and polymorphs correlate with clinical efficacy in rheumatoid arthritis. Clin Exp Rheumatol 17:313–320
Antonioli L, Pacher P, Vizi ES, Haskó G (2013) CD39 and CD73 in immunity and inflammation. Trends Mol Med 19:355–367. https://doi.org/10.1016/j.molmed.2013.03.005
Benito-Garcia E et al (2006) Dietary caffeine intake does not affect methotrexate efficacy in patients with rheumatoid arthritis. J Rheumatol 33:1275–1281
Bohanec Grabar P, Logar D, Lestan B, Dolzan V (2008) Genetic determinants of methotrexate toxicity in rheumatoid arthritis patients: a study of polymorphisms affecting methotrexate transport and folate metabolism. Eur J Clin Pharmacol 64:1057–1068. https://doi.org/10.1007/s00228-008-0521-7
Boissier MC, Semerano L, Challal S, Saidenberg-Kermanac’h N, Falgarone G (2012) Rheumatoid arthritis: from autoimmunity to synovitis and joint destruction. J Autoimmun 39:222–228. https://doi.org/10.1016/j.jaut.2012.05.021
Braun J et al (2008) Comparison of the clinical efficacy and safety of subcutaneous versus oral administration of methotrexate in patients with active rheumatoid arthritis: results of a six-month, multicenter, randomized, double-blind, controlled, phase IV trial. Arthritis Rheum 58:73–81. https://doi.org/10.1002/art.23144
Budzik GP, Colletti LM, Faltynek CR (2000) Effects of methotrexate on nucleotide pools in normal human T cells and the CEM T cell line. Life Sci 66:2297–2307
Chara L et al (2015) The number of circulating monocytes as biomarkers of the clinical response to methotrexate in untreated patients with rheumatoid arthritis. J Transl Med 13:2. https://doi.org/10.1186/s12967-014-0375-y
Cronstein BN (2005) Low-dose methotrexate: a mainstay in the treatment of rheumatoid arthritis. Pharmacol Rev 57:163–172. https://doi.org/10.1124/pr.57.2.3
Dervieux T (2009) Methotrexate pharmacogenomics in rheumatoid arthritis: introducing false-positive report probability. Rheumatology (Oxford) 48:597–598. https://doi.org/10.1093/rheumatology/kep060
Dhir V, Sandhu A, Gupta N, Dhawan V, Sharma S, Sharma A (2013) Low serum levels of myeloid progenitor inhibitory factor-1 predict good response to methotrexate in rheumatoid arthritis. ISRN Inflamm 2013:460469. https://doi.org/10.1155/2013/460469
Drozdzik M, Rudas T, Pawlik A, Kurzawski M, Czerny B, Gornik W, Herczynska M (2006) The effect of 3435C > T MDR1 gene polymorphism on rheumatoid arthritis treatment with disease-modifying antirheumatic drugs. Eur J Clin Pharmacol 62:933–937. https://doi.org/10.1007/s00228-006-0192-1
Fransen J, Kooloos WM, Wessels JA, Huizinga TW, Guchelaar HJ, van Riel PL, Barrera P (2012) Clinical pharmacogenetic model to predict response of MTX monotherapy in patients with established rheumatoid arthritis after DMARD failure. Pharmacogenomics 13:1087–1094. https://doi.org/10.2217/pgs.12.83
Furumiya M et al (2014) Noncompetitive inhibition of proton-coupled folate transporter by myricetin. Drug Metab Pharmacokinet 29:312–316
Gerards AH, de Lathouder S, de Groot ER, Dijkmans BA, Aarden LA (2003) Inhibition of cytokine production by methotrexate. Studies in healthy volunteers and patients with rheumatoid arthritis. Rheumatology (Oxford) 42:1189–1196. https://doi.org/10.1093/rheumatology/keg323
Haroon N, Srivastava R, Misra R, Aggarwal A (2008) A novel predictor of clinical response to methotrexate in patients with rheumatoid arthritis: a pilot study of in vitro T cell cytokine suppression. J Rheumatol 35:975–978
Hasko G, Linden J, Cronstein B, Pacher P (2008) Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov 7:759–770. https://doi.org/10.1038/nrd2638
Herman S, Zurgil N, Deutsch M (2005) Low dose methotrexate induces apoptosis with reactive oxygen species involvement in T lymphocytic cell lines to a greater extent than in monocytic lines. Inflamm Res 54:273–280. https://doi.org/10.1007/s00011-005-1355-8
Hooijberg JH et al (2014) Folates provoke cellular efflux and drug resistance of substrates of the multidrug resistance protein 1 (MRP1). Cancer Chemother Pharmacol 73:911–917. https://doi.org/10.1007/s00280-014-2421-0
Hryniuk WM, Brox LW, Henderson JF, Tamaoki T (1975) Consequences of methotrexate inhibition of purine biosynthesis in L5178Y cells. Cancer Res 35:1427–1432
Inoue K, Yuasa H (2014) Molecular basis for pharmacokinetics and pharmacodynamics of methotrexate in rheumatoid arthritis therapy. Drug Metab Pharmacokinet 29:12–19
Kato T, Hamada A, Mori S, Saito H (2012) Genetic polymorphisms in metabolic and cellular transport pathway of methotrexate impact clinical outcome of methotrexate monotherapy in Japanese patients with rheumatoid arthritis. Drug Metab Pharmacokinet 27:192–199
Klareskog L et al (2004) Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet 363:675–681. https://doi.org/10.1016/s0140-6736(04)15640-7
Kremer JM (2004) Toward a better understanding of methotrexate. Arthritis Rheum 50:1370–1382. https://doi.org/10.1002/art.20278
Kremer JM, Galivan J, Streckfuss A, Kamen B (1986) Methotrexate metabolism analysis in blood and liver of rheumatoid arthritis patients. Association with hepatic folate deficiency and formation of polyglutamates. Arthritis Rheum 29:832–835
Kulier R, Kapp N, Gulmezoglu AM, Hofmeyr GJ, Cheng L, Campana A (2011) Medical methods for first trimester abortion. Cochrane Database Syst Rev 2011:Cd002855. https://doi.org/10.1002/14651858.CD002855.pub4
Lima A, Bernardes M, Azevedo R, Medeiros R, Seabra V (2015) Pharmacogenomics of Methotrexate Membrane Transport Pathway: can Clinical Response to Methotrexate in Rheumatoid Arthritis Be Predicted? Int J Mol Sci 16:13760–13780. https://doi.org/10.3390/ijms160613760
Ma MH et al (2012) Remission in early rheumatoid arthritis: predicting treatment response. J Rheumatol 39:470–475. https://doi.org/10.3899/jrheum.110169
Maillefert JF et al (2010) Prediction of response to disease modifying antirheumatic drugs in rheumatoid arthritis. Joint Bone Spine 77:558–563. https://doi.org/10.1016/j.jbspin.2010.02.018
McInnes IB, Schett G (2011) The pathogenesis of rheumatoid arthritis. N Engl J Med 365:2205–2219. https://doi.org/10.1056/NEJMra1004965
Micsik T, Lorincz A, Gal J, Schwab R, Petak I (2015) MDR-1 and MRP-1 activity in peripheral blood leukocytes of rheumatoid arthritis patients. Diagn Pathol 10:216. https://doi.org/10.1186/s13000-015-0447-1
Mori S, Hirose J, Yonemura K (2010) Contribution of HLA-DRB1*04 alleles and anti-cyclic citrullinated antibodies to development of resistance to disease-modifying antirheumatic drugs in early rheumatoid arthritis. Clin Rheumatol 29:1357–1366. https://doi.org/10.1007/s10067-010-1454-y
Muralidharan N, Antony PT, Jain VK, Mariaselvam CM, Negi VS (2015) Multidrug resistance 1 (MDR1) 3435C > T gene polymorphism influences the clinical phenotype and methotrexate-induced adverse events in South Indian Tamil rheumatoid arthritis. Eur J Clin Pharmacol 71:959–965. https://doi.org/10.1007/s00228-015-1885-0
Nesher G, Mates M, Zevin S (2003) Effect of caffeine consumption on efficacy of methotrexate in rheumatoid arthritis. Arthritis Rheum 48:571–572. https://doi.org/10.1002/art.10766
Patro PS, Singh A, Misra R, Aggarwal A (2016) Myeloid-related Protein 8/14 Levels in Rheumatoid Arthritis: marker of Disease Activity and Response to Methotrexate. J Rheumatol 43:731–737. https://doi.org/10.3899/jrheum.150998
Peres RS et al (2015) Low expression of CD39 on regulatory T cells as a biomarker for resistance to methotrexate therapy in rheumatoid arthritis. Proc Natl Acad Sci U S A 112:2509–2514. https://doi.org/10.1073/pnas.1424792112
Ponchel F et al (2014) An immunological biomarker to predict MTX response in early RA. Ann Rheum Dis 73:2047–2053. https://doi.org/10.1136/annrheumdis-2013-203566
Prasad S, Tripathi D, Rai MK, Aggarwal S, Mittal B, Agarwal V (2014) Multidrug resistance protein-1 expression, function and polymorphisms in patients with rheumatoid arthritis not responding to methotrexate. Int J Rheum Dis 17:878–886. https://doi.org/10.1111/1756-185x.12362
Ranganathan P et al (2008) Methotrexate (MTX) pathway gene polymorphisms and their effects on MTX toxicity in Caucasian and African American patients with rheumatoid arthritis. J Rheumatol 35:572–579
Saevarsdottir S et al (2011a) Predictors of response to methotrexate in early DMARD naive rheumatoid arthritis: results from the initial open-label phase of the SWEFOT trial. Ann Rheum Dis 70:469–475. https://doi.org/10.1136/ard.2010.139212
Saevarsdottir S et al (2011b) Patients with early rheumatoid arthritis who smoke are less likely to respond to treatment with methotrexate and tumor necrosis factor inhibitors: observations from the Epidemiological Investigation of Rheumatoid Arthritis and the Swedish Rheumatology Register cohorts. Arthritis Rheum 63:26–36. https://doi.org/10.1002/art.27758
Sakowicz-Burkiewicz M, Kocbuch K, Grden M, Maciejewska I, Szutowicz A, Pawelczyk T (2012) Impact of adenosine receptors on immunoglobulin production by human peripheral blood B lymphocytes. J Physiol Pharmacol 63:661–668
Seitz M, Zwicker M, Villiger PM (2003) Pretreatment cytokine profiles of peripheral blood mononuclear cells and serum from patients with rheumatoid arthritis in different american college of rheumatology response groups to methotrexate. J Rheumatol 30:28–35
Sharma S et al (2008) Interaction of genes from influx-metabolism-efflux pathway and their influence on methotrexate efficacy in rheumatoid arthritis patients among Indians. Pharmacogenet Genomics 18:1041–1049
Sharma S et al (2009) Purine biosynthetic pathway genes and methotrexate response in rheumatoid arthritis patients among north Indians. Pharmacogenet Genomics 19:823–828
Shea B et al (2013) Folic acid and folinic acid for reducing side effects in patients receiving methotrexate for rheumatoid arthritis. Cochrane Database Syst Rev 2013:Cd000951. https://doi.org/10.1002/14651858.CD000951.pub2
Shurin GV, Tourkova IL, Kaneno R, Shurin MR (2009) Chemotherapeutic agents in noncytotoxic concentrations increase antigen presentation by dendritic cells via an IL-12-dependent mechanism. J Immunol 183:137–144. https://doi.org/10.4049/jimmunol.0900734
Singh JA et al (2016) 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res (Hoboken) 68:1–25. https://doi.org/10.1002/acr.22783
Smolen JS et al (2017) EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis 76:960–977. https://doi.org/10.1136/annrheumdis-2016-210715
Stamp LK et al (2010) Polymorphisms within the folate pathway predict folate concentrations but are not associated with disease activity in rheumatoid arthritis patients on methotrexate. Pharmacogenet Genomics 20:367–376. https://doi.org/10.1097/FPC.0b013e3283398a71
Stamp LK, Hazlett J, Highton J, Hessian PA (2013) Expression of methotrexate transporters and metabolizing enzymes in rheumatoid synovial tissue. J Rheumatol 40:1519–1522. https://doi.org/10.3899/jrheum.130066
Takatori R et al (2006) ABCB1 C3435T polymorphism influences methotrexate sensitivity in rheumatoid arthritis patients. Clin Exp Rheumatol 24:546–554
Tazoe Y et al (2015) Reduced folate carrier 1 gene expression levels are correlated with methotrexate efficacy in Japanese patients with rheumatoid arthritis. Drug Metab Pharmacokinet 30:227–230. https://doi.org/10.1016/j.dmpk.2015.02.001
Tian H, Cronstein BN (2007) Understanding the mechanisms of action of methotrexate: implications for the treatment of rheumatoid arthritis. Bull NYU Hosp Jt Dis 65:168–173
van der Heijden JW et al (2009) Involvement of breast cancer resistance protein expression on rheumatoid arthritis synovial tissue macrophages in resistance to methotrexate and leflunomide. Arthritis Rheum 60:669–677. https://doi.org/10.1002/art.24354
Visser K et al (2009) Multinational evidence-based recommendations for the use of methotrexate in rheumatic disorders with a focus on rheumatoid arthritis: integrating systematic literature research and expert opinion of a broad international panel of rheumatologists in the 3E Initiative. Ann Rheum Dis 68:1086–1093. https://doi.org/10.1136/ard.2008.094474
Weinblatt ME et al (1994) Methotrexate in rheumatoid arthritis. A five-year prospective multicenter study. Arthritis Rheum 37:1492–1498
Wessels JA et al (2007) A clinical pharmacogenetic model to predict the efficacy of methotrexate monotherapy in recent-onset rheumatoid arthritis. Arthritis Rheum 56:1765–1775. https://doi.org/10.1002/art.22640
Wevers-de Boer K et al (2012) Remission induction therapy with methotrexate and prednisone in patients with early rheumatoid and undifferentiated arthritis (the IMPROVED study). Ann Rheum Dis 71:1472–1477. https://doi.org/10.1136/annrheumdis-2011-200736
Wijngaarden S, van Roon JA, van de Winkel JG, Bijlsma JW, Lafeber FP (2005) Down-regulation of activating Fcgamma receptors on monocytes of patients with rheumatoid arthritis upon methotrexate treatment. Rheumatology (Oxford) 44:729–734. https://doi.org/10.1093/rheumatology/keh583
Witte T (2015) Methotrexate as combination partner of TNF inhibitors and tocilizumab. What is reasonable from an immunological viewpoint? Clin Rheumatol 34:629–634. https://doi.org/10.1007/s10067-015-2861-x
Wolf J, Stranzl T, Filipits M, Pohl G, Pirker R, Leeb B, Smolen JS (2005) Expression of resistance markers to methotrexate predicts clinical improvement in patients with rheumatoid arthritis. Ann Rheum Dis 64:564–568. https://doi.org/10.1136/ard.2003.014985
Woolf AD, Pfleger B (2003) Burden of major musculoskeletal conditions. Bull World Health Organ 81:646–656
Yu X, Wang C, Luo J, Zhao X, Wang L, Li X (2013) Combination with methotrexate and cyclophosphamide attenuated maturation of dendritic cells: inducing Treg skewing and Th17 suppression in vivo. Clin Dev Immunol 2013:238035. https://doi.org/10.1155/2013/238035
Yuasa H, Inoue K, Hayashi Y (2009) Molecular and functional characteristics of proton-coupled folate transporter. J Pharm Sci 98:1608–1616. https://doi.org/10.1002/jps.21515
Yue C et al (2010) The effects of adalimumab and methotrexate treatment on peripheral Th17 cells and IL-17/IL-6 secretion in rheumatoid arthritis patients. Rheumatol Int 30:1553–1557. https://doi.org/10.1007/s00296-009-1179-x
Zhu H, Deng FY, Mo XB, Qiu YH, Lei SF (2014) Pharmacogenetics and pharmacogenomics for rheumatoid arthritis responsiveness to methotrexate treatment: the 2013 update. Pharmacogenomics 15:551–566. https://doi.org/10.2217/pgs.14.25
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yu, M.B., Firek, A. & Langridge, W.H.R. Predicting methotrexate resistance in rheumatoid arthritis patients. Inflammopharmacol 26, 699–708 (2018). https://doi.org/10.1007/s10787-018-0459-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-018-0459-z