
Abstract
The gut-brain axis (GBA) is a crucial communication network linking the gastrointestinal (GI) tract and the central nervous system (CNS). The gut microbiota significantly influences metabolic, immune, and neural functions by generating a diverse array of bioactive compounds that modulate brain function and maintain homeostasis. A pivotal mechanism in this communication is the kynurenine pathway, which metabolises tryptophan into various derivatives, including neuroactive and neurotoxic compounds. Alterations in gut microbiota composition can increase gut permeability, triggering inflammation and neuroinflammation, and contributing to neuropsychiatric disorders. This review elucidates the mechanisms by which changes in gut permeability may lead to systemic inflammation and neuroinflammation, with a focus on the kynurenine pathway. We explore how probiotics can modulate the kynurenine pathway and reduce neuroinflammation, highlighting their potential as therapeutic interventions for neuropsychiatric disorders. The review integrates experimental data, discusses the balance between neurotoxic and neuroprotective kynurenine metabolites, and examines the role of probiotics in regulating inflammation, cognitive development, and gut-brain axis functions. The insights provided aim to guide future research and therapeutic strategies for mitigating GI complaints and their neurological consequences.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
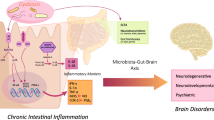
The gut-brain axis (GBA) is a critical communication network linking the gastrointestinal (GI) tract and the central nervous system (CNS). Acting as an anaerobic bioreactor, the gut microbiota significantly affects metabolic, immune, and neural functions by generating a diverse array of microbial metabolites, peptides, gut hormones, and neuroactive substances [15, 31]. These bioactive compounds travel via the ENS, circulatory system, vagus nerve, and immune system, thereby modulating brain function and contributing to the maintenance of homeostasis [5, 30, 64].
A key mechanism in gut-brain communication is the kynurenine pathway (KP), the main route for the metabolism of essential amino acid, tryptophan (TRP). Approximately 90% of dietary TRP is converted into kynurenine and its derivatives via this pathway, with the remainder metabolised into serotonin and indole compounds. The KP plays a significant role in the GBA and is involved in various physiological and pathological processes, including neuroinflammation and neurodegeneration. Alterations in gut microbiota composition and function can result in increased gut permeability, triggering inflammation and neuroinflammation. This process is implicated in the pathophysiology of numerous neuropsychiatric disorders [31, 64].
Advancements in high-throughput sequencing and metabolomic analysis have expanded our comprehension of the gut's role in systemic health, extending beyond its primary digestive and absorptive functions. It is now acknowledged that the GI tract plays a crucial role in immune regulation and systemic inflammation. Physiological stressors, such as reduced blood flow and pathogenic invasions, can disrupt GI homeostasis, leading to symptoms such as bloating, cramping, and diarrhoea [75], [117]. These GI dysfunctions can further impact the CNS via the GBA, affecting mood, cognitive functions, and stress responses.
This review elucidates the mechanisms by which changes in gut permeability may lead to systemic inflammation and subsequent neuroinflammation, focusing particularly on the KP. By delineating these mechanisms, we aim to provide insights into potential therapeutic strategies for mitigating GI complaints and their neurological consequences. Special attention will be given to the role of probiotics in modulating the KP and reducing neuroinflammation.
Molecular Mechanisms of the Kynurenine Pathway
The kynurenine pathway (KP) serves as the primary route for the metabolism of TRP, an essential amino acid integral to numerous physiological functions. This pathway plays a significant role in the GBA and is involved in a variety of physiological and pathological processes, including neuroinflammation and neurodegeneration.
The KP begins with the conversion of TRP to N-formylkynurenine (N-fKYN), a reaction catalysed by the rate-limiting enzymes indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO). Subsequently, N-fKYN is hydrolysed to kynurenine (KYN) by kynurenine formamidase. TDO primarily facilitates the basal metabolism of TRP in the liver, whereas IDO is predominantly active in immune cells and can be induced by pro-inflammatory cytokines such as interferon-gamma (IFN-γ), interleukin-1 (IL-1), IL-6, and tumour necrosis factor-alpha (TNF-α)([21, 22, 52]) (Fig. 1).

The tryptophan-kynurenine metabolic pathway.
KYN serves as a crucial metabolite within the pathway, branching into several important metabolites. The first branch involves the conversion of KYN into kynurenic acid (KYNA) via the enzyme kynurenine aminotransferase (KAT). KYNA is recognised for its neuroprotective properties, acting as an antagonist to the N-methyl-D-aspartate (NMDA) receptor. The second branch converts KYN into anthranilic acid (AA) through the action of kynureninase (KYNU). The third branch transforms KYN into 3-hydroxykynurenine (3-HK) via kynurenine 3-monooxygenase (KMO) [106].
3-HK can be further metabolised into 3-hydroxyanthranilic acid (3-HAA) by KYNU or into xanthurenic acid (XA) by KAT. In the brain, AA is efficiently converted into 3-HAA, which can subsequently form cinnabarinic acid (CA) or 2-amino-3-carboxymuconate-6-semialdehyde (ACMS). ACMS has several metabolic fates: it can be converted to quinolinic acid (QUIN), an NMDA receptor agonist and neurotoxin, or to picolinic acid (PIC) via 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase (ACMSD). QUIN is particularly noteworthy as it can further metabolise into nicotinamide adenine dinucleotide (NAD +) via quinolinate phosphoribosyltransferase (QPRT), underscoring the pathway's role in cellular energy metabolism [4, 64].
Neuroinflammation
QUIN is responsible for metabolising more than 95% of L-tryptophan in the GI tract. QUIN facilitates the entry of L-tryptophan into the bloodstream and its passage through the blood–brain barrier (BBB), a critical factor for central serotonergic signalling. Consequently, the QUIN pathway is linked to neurodegenerative processes due to its essential role in the biosynthesis of several neuroactive intermediates, such as kynurenic acid, niacin, and NAD + . Additionally, QUIN is involved in the production of neurotoxic intermediates, promoting excessive stimulation of the NR2A and NR2B subunits of the NMDA receptor. This excessive stimulation leads to increased calcium influx into neurons, contributing to the generation of reactive oxygen species (ROS) and free radicals [52]. This process ultimately results in neuronal damage and death through mechanisms such as lipid peroxidation, which compromises membrane fluidity and permeability [82, 96].
Experimental evidence demonstrates that QUIN impairs blood–brain barrier (BBB) function by inducing the production of nitric oxide. This nitric oxide production triggers the hyperphosphorylation of cytoskeletal intermediate protein filaments in astrocytes and neurons, leading to significant cellular and molecular disruptions. These disruptions contribute to increased BBB permeability and promote neuroinflammatory processes [12]. The effects of QUIN are particularly pronounced in brain regions with high neuronal susceptibility, such as the cortex, striatum, and hippocampus, which are commonly affected in neurodegenerative diseases like Alzheimer's disease (AD). At low concentrations, QUIN can induce stem cell proliferation and serves as an intermediate in the synthesis of NAD + in human brain cells. However, the damage induced by QUIN varies depending on the brain region, with cortical, striatal, and hippocampal neurons being especially sensitive. This variability in neuronal sensitivity may partly explain the elevated levels of neurodegeneration observed in these regions in AD patients, which correlates with increased QUIN levels and associated inflammatory processes [83].
3-HK also induces oxidative stress and neuronal apoptosis through its interaction with xanthine oxidase, leading to the production of ROS such as superoxide radicals (O2-) and hydroxyl radicals (OH-) [84]. These ROS can cleave DNA and promote apoptosis, resulting in neuronal damage and cognitive and motor dysfunctions. Both L-kynurenine and 3-HK exhibit similar physiological characteristics and distribution within the CNS, with higher concentrations observed in the cerebral cortex, striatum, and hippocampus ([120]). Despite the relatively low number of in vivo studies, high levels of 3-HK have been associated with neuroinflammation, demonstrating both antioxidant and pro-oxidant properties depending on concentration. Low concentrations of 3-HK are linked to strong pro-oxidant activity and neuronal toxicity, whereas higher concentrations increase resistance to oxidation [47].
The physiological balance between neurotoxic kynurenines, such as 3-HK and QUIN, and neuroprotective kynurenines, such as KYNA, is crucial for CNS homeostasis, contributing significantly to neuroprotection against oxidative stress and ROS production [23]. As previously mentioned, under normal physiological conditions, astrocytic QUIN produces KYNA, a neuroprotective agent, while neuronal QUIN synthesises NAD + , thereby improving cellular energy status. In contrast, under pathological conditions, inflammatory signals stimulate the QUIN pathway in macrophages, microglia, and dendritic cells, leading to the production of high amounts of QUIN. This necessitates a regulatory balance controlled by IDO, which activates L-tryptophan catabolism via the QUIN pathway instead of the alternative serotonin production pathway. Inflammation triggers the production of metabolites like 3-HK and QUIN, affecting cognitive function and promoting neurodegeneration [101].
The imbalance among neurotoxic, neuroprotective, and immunomodulatory QUIN metabolites is reported in several neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD). The KP is critical in these conditions due to its role in tryptophan metabolism and the production of neuroactive compounds like serotonin, KYN, and KYNA, which significantly impact brain function and fatigue perception. In AD, disturbances in the KP result in elevated levels of neurotoxic QUIN and reduced levels of neuroprotective KYNA, contributing to neuroinflammation and the formation of amyloid plaques and tau tangles, hallmark features of AD pathology [59]. Similarly, in PD, KP dysregulation leads to increased levels of 3-HK and QUIN, inducing oxidative stress and mitochondrial dysfunction, which contribute to neuronal death and motor dysfunction. In MS, elevated levels of QUIN during active disease phases contribute to the inflammatory milieu and neuronal damage observed in MS lesions, exacerbating disease progression [38]. ALS, marked by the progressive degeneration of motor neurons, shows KP dysregulation with elevated levels of QUIN associated with excitotoxicity and oxidative stress, leading to motor neuron death [38]. Lastly, in HD, increased production of neurotoxic metabolites like QUIN results in neuronal loss and characteristic motor and cognitive dysfunctions, with elevated QUIN levels correlating with disease severity and progression [38].
The KP's involvement in neuroinflammation and its impact on neurotransmitter systems, including glutamatergic, GABAergic, dopaminergic, and noradrenergic neurotransmissions, underscores its importance in brain function and the pathogenesis of disorders such as depression and neurodegeneration [48, 81, 114].
Gut Permeability and Neuroinflammation
The immune system, co-evolving with commensal microorganisms, has established a relationship characterised by mutualism and homeostasis. Effective host immunity prevents commensal microbes from overexploiting resources while maintaining tolerance to harmless stimuli [1], [117]. Disruptions induced by antibiotics, dietary changes, or environmental pollutants can destabilise the gut microbiome ([18]). This destabilisation may impair the interfaces between host and microbiome, altering immune responses and potentially leading to systemic spread of commensal microbes, increased susceptibility to pathogenic invasion, and inappropriate immune reactions [25, 42].
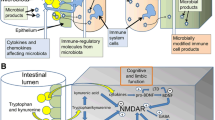
The microbiota employs various defence mechanisms against colonisation, pathogen overgrowth, and resultant damage or infection. One such mechanism is colonisation resistance, where both commensal and pathogenic microorganisms compete for resources and functional space, often mediated by quorum sensing [42], [123, 124]. Integral to this defence are the intestinal epithelial cells (IECs), primarily enterocytes, which form the gut lining and regulate the trans-epithelial movement of substances. This lining is reinforced by a complex array of junction proteins, including tight junctions (TJPs), adherens junctions, gap junctions, and desmosomes, ensuring structural integrity and function [112].
The epithelial barrier (Fig. 2), functioning as a selectively permeable membrane, is regulated via two primary pathways: paracellular and transcellular transport. Paracellular transport allows the passive flow of ions and small molecules between epithelial cells, controlled by tight junctions involving proteins such as claudins, occludins, and junctional adhesion molecules [80]. In contrast, the transcellular pathway involves the active transport of substances across cell membranes, mediated by various transporters and channels, and requiring energy [91]. IECs, along with a protective mucus barrier, are pivotal in controlling the passage of substances. Tight junction proteins such as claudins, occludins, and ZO-1 regulate this permeability ([71]. IECs include various cell types: goblet cells produce mucus essential for maintaining the mucus barrier,enteroendocrine cells release hormones in response to luminal signals; sensory tuft cells are involved in immune responses; and Paneth cells contribute to mucosal defence through the secretion of antimicrobial peptides [29, 50, 61].

The gut epithelial barrier consists of the apical plasma membrane of enterocytes, held together by tight junction proteins (claudin and occludin) and adherens junction proteins (E-cadherin and catenin), as well as the zonula occludens proteins ZO1 and ZO2, which are adaptor proteins necessary for the structural and regulatory functions of tight junctions. a | Upregulation of junctional proteins can be induced by microbiota metabolites including polyphenols, indole and indole derivatives, short-chain fatty acids (SCFAs) and polyamines. b | Downregulation of junctional proteins is mediated by: lipopolysaccharides (LPS) through binding to Toll-like receptor 4 (TLR4); by zonulin, a protein that activates the EGF receptor (EGFR) through transactivation of the proteinase-activated receptor 2 (PAR2), thereby inducing protein kinase C (PKC) phosphorylation; and by pro-inflammatory cytokines, including IL-1β, interferon-γ (IFNγ) and tumour necrosis factor (TNF). JAM, junctional adhesion molecules.
The human gut harbours a high concentration of gram-negative bacteria containing endotoxin, particularly in the lower intestine. These bacteria are also found in saliva, dental plaque, skin, lungs, respiratory tract, and urinary tract. Humans are highly sensitive to endotoxin, with significantly lower tolerance levels compared to other mammals. Recent studies have highlighted the role of tight junctions in maintaining epithelial permeability and their regulation by gut microbiota interactions. Dysbiosis, characterised by an imbalance in the gut microbial community, often leads to augmented intestinal permeability and subsequent chronic inflammation. This dysregulation is implicated in various diseases, including inflammatory bowel disease (IBD), metabolic disorders, and neurological conditions [3, 89, 121]. Furthermore, research has shown that endotoxin can significantly impact the kynurenine pathway, influencing neurological functions and contributing to conditions like Alzheimer's disease and Parkinson's disease [38, 79, 93].
Disruptions in the functionality of tight junctions or transporter activities can lead to increased epithelial permeability, allowing luminal antigens, pathogens, and toxins to translocate into the submucosa and systemic circulation. Such alterations can trigger immune responses and inflammatory cascades. GI barrier dysfunction enables endotoxins, such as LPS from gram-negative bacteria, to pass through the intestinal barrier into systemic circulation, a process termed endotoxemia. This passage of LPS can significantly impact the kynurenine pathway and subsequently influence neurological conditions [43]. LPS, consisting of lipid A, a short sugar chain core, and an O-antigen, is a major component of the outer membrane of gram-negative bacteria. Soluble endotoxin is released when bacteria are destroyed or physiologically as outer membrane vesicles. Different species of gram-negative bacteria have varying endotoxin structures, mainly due to differences in the O-antigen and lipid A, which are detected by the MD2/TLR4 receptor complex, determining the inflammation and toxicity of the endotoxin (Brown, 2019,Bryant et al., 2010).
Endotoxemia activates the local immune response, leading to the secretion of soluble factors such as soluble CD14 (sCD14) via the activation of T-lymphocytes, monocytes, and tissue macrophages. LPS binds to serum lipopolysaccharide-binding protein (LBP) to form an LPS-LBP complex, which then binds to the CD14 receptor on immune cells, initiating the production and release of pro-inflammatory cytokines such as Tumour Necrosis Factor Alpha (TNF-α) and Interleukin-6 (IL-6). This response disrupts GI barrier tight junction proteins, exacerbating intestinal permeability [43, 113].
The inflammatory response induced by endotoxin primarily involves the activation of Toll-like receptor 4 (TLR4) with its co-receptor MD2 on immune cells. This interaction initiates intracellular signalling cascades involving MyD88, TRAF6, and the IκB kinase (IKK) complex, leading to the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). NF-κB translocates into the nucleus, upregulating genes encoding pro-inflammatory cytokines such as IL-6 and TNF-α [49, 88, 111]. IL-6 and TNF-α exert their effects by binding to their respective receptors, IL-6R and TNFR1/2, on target cells. IL-6 primarily activates the Janus tyrosine kinase and signal transducers and activators of transcription (JAK-STAT) pathway, leading to the phosphorylation and activation of STAT3, which regulates inflammatory gene transcription. TNF-α activates various signalling pathways, including NF-κB, Jun N-terminal kinase (JNK), and Mitogen-activated protein kinases (MAPK) pathways. Additionally, interferon-gamma (IFN-γ), another pro-inflammatory cytokine, upregulates the enzyme IDO, catalysing the conversion of tryptophan into N-formylkynurenine, initiating the KP. KMO further converts kynurenine into 3-hydroxykynurenine, an intermediate in the quinolinic acid pathway [64, 67]. Moreover, pro-inflammatory cytokines like IFN-γ can stimulate KMO, potentially promoting neurotoxicity by favouring the quinolinic acid pathway [52]. These inflammatory signals can affect neural drive, contributing to feelings of sadness and increased perceptions of fatigue, potentially leading to neuroinflammatory and neuropsychiatric conditions. Neuroinflammation mediated by the kynurenine pathway is implicated in the pathogenesis of various neurological disorders, including depression, Alzheimer's disease, and Parkinson's disease [6, 79, 115].
There are several mechanisms by which increased levels of cytokines in the periphery can reach and affect the brain. These include passage through leaky regions in the blood–brain barrier (BBB) such as circumventricular organs, active transport through transport molecules, activation of cells lining the cerebral vasculature (endothelial cells and perivascular macrophages), binding to cytokine receptors associated with the vagus nerve, stimulating the hypothalamic–pituitary–adrenal (HPA) axis at the anterior pituitary or hypothalamus, and recruitment of activated cells such as monocytes/macrophages from the periphery to the brain [10, 68, 69]. Through activation of the intracellular signalling pathway mitogen-activated protein kinase, cytokines can increase the number and function of the reuptake pumps for serotonin, noradrenaline, and dopamine, which in turn can reduce the availability of these neurotransmitters within the synaptic cleft. Preclinical studies have demonstrated that increased inflammatory cytokines reduce central levels of brain-derived neurotrophic factor (BDNF) and neurogenesis, leading to depressive-like behaviour [27]. However, the relationship between peripheral and central inflammatory markers and antidepressants is complex and it remains unclear which pathways are most relevant for cytokine signal transmission in stress-related disorders such as depression [10, 39], [56].
There is some evidence, albeit from small studies of short duration, suggesting that anti-inflammatory agents such as non-steroidal anti-inflammatory drugs (NSAIDs) and cytokine inhibitors reduce depressive symptoms [10]. For depressed patients with raised inflammatory markers, this raises the prospect of whether reducing low-grade inflammation could alleviate depressive symptoms. Although a randomised controlled trial of the monoclonal antibody infliximab, a TNF-α antagonist, was not superior to placebo in reducing depressive symptoms overall, in patients with high baseline CRP levels there were greater reductions in depressive symptoms than in those with low CRP levels [74]. Another study showed that CRP level at baseline differentially predicted treatment outcome with escitalopram or nortriptyline [41]. These studies provide the impetus for stratification of depressed patients based on inflammatory profiles to advance personalised medicine. Developing more nuanced profiles of inflammatory proteins and gene expression, as well as cellular immune parameters, likely represents the future for predictors and targets of response to anti-inflammatory therapies.
The brain regions most reliably identified as being most affected by the administration of inflammatory stimuli include the basal ganglia and the dorsal anterior cingulate cortex (dACC). The dACC, part of the brain’s limbic system, is involved in cognitive and emotional processing. Cytokines can induce increases in neural activity most strongly in either the subgenual or the dorsal area of the dACC and have been associated with the development of mood and anxiety symptoms ([56]). Cytokines can impair basal ganglia functioning through known inhibitory effects on dopamine signalling in the CNS [68]. Reductions in basal ganglia activity have been noted in more posterior regions, where they are associated with fatigue, and in more ventral regions (such as the nucleus accumbens), where they have been linked to the development of anhedonia [19, 40].
Microglia are central to the inflammatory process and a source of cytokines [16]. These phagocytic innate immune cells account for approximately 10% of cells in the brain and contribute to the plasticity of neural circuits by modulating synaptic architecture and function [35]. Microglial process motility can be modulated by glutamatergic and GABAergic neurotransmission. Preclinical studies have shown that acute stress results in microglia activation and increased levels of pro-inflammatory cytokines in areas such as the hippocampus and hypothalamus [26]. Most studies show increases in activated microglia in response to chronic stress. Preliminary changes in the microenvironment of the microglia may result in a susceptibility to a secondary inflammatory stimulus. This concept of microglia priming may be of relevance to depression, which often requires multiple environmental “hits” [107]. In an environmental two-hit rodent model in which the first experimental manipulation targeted pregnant dams, and the second manipulation was given to the resulting offspring, exposure to prenatal immune challenge and peripubertal stress synergistically induced pathological effects on adult behavioural functions and neurochemistry [44]. Thus, early-life stress primes microglia, leading to a potentiated response to subsequent stress. Interestingly, the microbiota regulates microglia maturation and function. Clinically, microglial activation in the prefrontal cortex (PFC), anterior cingulate cortex (ACC), and insula in medication-free depressed patients has been demonstrated using translocator protein density measured by distribution volume in a positron emission tomography study [28, 107].
Gut-Brain Axis and Cognitive Function
The GBA plays a crucial role in mental and cognitive health, with mechanisms under extensive research (Berding, Vlckova, et al., 2021; [15, 31, 46, 75]). This axis comprises a bidirectional communication network between the CNS and ENS, integrating emotional and cognitive brain centres with intestinal functions [92, 110]. Alterations in the gut microbiota may influence the peripheral and CNS, potentially affecting brain function and cognitive processes [42, 85, 102], [126]. This communication involves both direct and indirect signalling through chemical transmitters, neuronal pathways, and the immune system [110]. Chemical signalling includes the production of neuroactive compounds by the gut microbiota, such as γ-aminobutyric acid (GABA, noradrenaline, dopamine, serotonin, and amino acids like tyramine and tryptophan (Berding, Vlckova, et al., 2021 [14, 64],). Microbial metabolites, notably short-chain fatty acids (SCFAs) such as butyrate, propionate, and acetate, are by-products of dietary fibre fermentation by intestinal microorganisms [77, 100]. These compounds traverse the portal circulation, influencing the host’s immune system, metabolism, and neuronal cells within the ENS and vagus nerve pathways [78, 100]. SCFAs have been demonstrated to affect CNS function by regulating neuroplasticity, gene expression, and immune responses, with butyrate notably modulating the expression of brain-derived neurotrophic factor (BDNF and attenuating depressive-like behaviours in animal models (Berding, Carbia, et al., 2021 [53],).
Neuronal pathways, particularly the vagus nerve, play a pivotal role in the GBA by conveying sensory signals from the gut to the CNS. This transmission involves the activation of mechanoreceptors and chemoreceptors responsive to various chemical stimuli [15]. The ENS, often described as the "second brain," contains an extensive neuronal network that regulates gut functions and is influenced by the gut microbiota, impacting gut motility and intestinal barrier function [5, 15]. Furthermore, the gut microbiota directly influences and is influenced by the immune system [42]. It plays a significant role in the development and function of the peripheral immune system and is integral to the healthy development, maturation, and activation of microglia, the innate immune cells in the brain ([18]). Signals from microbial metabolism are crucial for microglial function, as shown in studies where the restoration of microglial morphology and function in germ-free (GF) mice treated with bacterial-derived SCFAs was noted [35].
Additionally, the gut microbiota's interaction with the brain is mediated through the systemic immune system via circulating cytokines, which can alter immune signalling within the brain, potentially leading to symptoms such as loss of appetite, irritability, and low mood([20], [56, 95]). Research has also suggested that the gut microbiota influences the permeability of the BBB, with GF mice exhibiting increased BBB permeability partly due to reduced expression of tight-junction proteins such as occludin and claudin 5 [13].
Gut Microbiota and Neuroimmune Pathways
The metabolism of the kynurenine pathway is intricately regulated by inflammatory mediators and immunoresponsive enzymes([17]). The gut microbiota plays a vital role in educating and regulating the host's immune system throughout life [34]. This regulatory function is evidenced not only in germ-free (GF) animals but also in those with depleted gut microbiota due to antibiotic treatment, which exhibit compromised immune responses to infections [54]. Conversely, the immune system shapes the composition and diversity of the intestinal microbiota [55].
GF animals display an immature immune system, potentially explaining the reduced kynurenine pathway metabolism observed in these animals [24]. Upon colonisation post-weaning, normal metabolic functions are restored, aligning with the reinstatement of immune system function following the introduction of intestinal microbiota [24, 90]. These findings have translational relevance as low-grade immune activation in IBS correlates with alterations in gut microbiota and increased KP metabolism [2]. The aryl hydrocarbon receptor, which responds to both exogenous and endogenous stimuli, modulates immune responses and maintains host-microbe homeostasis. Indole, produced from tryptophan by microbes, acts as a ligand for this receptor [9, 116]. Although kynurenine was traditionally seen as an inert precursor, it activates the aryl hydrocarbon receptor [58], which in turn regulates IDO and TDO expression [9, 60].
The complex interplay between the gut microbiota, kynurenine pathway metabolism, and the immune response is exemplified by increased kynurenic acid levels in the absence of aryl hydrocarbon receptors in mice and aryl hydrocarbon receptor activation in the brain following experimental stroke [9]. Additionally, astrocyte activity and CNS inflammation are modulated by type I interferons and tryptophan metabolites via the aryl hydrocarbon receptor, and administration of an aryl hydrocarbon receptor agonist can attenuate intestinal inflammation in mouse models of colitis ([21, 22]).
Microbial metabolites, such as SCFAs, also influence intestinal barrier integrity and systemic inflammation, leading to alterations in kynurenine pathway metabolism [108], [123, 124]. Notably, the gut microbiota regulates microglia maturation and function [35], yet KP metabolites in the CNS have not been reported in microbiota-deficient animals. Elevated levels of kynurenine and its metabolites have been observed in the brains of Toxoplasma gondii-infected mice, with reactivation linked to brain IDO activation via IFN-γ dependent mechanisms [33], [118]
The relationship between tryptophan metabolism and gut microbiota composition is supported by preclinical studies demonstrating increased circulating tryptophan levels in GF animals [24, 76]. Despite this, kynurenine pathway metabolism and circulating serotonin concentrations are decreased [24]. This aligns with findings that GI serotonin synthesis, influenced by microbial metabolites such as SCFAs or tryptophan-derived indole metabolites, modulates circulating levels [65, 92].
Infection with Trichuris muris increases the kynurenine/tryptophan ratio [97]. Preclinical studies highlight total tryptophan concentrations' role in brain uptake, although the dynamics of tryptophan flux down the KP warrant further investigation [83]. Increased circulating tryptophan levels in GF animals result in higher hippocampal serotonin concentrations [24]. However, it remains unclear whether reduced circulating kynurenine availability in microbiota-deficient animals affects CNS kynurenine and downstream metabolites.
Tryptophan metabolism via the KP has significant implications for neurogastroenterology due to its effects on GI and CNS functions and GBA signalling. IBS, characterised by altered tryptophan metabolism, is linked to GI symptoms and co-morbid mood and anxiety disorders [2, 51]. Mucosal kynurenic acid and 5-HT levels correlate with anxiety and depression scores in IBS patients [63]. Acute tryptophan depletion studies demonstrate the impact of peripheral tryptophan levels on CNS and ENS function, highlighting altered tryptophan metabolism in GBA dysregulation in IBS ([21, 22, 100]).
Mood and anxiety disorders are common in IBS, linked to inflammatory-mediated tryptophan metabolism along the KP ([21, 22, 92]). Dysregulated brain-gut communication impacts peripheral and central symptoms in IBS [51]. The GBA plays a crucial role in mental and cognitive health, integrating emotional and cognitive brain centres with intestinal functions through direct and indirect signalling pathways [5, 15]. Microbial metabolites like SCFAs influence CNS function, regulating neuroplasticity and gene expression, while the gut microbiota affects BBB permeability and immune signalling within the brain [94].
The Role of Probiotics in Regulating Inflammation, Cognitive Development, and the Kynurenine Pathway
Probiotics are living, non-pathogenic bacteria and yeasts that, when administered in adequate amounts, confer health benefits by promoting microbial balance, particularly in the digestive system [62]. They primarily include Lactobacillus and Bifidobacterium species or Saccharomyces boulardii [70]. These probiotic strains engage in various physiological activities, such as reducing the pH of the intestine, cell-to-cell signalling, inhibiting the colonisation of pathogenic microbes, and regulating the host's immune response [87]. A distinct category of probiotics known as "psychobiotics" has been identified for their potential to improve psychological and mental health, affecting mood, anxiety, focus, memory, and cognition [32, 105].
The gut microbiota (GM), comprising a complex community of microbes, their genomes, and metabolic products, plays an important role in maintaining host health [86]. The dominant bacterial phyla in the GM include Bacteroidetes, Proteobacteria, and Actinobacteria, with common genera being Streptococcus, Pseudomonas, Bacteroides, Fusobacteria, Clostridium, and Lactobacillus [62]. These gut bacteria contribute to chronic inflammation and defence mechanisms, preserve the mucosal barrier, and assist in metabolism [70, 77]. The GM is also involved in producing GI hormones, short-chain fatty acids, vitamins, and medication absorption [45, 108].
Probiotics can modulate the composition of the GM and restore gut ecosystem balance, offering potential therapeutic approaches for cognitive deficits [70, 98]. For instance, a probiotic mixture containing Lactobacillus acidophilus, L. rhamnosus, and Bifidobacteria longum administered for three months improved Bifidobacteria and Lactobacilli levels and symptoms of autism [109]. Similarly, the supplementation of Bifidobacterium breve strain A1 facilitated hippocampal learning and memory in a Parkinson’s disease mouse model by recovering the expression of synaptophysin and postsynaptic density protein-95 [93].
Probiotics exert their beneficial effects through various mechanisms. They produce antioxidant enzymes (catalase, superoxide dismutase) and antioxidants (butyrate, folate, glutathione), and chelate metal ions, reducing oxidative stress [73, 99], [119]. Additionally, probiotics can inhibit TLR activation, reducing inflammatory responses, enhancing BBB integrity, and improving neurological functions ([119]). Probiotics also influence cognitive function by upregulating brain-derived neurotrophic factor (BDNF), increasing monoamine levels, and enhancing neuroplasticity, potentially ameliorating depression ([57, 73]).
Experimental support for the use of probiotics as therapeutic targets comes from various animal studies. For instance, L. rhamnosus JB-1 modulates GABA receptor expression, resulting in reduced anxiety-like symptoms by activating the vagus nerve [14]. Probiotics have also shown to decrease pro-inflammatory cytokines (e.g., IL-6, TNF-α) and increase anti-inflammatory cytokines (e.g., IL-10, TGF-β) in the brain, improving the gut barrier and reducing LPS levels in the bloodstream, which can mitigate neuroinflammation [80].
Probiotics have been found to modulate the KP, reducing neuroinflammation and promoting cognitive health [78]. As mentioned previously, under normal physiological conditions, TRP is metabolised by TDO, maintaining the KP in equilibrium. However, inflammatory factors increase the activity of IDO, leading to a higher production of QUIN, which can disrupt cognitive functions [100]. Probiotics can potentially modulate the KP by influencing TRP metabolism. For example, supplementation with specific strains like Lactobacillus reuteri and Bifidobacterium infantis has been shown to reduce the levels of neurotoxic metabolites, promoting neuroprotection. A study by Rudzki and colleagues demonstrated that the administration of probiotics altered the gut microbiota composition, resulting in decreased levels of QUIN and increased levels of KYNA in the brain, which is associated with improved cognitive function [103].
Additionally, combination probiotic therapy, such as Bifidobacterium lactis, B. bifidum, Lactobacillus casei, and L. acidophilus in aging mice, mitigates age-related disruption of the blood–brain barrier and intestinal barrier integrity, thereby reducing plasma and cerebral LPS and pro-inflammatory cytokines like IL-6, TNF-α, TLR4, and NF-κB translocation in the brain [36]. This improvement in microbial composition is accompanied by enhanced memory functions and reduced neuronal and synaptic injuries, as well as decreased microglia activation in the brain [36].
Clinical trials and observational studies are essential to evaluate the efficacy of probiotics in cognitive health. For instance, supplementation with Lactiplantibacillus plantarum OLL 2712 for 12 weeks reduced inflammation and improved memory in elderly adults [104]. Probiotics such as Lactobacillus casei Shirota have been shown to alleviate constipation and abdominal pain in Parkinson’s disease patients (Cassani et al., 2011). Probiotic supplementation has also improved verbal memory and cognitive performance in elderly subjects [66].
A meta-analysis by Zhu and colleagues ([125]) reported that probiotics significantly improved cognitive functions, particularly in mild cognitive impairment. Another meta-analysis suggested that probiotics could improve insulin resistance, lipid metabolism, and cognitive and GI health in patients with Alzheimer’s disease, mild cognitive impairment, and Parkinson’s disease [122]
While probiotics offer health benefits, side effects, albeit rare, may occur including systemic infections, GI side effects, and immune stimulation ([72]). Probiotic-induced d-lactic acidosis can occur in individuals with short bowel syndrome, leading to neurological symptoms such as memory loss and delirium [11].
To ensure safe probiotic use, microbiome profiling is recommended to identify factors affecting individual responses. Manufacturers should re-evaluate older strains for antibiotic resistance and disclose each probiotic strain’s antibiogram. Research into animal models is encouraged to detect potential long-term impacts of probiotics, particularly next-generation strains. Companies must monitor and report adverse events in compliance with regulatory regulations [62].
Future Perspectives
Integrating Gut Permeability, the Kynurenine Pathway, and Neuroinflammation
The GM actively communicates with the CNS through neural, endocrine, and immune pathways. Preclinical studies utilising germ-free animals and faecal microbiota transplantation have demonstrated the impact of GM on neuroinflammatory responses, providing insights into the potential therapeutic applications of GM modulation in neuroinflammatory diseases. Probiotics are live beneficial bacteria, while prebiotics are substances that selectively promote the growth of beneficial gut microbiota. Targeted administration of specific probiotics or prebiotics holds promise for modulating the GM and reducing neuroinflammation. Further research is needed to identify specific strains and combinations that effectively modulate neuroinflammatory processes. Postbiotics are the metabolic byproducts of probiotic bacteria, including short-chain fatty acids, antimicrobial peptides, and bioactive molecules. These postbiotics exhibit immunomodulatory and anti-inflammatory properties, offering potential therapeutic avenues for neuroinflammatory disorders. Understanding the mechanisms of action and developing strategies to enhance the production and delivery of beneficial postbiotics are essential areas for future investigation. Faecal microbiota transplantation, which involves transferring faecal material from a healthy donor to a recipient with a dysbiotic GM, has shown promising results in the treatment of various GI disorders and is now being explored as a potential therapy for neuroinflammatory diseases.
Conclusion
The kynurenine pathway (KP) plays a pivotal role in gut permeability and inflammation, significantly impacting the gut-brain axis (GBA) and contributing to various neuropsychiatric disorders. Our review has elucidated the mechanisms by which alterations in gut microbiota composition can increase gut permeability, triggering systemic inflammation and neuroinflammation. The KP, which metabolises tryptophan into neuroactive and neurotoxic compounds, serves as a critical mediator in this process. We have highlighted the dual nature of kynurenine metabolites, balancing neurotoxic effects of compounds like quinolinic acid with the neuroprotective properties of kynurenic acid.
Probiotics emerge as promising therapeutic interventions, capable of modulating the KP and reducing neuroinflammation. Experimental data indicate that specific probiotic strains can shift the balance towards neuroprotective metabolites, thereby mitigating cognitive and emotional disturbances associated with increased gut permeability and systemic inflammation. These findings not only underscore the importance of maintaining a healthy gut microbiota but also open avenues for novel therapeutic strategies targeting the GBA.
Future research should focus on delineating the specific probiotic strains and combinations that are most effective in modulating the KP and reducing neuroinflammation. Additionally, the development of advanced analytical methods for measuring KP metabolites in clinical settings will enhance our understanding of their role in neuropsychiatric conditions. Integrating gut permeability, the kynurenine pathway, and neuroinflammation into a cohesive framework will provide deeper insights into the pathophysiology of neuropsychiatric disorders and guide the development of targeted interventions. Thus, the modulation of the gut microbiota holds great potential for therapeutic advancements in treating both gastrointestinal and neurological conditions, paving the way for improved mental and cognitive health.
Data Availability
No datasets were generated or analysed during the current study.
Abbreviations
- GBA:
-
Gut-Brain Axis
- GI:
-
Gastrointestinal
- CNS:
-
Central Nervous System
- KP:
-
Kynurenine Pathway
- Trp:
-
Tryptophan
- IDO:
-
Indoleamine 2,3-Dioxygenase
- TDO:
-
Tryptophan 2,3-Dioxygenase
- N-fKYN:
-
N-formylkynurenine
- KYN:
-
Kynurenine
- KAT:
-
Kynurenine Aminotransferase
- KYNA:
-
Kynurenic Acid
- KYNU:
-
Kynureninase
- 3-HK:
-
3-Hydroxykynurenine
- 3-HAA:
-
3-Hydroxyanthranilic Acid
- QA:
-
Quinolinic Acid
- QPRT:
-
Quinolinate Phosphoribosyltransferase
- NAD + :
-
Nicotinamide Adenine Dinucleotide
- AA:
-
Anthranilic Acid
- XA:
-
Xanthurenic Acid
- ACMS:
-
2-Amino-3-carboxymuconate-6-semialdehyde
- PIC:
-
Picolinic Acid
- BBB:
-
Blood-Brain Barrier
- ROS:
-
Reactive Oxygen Species
- NMDA:
-
N-methyl-D-aspartate
- SCFA:
-
Short-Chain Fatty Acid
- ENS:
-
Enteric Nervous System
- LPS:
-
Lipopolysaccharide
- TLR4:
-
Toll-Like Receptor 4
- TNF-α:
-
Tumour Necrosis Factor Alpha
- IL-6:
-
Interleukin-6
- sCD14:
-
Soluble CD14
- NF-κB:
-
Nuclear Factor Kappa-light-chain-enhancer of Activated B Cells
- JNK:
-
Jun N-terminal Kinase
- MAPK:
-
Mitogen-Activated Protein Kinases
- IFN-γ:
-
Interferon-gamma
- HPA:
-
Hypothalamic-Pituitary-Adrenal
- BDNF:
-
Brain-Derived Neurotrophic Factor
- NSAIDs:
-
Non-Steroidal Anti-Inflammatory Drugs
- CRP:
-
C-Reactive Protein
- GF:
-
Germ-Free
- GM:
-
Gut Microbiota
- PFC:
-
Prefrontal Cortex
- ACC:
-
Anterior Cingulate Cortex
- SCFAs:
-
Short-Chain Fatty Acids
- GABA:
-
Gamma-Aminobutyric Acid
- PI3K/Akt:
-
Phosphoinositide 3-Kinase/Protein Kinase B
- ERK:
-
Extracellular Signal-Regulated Kinase
- TGF-β:
-
Transforming Growth Factor Beta
References
Abt, M. C., Osborne, L. C., Monticelli, L. A., Doering, T. A., Alenghat, T., Sonnenberg, G. F., Paley, M. A., Antenus, M., Williams, K. L., Erikson, J., Wherry, E. J., & Artis, D. (2012). Commensal Bacteria Calibrate the Activation Threshold of Innate Antiviral Immunity. Immunity, 37(1). https://doi.org/10.1016/j.immuni.2012.04.011
Agnello, M., Carroll, L. N., Imam, N., Pino, R., Palmer, C., Varas, I., Greene, C., Hitschfeld, M., Gupta, S., Almonacid, D. E., & Hoaglin, M. C. (2020). Gut microbiome composition and risk factors in a large cross-sectional IBS cohort. BMJ Open Gastroenterology, 7(1). https://doi.org/10.1136/bmjgast-2019-000345
Allam-Ndoul, B., Castonguay-Paradis, S., & Veilleux, A. (2020). Gut microbiota and intestinal trans-epithelial permeability. In International Journal of Molecular Sciences (Vol. 21, Issue 17). https://doi.org/10.3390/ijms21176402
Anderson, E. W., Fishbein, J., Hong, J., Roeser, J., Furie, R. A., Aranow, C., Volpe, B. T., Diamond, B., & Mackay, M. (2021). Quinolinic acid, a kynurenine/tryptophan pathway metabolite, associates with impaired cognitive test performance in systemic lupus erythematosus. Lupus Science and Medicine, 8(1). https://doi.org/10.1136/lupus-2021-000559
Appleton, J. (2018). The gut-brain axis: Influence of microbiota on mood and mental health. In Integrative Medicine (Boulder) (Vol. 17, Issue 4).
Bay-Richter, C., & Wegener, G. (2022). Antidepressant Effects of NSAIDs in Rodent Models of Depression—A Systematic Review. In Frontiers in Pharmacology (Vol. 13). https://doi.org/10.3389/fphar.2022.909981
Berding, K., Carbia, C., & Cryan, J. F. (2021). Going with the grain: Fiber, cognition, and the microbiota-gut-brain-axis. In Experimental Biology and Medicine (Vol. 246, Issue 7). https://doi.org/10.1177/1535370221995785
Berding, K., Vlckova, K., Marx, W., Schellekens, H., Stanton, C., Clarke, G., Jacka, F., Dinan, T. G., & Cryan, J. F. (2021). Diet and the Microbiota-Gut-Brain Axis: Sowing the Seeds of Good Mental Health. In Advances in Nutrition (Vol. 12, Issue 4). https://doi.org/10.1093/advances/nmaa181
Bessede, A., Gargaro, M., Pallotta, M. T., Matino, D., Servillo, G., Brunacci, C., Bicciato, S., Mazza, E. M. C., Macchiarulo, A., Vacca, C., Iannitti, R., Tissi, L., Volpi, C., Belladonna, M. L., Orabona, C., Bianchi, R., Lanz, T. V., Platten, M., Della Fazia, M. A., … Puccetti, P. (2014). Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature, 511(7508). https://doi.org/10.1038/nature13323
Bhatt, S., Devadoss, T., Jha, N. K., Baidya, M., Gupta, G., Chellappan, D. K., Singh, S. K., & Dua, K. (2023). Targeting inflammation: a potential approach for the treatment of depression. In Metabolic Brain Disease (Vol. 38, Issue 1). https://doi.org/10.1007/s11011-022-01095-1
Boyle, R. J., Robins-Browne, R. M., & Tang, M. L. K. (2006). Probiotic use in clinical practice: What are the risks? In American Journal of Clinical Nutrition (Vol. 83, Issue 6). https://doi.org/10.1093/ajcn/83.6.1256
Braidy, N., Grant, R., Adams, S., Brew, B. J., & Guillemin, G. J. (2009). Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotoxicity Research, 16(1). https://doi.org/10.1007/s12640-009-9051-z
Braniste, V., Al-Asmakh, M., Kowal, C., Anuar, F., Abbaspour, A., Tóth, M., Korecka, A., Bakocevic, N., Guan, N. L., Kundu, P., Gulyás, B., Halldin, C., Hultenby, K., Nilsson, H., Hebert, H., Volpe, B. T., Diamond, B., & Pettersson, S. (2014). Erratum: Erratum for the Research Article: The gut microbiota influences blood-brain barrier permeability in mice (Sci. Transl. Med. 6 266er7 (2014)). In Science Translational Medicine (Vol. 6, Issue 266). https://doi.org/10.1126/scitranslmed.aaa4288
Bravo, J. A., Forsythe, P., Chew, M. V., Escaravage, E., Savignac, H. M., Dinan, T. G., Bienenstock, J., & Cryan, J. F. (2011). Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proceedings of the National Academy of Sciences of the United States of America, 108(38). https://doi.org/10.1073/pnas.1102999108
Breit, S., Kupferberg, A., Rogler, G., & Hasler, G. (2018). Vagus nerve as modulator of the brain-gut axis in psychiatric and inflammatory disorders. In Frontiers in Psychiatry (Vol. 9, Issue MAR). https://doi.org/10.3389/fpsyt.2018.00044
Calcia, M. A., Bonsall, D. R., Bloomfield, P. S., Selvaraj, S., Barichello, T., & Howes, O. D. (2016). Stress and neuroinflammation: A systematic review of the effects of stress on microglia and the implications for mental illness. In Psychopharmacology (Vol. 233, Issue 9). https://doi.org/10.1007/s00213-016-4218-9
Campbell, B. M., Charych, E., Lee, A. W., & Möller, T. (2014). Kynurenines in CNS disease: Regulation by inflammatory cytokines. In Frontiers in Neuroscience (Issue 8 FEB). https://doi.org/10.3389/fnins.2014.00012
Campbell, C., Kandalgaonkar, M. R., Golonka, R. M., Yeoh, B. S., Vijay-Kumar, M., & Saha, P. (2023). Crosstalk between Gut Microbiota and Host Immunity: Impact on Inflammation and Immunotherapy. In Biomedicines (Vol. 11, Issue 2). https://doi.org/10.3390/biomedicines11020294
Capuron, L., & Miller, A. H. (2011). Immune system to brain signaling: Neuropsychopharmacological implications. In Pharmacology and Therapeutics (Vol. 130, Issue 2). https://doi.org/10.1016/j.pharmthera.2011.01.014
Chen, L., Li, H., Li, J., Chen, Y., & Yang, Y. (2019). Lactobacillus rhamnosus GG treatment improves intestinal permeability and modulates microbiota dysbiosis in an experimental model of sepsis. International Journal of Molecular Medicine, 43(3). https://doi.org/10.3892/ijmm.2019.4050
Chen, L. M., Bao, C. H., Wu, Y., Liang, S. H., Wang, D., Wu, L. Y., Huang, Y., Liu, H. R., & Wu, H. G. (2021). Tryptophan-kynurenine metabolism: a link between the gut and brain for depression in inflammatory bowel disease. In Journal of Neuroinflammation (Vol. 18, Issue 1). https://doi.org/10.1186/s12974-021-02175-2
Chen, Y., Jin, Y., Stanton, C., Paul Ross, R., Zhao, J., Zhang, H., Yang, B., & Chen, W. (2021). Alleviation effects of Bifidobacterium breve on DSS-induced colitis depends on intestinal tract barrier maintenance and gut microbiota modulation. European Journal of Nutrition, 60(1). https://doi.org/10.1007/s00394-020-02252-x
Ciapała, K., Mika, J., & Rojewska, E. (2021). The kynurenine pathway as a potential target for neuropathic pain therapy design: From basic research to clinical perspectives. In International Journal of Molecular Sciences (Vol. 22, Issue 20). https://doi.org/10.3390/ijms222011055
Clarke, G., Grenham, S., Scully, P., Fitzgerald, P., Moloney, R. D., Shanahan, F., Dinan, T. G., & Cryan, J. F. (2013). The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Molecular Psychiatry, 18(6). https://doi.org/10.1038/mp.2012.77
Colbey, C., Cox, A. J., Pyne, D. B., Zhang, P., Cripps, A. W., & West, N. P. (2018). Upper Respiratory Symptoms, Gut Health and Mucosal Immunity in Athletes. In Sports Medicine (Vol. 48). https://doi.org/10.1007/s40279-017-0846-4
Coxon, J. P., Cash, R. F. H., Hendrikse, J. J., Rogasch, N. C., Stavrinos, E., Suo, C., & Yücel, M. (2018). GABA concentration in sensorimotor cortex following high-intensity exercise and relationship to lactate levels. Journal of Physiology, 596(4). https://doi.org/10.1113/JP274660
Dantzer, R. (2009). Cytokine, Sickness Behavior, and Depression. In Immunology and Allergy Clinics of North America (Vol. 29, Issue 2). https://doi.org/10.1016/j.iac.2009.02.002
Deng, Y., Gong, P., Han, S., Zhang, J., Zhang, S., Zhang, B., Lin, Y., Xu, K., Wen, G., & Liu, K. (2023). Reduced cerebral cortex thickness is related to overexpression of exosomal miR-146a-5p in medication-free patients with major depressive disorder. Psychological Medicine, 53(13). https://doi.org/10.1017/S0033291722003567
Di Tommaso, N., Gasbarrini, A., & Ponziani, F. R. (2021). Intestinal barrier in human health and disease. In International Journal of Environmental Research and Public Health (Vol. 18, Issue 23). https://doi.org/10.3390/ijerph182312836
Dinan, T. G., & Cryan, J. F. (2017a). Gut instincts: microbiota as a key regulator of brain development, ageing and neurodegeneration. Journal of Physiology, 595(2). https://doi.org/10.1113/JP273106
Dinan, T. G., & Cryan, J. F. (2017b). The Microbiome-Gut-Brain Axis in Health and Disease. In Gastroenterology Clinics of North America (Vol. 46, Issue 1). https://doi.org/10.1016/j.gtc.2016.09.007
Dinan, T. G., Stanton, C., & Cryan, J. F. (2013). Psychobiotics: A novel class of psychotropic. In Biological Psychiatry (Vol. 74, Issue 10). https://doi.org/10.1016/j.biopsych.2013.05.001
Donley, D. W., Olson, A. R., Raisbeck, M. F., Fox, J. H., & Gigley, J. P. (2016). Huntingtons disease mice infected with toxoplasma gondii demonstrate early kynurenine pathway activation, altered CD8+ T-Cell responses, and premature mortality. PLoS ONE, 11(9). https://doi.org/10.1371/journal.pone.0162404
El Aidy, S., Dinan, T. G., & Cryan, J. F. (2015). Gut Microbiota: The Conductor in the Orchestra of Immune-Neuroendocrine Communication. In Clinical Therapeutics (Vol. 37, Issue 5). https://doi.org/10.1016/j.clinthera.2015.03.002
Erny, D., De Angelis, A. L. H., Jaitin, D., Wieghofer, P., Staszewski, O., David, E., Keren-Shaul, H., Mahlakoiv, T., Jakobshagen, K., Buch, T., Schwierzeck, V., Utermöhlen, O., Chun, E., Garrett, W. S., Mccoy, K. D., Diefenbach, A., Staeheli, P., Stecher, B., Amit, I., & Prinz, M. (2015). Host microbiota constantly control maturation and function of microglia in the CNS. Nature Neuroscience, 18(7). https://doi.org/10.1038/nn.4030
Fang, X., Yue, M., Wei, J., Wang, Y., Hong, D., Wang, B., Zhou, X., & Chen, T. (2021). Evaluation of the Anti-Aging Effects of a Probiotic Combination Isolated From Centenarians in a SAMP8 Mouse Model. Frontiers in Immunology, 12. https://doi.org/10.3389/fimmu.2021.792746
FARQUHAR, M. G., & PALADE, G. E. (1963). Junctional complexes in various epithelia. The Journal of Cell Biology, 17. https://doi.org/10.1083/jcb.17.2.375
Fathi, M., Vakili, K., Yaghoobpoor, S., Tavasol, A., Jazi, K., Hajibeygi, R., Shool, S., Sodeifian, F., Klegeris, A., McElhinney, A., Tavirani, M. R., & Sayehmiri, F. (2022). Dynamic changes in metabolites of the kynurenine pathway in Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: A systematic Review and meta-analysis. In Frontiers in Immunology (Vol. 13). https://doi.org/10.3389/fimmu.2022.997240
Felger, J. C. (2017). The role of dopamine in inflammation-associated depression: Mechanisms and therapeutic implications. In Current Topics in Behavioral Neurosciences (Vol. 31). https://doi.org/10.1007/7854_2016_13
Felger, J. C., & Treadway, M. T. (2017). Inflammation Effects on Motivation and Motor Activity: Role of Dopamine. In Neuropsychopharmacology (Vol. 42, Issue 1). https://doi.org/10.1038/npp.2016.143
Fourrier, C., Sampson, E., Mills, N. T., & Baune, B. T. (2018). Anti-inflammatory treatment of depression: Study protocol for a randomised controlled trial of vortioxetine augmented with celecoxib or placebo. Trials, 19(1). https://doi.org/10.1186/s13063-018-2829-7
Fröhlich, E. E., Farzi, A., Mayerhofer, R., Reichmann, F., Jačan, A., Wagner, B., Zinser, E., Bordag, N., Magnes, C., Fröhlich, E., Kashofer, K., Gorkiewicz, G., & Holzer, P. (2016). Cognitive impairment by antibiotic-induced gut dysbiosis: Analysis of gut microbiota-brain communication. Brain, Behavior, and Immunity, 56. https://doi.org/10.1016/j.bbi.2016.02.020
Ghosh, S. S., Wang, J., Yannie, P. J., & Ghosh, S. (2020). Intestinal barrier dysfunction, LPS translocation, and disease development. Journal of the Endocrine Society, 4(2). https://doi.org/10.1210/jendso/bvz039
Giovanoli, S., Engler, H., Engler, A., Richetto, J., Voget, M., Willi, R., Winter, C., Riva, M. A., Mortensen, P. B., Schedlowski, M., & Meyer, U. (2013). Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. In Science (Vol. 339, Issue 6123). https://doi.org/10.1126/science.1228261
Gómez-Eguílaz, M., Ramón-Trapero, J. L., Pérez-Martínez, L., & Blanco, J. R. (2019). The microbiota-gut-brain axis and its great projections. Revista de Neurologia, 68(3). https://doi.org/10.33588/rn.6803.2018223
Gomez-Eguilaz, M., Ramon-Trapero, J. L., Perez-Martinez, L., & Blanco, J. R. (2019). [The microbiota-gut-brain axis and its great projections]. TT - El eje microbiota-intestino-cerebro y sus grandes proyecciones. Rev Neurol, 68(3).
González-Sánchez, M., Jiménez, J., Narváez, A., Antequera, D., Llamas-Velasco, S., Martín, A. H. S., Arjona, J. A. M., Munain, A. L. de, Bisa, A. L., Marco, M. P., Rodríguez-Núñez, M., Pérez-Martínez, D. A., Villarejo-Galende, A., Bartolome, F., Domínguez, E., & Carro, E. (2020). Kynurenic acid levels are increased in the CSF of Alzheimer’s disease patients. Biomolecules, 10(4). https://doi.org/10.3390/biom10040571
Guillemin, G. J., & Brew, B. J. (2002). Implications of the kynurenine pathway and quinolinic acid in Alzheimer’s disease. In Redox Report (Vol. 7, Issue 4). https://doi.org/10.1179/135100002125000550
Han, H., You, Y., Cha, S., Kim, T. R., Sohn, M., & Park, J. (2023). Multi-Species Probiotic Strain Mixture Enhances Intestinal Barrier Function by Regulating Inflammation and Tight Junctions in Lipopolysaccharides Stimulated Caco-2 Cells. Microorganisms, 11(3). https://doi.org/10.3390/microorganisms11030656
Haroun, E., Kumar, P. A., Saba, L., Kassab, J., Ghimire, K., Dutta, D., & Lim, S. H. (2023). Intestinal barrier functions in hematologic and oncologic diseases. In Journal of Translational Medicine (Vol. 21, Issue 1). https://doi.org/10.1186/s12967-023-04091-w
Heenan, P. E., Keenan, J. I., Bayer, S., Simon, M., & Gearry, R. B. (2020). Irritable bowel syndrome and the gut microbiota. In Journal of the Royal Society of New Zealand (Vol. 50, Issue 3). https://doi.org/10.1080/03036758.2019.1695635
Hestad, K., Alexander, J., Rootwelt, H., & Aaseth, J. O. (2022). The Role of Tryptophan Dysmetabolism and Quinolinic Acid in Depressive and Neurodegenerative Diseases. In Biomolecules (Vol. 12, Issue 7). https://doi.org/10.3390/biom12070998
Holscher, H. D. (2017). Dietary fiber and prebiotics and the gastrointestinal microbiota. In Gut Microbes (Vol. 8, Issue 2). https://doi.org/10.1080/19490976.2017.1290756
Holzscheiter, M., Layland, L. E., Loffredo-Verde, E., Mair, K., Vogelmann, R., Langer, R., Wagner, H., & Prazeres da Costa, C. (2014). Lack of host gut microbiota alters immune responses and intestinal granuloma formation during schistosomiasis. Clinical and Experimental Immunology, 175(2). https://doi.org/10.1111/cei.12230
Hooper, L. V., Littman, D. R., & Macpherson, A. J. (2012). Interactions between the microbiota and the immune system. In Science (Vol. 336, Issue 6086). https://doi.org/10.1126/science.1223490
Huang, F., & Wu, X. (2021). Brain Neurotransmitter Modulation by Gut Microbiota in Anxiety and Depression. In Frontiers in Cell and Developmental Biology (Vol. 9). https://doi.org/10.3389/fcell.2021.649103
Huang, R., Wang, K., & Hu, J. (2016). Effect of probiotics on depression: A systematic review and meta-analysis of randomized controlled trials. In Nutrients (Vol. 8, Issue 8). https://doi.org/10.3390/nu8080483
Hubbard, T. D., Murray, I. A., & Perdew, G. H. (2015). Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metabolism and Disposition, 43(10). https://doi.org/10.1124/dmd.115.064246
Hughes, T. D., Güner, O. F., Iradukunda, E. C., Phillips, R. S., & Bowen, J. P. (2022). The Kynurenine Pathway and Kynurenine 3-Monooxygenase Inhibitors. In Molecules (Vol. 27, Issue 1). https://doi.org/10.3390/molecules27010273
Jaronen, M., & Quintana, F. J. (2014). Immunological relevance of the coevolution of IDO1 and AHR. In Frontiers in Immunology (Vol. 5, Issue OCT). https://doi.org/10.3389/fimmu.2014.00521
Johansson, M. E. V., & Hansson, G. C. (2016). Immunological aspects of intestinal mucus and mucins. In Nature Reviews Immunology (Vol. 16, Issue 10). https://doi.org/10.1038/nri.2016.88
Kaistha, S. D., & Deshpande, N. (2021). Traditional Probiotics, Next-Generation Probiotics and Engineered Live Biotherapeutic Products in Chronic Wound Healing. In Wound Healing Research: Current Trends and Future Directions. https://doi.org/10.1007/978-981-16-2677-7_8
Kennedy, P. J., Clarke, G., Quigley, E. M. M., Groeger, J. A., Dinan, T. G., & Cryan, J. F. (2012). Gut memories: Towards a cognitive neurobiology of irritable bowel syndrome. In Neuroscience and Biobehavioral Reviews (Vol. 36, Issue 1). https://doi.org/10.1016/j.neubiorev.2011.07.001
Kennedy, P. J., Cryan, J. F., Dinan, T. G., & Clarke, G. (2017). Kynurenine pathway metabolism and the microbiota-gut-brain axis. In Neuropharmacology (Vol. 112). https://doi.org/10.1016/j.neuropharm.2016.07.002
Keszthelyi, D., Troost, F. J., Jonkers, D. M., Van Donkelaar, E. L., Dekker, J., Buurman, W. A., & Masclee, A. A. (2012). Does acute tryptophan depletion affect peripheral serotonin metabolism in the intestine? American Journal of Clinical Nutrition, 95(3). https://doi.org/10.3945/ajcn.111.028589
Kim, C. S., Cha, L., Sim, M., Jung, S., Chun, W. Y., Baik, H. W., & Shin, D. M. (2021). Probiotic supplementation improves cognitive function and mood with changes in gut microbiota in community- dwelling older adults: A randomized, double-blind, placebo-controlled, multicenter trial. Journals of Gerontology - Series A Biological Sciences and Medical Sciences, 76(1). https://doi.org/10.1093/GERONA/GLAA090
Köhler, A., Delbauve, S., Smout, J., Torres, D., & Flamand, V. (2021). Very early-life exposure to microbiota-induced TNF drives the maturation of neonatal pre-cDC1. Gut, 70(3). https://doi.org/10.1136/gutjnl-2019-319700
Konsman, J. P. (2022). Cytokines in the Brain and Neuroinflammation: We Didn’t Starve the Fire! In Pharmaceuticals (Vol. 15, Issue 2). https://doi.org/10.3390/ph15020140
Kvichansky, A. A., Volobueva, M. N., Spivak, Y. S., Tret’yakova, L. V., Gulyaeva, N. V., & Bolshakov, A. P. (2019). Expression of mRNAs for IL-1β, IL-6, IL-10, TNFα, CX3CL1, and TGFβ1 Cytokines in the Brain Tissues: Assessment of Contribution of Blood Cells with and without Perfusion. Biochemistry (Moscow), 84(8). https://doi.org/10.1134/S0006297919080066
Latif, A., Shehzad, A., Niazi, S., Zahid, A., Ashraf, W., Iqbal, M. W., Rehman, A., Riaz, T., Aadil, R. M., Khan, I. M., Özogul, F., Rocha, J. M., Esatbeyoglu, T., & Korma, S. A. (2023). Probiotics: mechanism of action, health benefits and their application in food industries. In Frontiers in Microbiology (Vol. 14). https://doi.org/10.3389/fmicb.2023.1216674
Lee, B., Moon, K. M., & Kim, C. Y. (2018). Tight junction in the intestinal epithelium: Its association with diseases and regulation by phytochemicals. In Journal of Immunology Research (Vol. 2018). https://doi.org/10.1155/2018/2645465
Lee, Y. Y., Leow, A. H. R., Chai, P. F., Raja Ali, R. A., Lee, W. S., & Goh, K. L. (2021). Use of probiotics in clinical practice with special reference to diarrheal diseases: A position statement of the Malaysian Society of Gastroenterology and Hepatology. In JGH Open (Vol. 5, Issue 1). https://doi.org/10.1002/jgh3.12469
Li, H. Y., Zhou, D. D., Gan, R. Y., Huang, S. Y., Zhao, C. N., Shang, A., Xu, X. Y., & Li, H. Bin. (2021). Effects and mechanisms of probiotics, prebiotics, synbiotics, and postbiotics on metabolic diseases targeting gut microbiota: A narrative review. In Nutrients (Vol. 13, Issue 9). https://doi.org/10.3390/nu13093211
Liu, H., Wu, X., Wang, Y., Liu, X., Peng, D., Wu, Y., Chen, J., Su, Y., Xu, J., Ma, X., Li, Y., Shi, J., Yang, X., Rong, H., Forti, M. Di, & Fang, Y. (2022). TNF-α, IL-6 and hsCRP in patients with melancholic, atypical and anxious depression: an antibody array analysis related to somatic symptoms. General Psychiatry, 35(4). https://doi.org/10.1136/gpsych-2022-100844
Louwies, T., Johnson, A. C., Orock, A., Yuan, T., & Greenwood-Van Meerveld, B. (2020). The microbiota-gut-brain axis: An emerging role for the epigenome. In Experimental Biology and Medicine (Vol. 245, Issue 2). https://doi.org/10.1177/1535370219891690
Mardinoglu, A., Boren, J., & Smith, U. (2016). Confounding Effects of Metformin on the Human Gut Microbiome in Type 2 Diabetes. In Cell Metabolism (Vol. 23, Issue 1). https://doi.org/10.1016/j.cmet.2015.12.012
Markowiak, P., & Ślizewska, K. (2017). Effects of probiotics, prebiotics, and synbiotics on human health. In Nutrients (Vol. 9, Issue 9). https://doi.org/10.3390/nu9091021
Marx, W., Scholey, A., Firth, J., D’Cunha, N. M., Lane, M., Hockey, M., Ashton, M. M., Cryan, J. F., O’Neil, A., Naumovski, N., Berk, M., Dean, O. M., & Jacka, F. (2020). Prebiotics, probiotics, fermented foods and cognitive outcomes: A meta-analysis of randomized controlled trials. In Neuroscience and Biobehavioral Reviews (Vol. 118). https://doi.org/10.1016/j.neubiorev.2020.07.036
Megur, A., Baltriukienė, D., Bukelskienė, V., & Burokas, A. (2021). The microbiota–gut–brain axis and Alzheimer’s disease: Neuroinflammation is to blame? In Nutrients (Vol. 13, Issue 1). https://doi.org/10.3390/nu13010037
Min, S., Than, N., Shin, Y. C., Hu, G., Shin, W., Ambrosini, Y. M., & Kim, H. J. (2022). Live probiotic bacteria administered in a pathomimetic Leaky Gut Chip ameliorate impaired epithelial barrier and mucosal inflammation. Scientific Reports, 12(1). https://doi.org/10.1038/s41598-022-27300-w
Mithaiwala, M. N., Santana-Coelho, D., Porter, G. A., & O’connor, J. C. (2021). Neuroinflammation and the Kynurenine pathway in CNS disease: Molecular mechanisms and therapeutic implications. In Cells (Vol. 10, Issue 6). https://doi.org/10.3390/cells10061548
M.M.A., A., C., G., Y.G., X., Y., C., & H., S. (2016). Factors controlling permeability of the blood-brain barrier. Cellular and Molecular Life Sciences, 73(1).
Moffett, J. R., Arun, P., Puthillathu, N., Vengilote, R., Ives, J. A., Badawy, A. A. B., & Namboodiri, A. M. (2020). Quinolinate as a Marker for Kynurenine Metabolite Formation and the Unresolved Question of NAD+ Synthesis During Inflammation and Infection. Frontiers in Immunology, 11. https://doi.org/10.3389/fimmu.2020.00031
Mor, A., Tankiewicz-Kwedlo, A., Krupa, A., & Pawlak, D. (2021). Role of kynurenine pathway in oxidative stress during neurodegenerative disorders. In Cells (Vol. 10, Issue 7). https://doi.org/10.3390/cells10071603
Mörkl, S., Butler, M. I., Holl, A., Cryan, J. F., & Dinan, T. G. (2020). Correction to: Probiotics and the Microbiota-Gut-Brain Axis: Focus on Psychiatry (Current Nutrition Reports, (2020), 9, 3, (171–182), https://doi.org/10.1007/s13668-020-00313-5). In Current Nutrition Reports (Vol. 9, Issue 3). https://doi.org/10.1007/s13668-020-00319-z
Mujagic, Z., De Vos, P., Boekschoten, M. V., Govers, C., Pieters, H. J. H. M., De Wit, N. J. W., Bron, P. A., Masclee, A. A. M., & Troost, F. J. (2017). The effects of Lactobacillus plantarum on small intestinal barrier function and mucosal gene transcription; A randomized double-blind placebo controlled trial. Scientific Reports, 7. https://doi.org/10.1038/srep40128
Nettleton, J. E., Klancic, T., Schick, A., Choo, A. C., Cheng, N., Shearer, J., Borgland, S. L., Rho, J. M., & Reimer, R. A. (2021). Prebiotic, probiotic, and synbiotic consumption alter behavioral variables and intestinal permeability and microbiota in btbr mice. Microorganisms, 9(9). https://doi.org/10.3390/microorganisms9091833
Nishida, A., Inoue, R., Inatomi, O., Bamba, S., Naito, Y., & Andoh, A. (2018). Gut microbiota in the pathogenesis of inflammatory bowel disease. In Clinical Journal of Gastroenterology (Vol. 11, Issue 1). https://doi.org/10.1007/s12328-017-0813-5
Odenwald, M. A., & Turner, J. R. (2017). The intestinal epithelial barrier: A therapeutic target? In Nature Reviews Gastroenterology and Hepatology (Vol. 14, Issue 1). https://doi.org/10.1038/nrgastro.2016.169
O’Hara, A. M., & Shanahan, F. (2006). The gut flora as a forgotten organ. In EMBO Reports (Vol. 7, Issue 7). https://doi.org/10.1038/sj.embor.7400731
Okumura, R., & Takeda, K. (2017). Roles of intestinal epithelial cells in the maintenance of gut homeostasis. In Experimental and Molecular Medicine (Vol. 49, Issue 5). https://doi.org/10.1038/emm.2017.20
O’Mahony, S. M., Clarke, G., Borre, Y. E., Dinan, T. G., & Cryan, J. F. (2015). Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. In Behavioural Brain Research (Vol. 277). https://doi.org/10.1016/j.bbr.2014.07.027
Park, J. M., Lee, S. C., Ham, C., & Kim, Y. W. (2023). Effect of probiotic supplementation on gastrointestinal motility, inflammation, motor, non-motor symptoms and mental health in Parkinson’s disease: a meta-analysis of randomized controlled trials. Gut Pathogens, 15(1). https://doi.org/10.1186/s13099-023-00536-1
Parker, A., Fonseca, S., & Carding, S. R. (2020). Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. In Gut Microbes (Vol. 11, Issue 2). https://doi.org/10.1080/19490976.2019.1638722
Peirce, J. M., & Alviña, K. (2019). The role of inflammation and the gut microbiome in depression and anxiety. In Journal of Neuroscience Research (Vol. 97, Issue 10). https://doi.org/10.1002/jnr.24476
Peng, X., Luo, Z., He, S., Zhang, L., & Li, Y. (2021). Blood-Brain Barrier Disruption by Lipopolysaccharide and Sepsis-Associated Encephalopathy. In Frontiers in Cellular and Infection Microbiology (Vol. 11). https://doi.org/10.3389/fcimb.2021.768108
Pinto-Sanchez, M. I., Hall, G. B., Ghajar, K., Nardelli, A., Bolino, C., Lau, J. T., Martin, F. P., Cominetti, O., Welsh, C., Rieder, A., Traynor, J., Gregory, C., De Palma, G., Pigrau, M., Ford, A. C., Macri, J., Berger, B., Bergonzelli, G., Surette, M. G., … Bercik, P. (2017). Probiotic Bifidobacterium longum NCC3001 Reduces Depression Scores and Alters Brain Activity: A Pilot Study in Patients With Irritable Bowel Syndrome. Gastroenterology, 153(2). https://doi.org/10.1053/j.gastro.2017.05.003
Plaza-Diaz, J., Ruiz-Ojeda, F. J., Gil-Campos, M., & Gil, A. (2019). Mechanisms of Action of Probiotics. Advances in Nutrition, 10. https://doi.org/10.1093/advances/nmy063
Plaza-Díaz, J., Ruiz-Ojeda, F. J., Vilchez-Padial, L. M., & Gil, A. (2017). Evidence of the anti-inflammatory effects of probiotics and synbiotics in intestinal chronic diseases. In Nutrients (Vol. 9, Issue 6). https://doi.org/10.3390/nu9060555
Purton, T., Staskova, L., Lane, M. M., Dawson, S. L., West, M., Firth, J., Clarke, G., Cryan, J. F., Berk, M., O’Neil, A., Dean, O., Hadi, A., Honan, C., & Marx, W. (2021). Prebiotic and probiotic supplementation and the tryptophan-kynurenine pathway: A systematic review and meta analysis. In Neuroscience and Biobehavioral Reviews (Vol. 123). https://doi.org/10.1016/j.neubiorev.2020.12.026
Pykhtina, V. S. (2023). The role of kynurenine pathway metabolites in the development of frailty in older adults. Problems of Geroscience, 1. https://doi.org/10.37586/2949-4745-1-2023-15-24
Rogers, G. B., Keating, D. J., Young, R. L., Wong, M. L., Licinio, J., & Wesselingh, S. (2016). From gut dysbiosis to altered brain function and mental illness: Mechanisms and pathways. In Molecular Psychiatry (Vol. 21, Issue 6). https://doi.org/10.1038/mp.2016.50
Rudzki, L., Ostrowska, L., Pawlak, D., Małus, A., Pawlak, K., Waszkiewicz, N., & Szulc, A. (2019). Probiotic Lactobacillus Plantarum 299v decreases kynurenine concentration and improves cognitive functions in patients with major depression: A double-blind, randomized, placebo controlled study. Psychoneuroendocrinology, 100. https://doi.org/10.1016/j.psyneuen.2018.10.010
Sakurai, K., Toshimitsu, T., Okada, E., Anzai, S., Shiraishi, I., Inamura, N., Kobayashi, S., Sashihara, T., & Hisatsune, T. (2022). Effects of Lactiplantibacillus plantarum OLL2712 on Memory Function in Older Adults with Declining Memory: A Randomized Placebo-Controlled Trial. Nutrients, 14(20). https://doi.org/10.3390/nu14204300
Sarkar, A., Lehto, S. M., Harty, S., Dinan, T. G., Cryan, J. F., & Burnet, P. W. J. (2016). Psychobiotics and the Manipulation of Bacteria–Gut–Brain Signals. In Trends in Neurosciences (Vol. 39, Issue 11). https://doi.org/10.1016/j.tins.2016.09.002
Schwartz, C. E. (2014). Aberrant tryptophan metabolism: The unifying biochemical basis for autism spectrum disorders? In Biomarkers in Medicine (Vol. 8, Issue 3). https://doi.org/10.2217/bmm.14.11
Setiawan, E., Wilson, A. A., Mizrahi, R., Rusjan, P. M., Miler, L., Rajkowska, G., Suridjan, I., Kennedy, J. L., Rekkas, P. V., Houle, S., & Meyer, J. H. (2015). Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry, 72(3). https://doi.org/10.1001/jamapsychiatry.2014.2427
Silva, Y. P., Bernardi, A., & Frozza, R. L. (2020). The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. In Frontiers in Endocrinology (Vol. 11). https://doi.org/10.3389/fendo.2020.00025
Sivamaruthi, B. S., Suganthy, N., Kesika, P., & Chaiyasut, C. (2020). The role of microbiome, dietary supplements, and probiotics in autism spectrum disorder. In International Journal of Environmental Research and Public Health (Vol. 17, Issue 8). https://doi.org/10.3390/ijerph17082647
Sound, R. 2021. The gut microbiota–brain axis and role of probiotics. In Nutraceuticals in Brain Health and Beyond. https://doi.org/10.1016/b978-0-12-820593-8.00013-6.
Sturgeon, C., & Fasano, A. (2016). Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases. In Tissue Barriers (Vol. 4, Issue 4). https://doi.org/10.1080/21688370.2016.1251384
Suzuki, T. (2020). Regulation of the intestinal barrier by nutrients: The role of tight junctions. In Animal Science Journal (Vol. 91, Issue 1). https://doi.org/10.1111/asj.13357
Takiishi, T., Fenero, C. I. M., & Câmara, N. O. S. (2017). Intestinal barrier and gut microbiota: Shaping our immune responses throughout life. In Tissue Barriers (Vol. 5, Issue 4). https://doi.org/10.1080/21688370.2017.1373208
Vécsei, L., Szalárdy, L., Fülöp, F., & Toldi, J. (2013). Kynurenines in the CNS: Recent advances and new questions. In Nature Reviews Drug Discovery (Vol. 12, Issue 1). https://doi.org/10.1038/nrd3793
Venkatesan, D., Iyer, M., Narayanasamy, A., Siva, K., & Vellingiri, B. (2020). Kynurenine pathway in Parkinson’s disease—An update. In eNeurologicalSci (Vol. 21). https://doi.org/10.1016/j.ensci.2020.100270
Vogel, C. F. A., Goth, S. R., Dong, B., Pessah, I. N., & Matsumura, F. (2008). Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochemical and Biophysical Research Communications, 375(3). https://doi.org/10.1016/j.bbrc.2008.07.156
Wang, C., Li, Q., & Ren, J. (2019). Microbiota-immune interaction in the pathogenesis of gut-derived infection. In Frontiers in Immunology (Vol. 10, Issue AUG). https://doi.org/10.3389/fimmu.2019.01873
Wang, G., Efstratiou, A., Adjou Moumouni, P. F., Liu, M., Jirapattharasate, C., Guo, H., Gao, Y., Cao, S., Zhou, M., Suzuki, H., Igarashi, I., & Xuan, X. (2016). Primary Babesia rodhaini infection followed by recovery confers protective immunity against B. rodhaini reinfection and Babesia microti challenge infection in mice. Experimental Parasitology, 169. https://doi.org/10.1016/j.exppara.2016.07.003
Wang, S., Wang, B., Mishra, S., Jain, S., Ding, J., Krtichevsky, S., Kitzman, D., & Yadav, H. (2021). A novel probiotics therapy for aging-related leaky gut and inflammation. Innovation in Aging, 5(Supplement_1), 668–669. https://doi.org/10.1093/geroni/igab046.2521
Wang, X., Wang, Z., Cao, J., Dong, Y., & Chen, Y. (2023). Gut microbiota-derived metabolites mediate the neuroprotective effect of melatonin in cognitive impairment induced by sleep deprivation. Microbiome, 11(1). https://doi.org/10.1186/s40168-022-01452-3
Wells, J. M., Brummer, R. J., Derrien, M., MacDonald, T. T., Troost, F., Cani, P. D., Theodorou, V., Dekker, J., Méheust, A., De Vos, W. M., Mercenier, A., Nauta, A., & Garcia-Rodenas, C. L. (2017). Homeostasis of the gut barrier and potential biomarkers. In American Journal of Physiology - Gastrointestinal and Liver Physiology (Vol. 312, Issue 3). https://doi.org/10.1152/ajpgi.00048.2015
Xiang, S., Ji, J. L., Li, S., Cao, X. P., Xu, W., Tan, L., & Tan, C. C. (2022). Efficacy and Safety of Probiotics for the Treatment of Alzheimer’s Disease, Mild Cognitive Impairment, and Parkinson’s Disease: A Systematic Review and Meta-Analysis. In Frontiers in Aging Neuroscience (Vol. 14). https://doi.org/10.3389/fnagi.2022.730036
Zheng, D., Liwinski, T., & Elinav, E. (2020). Interaction between microbiota and immunity in health and disease. In Cell Research (Vol. 30, Issue 6). https://doi.org/10.1038/s41422-020-0332-7
Zheng, N., Gao, Y., Zhu, W., Meng, D., & Allan Walker, W. (2020). Short chain fatty acids produced by colonizing intestinal commensal bacterial interaction with expressed breast milk are anti-inflammatory in human immature enterocytes. PLoS ONE, 15(2). https://doi.org/10.1371/journal.pone.0229283
Zhu, G., Zhao, J., Zhang, H., Chen, W., & Wang, G. (2021). Probiotics for mild cognitive impairment and alzheimer’s disease: A systematic review and meta-analysis. In Foods (Vol. 10, Issue 7). https://doi.org/10.3390/foods10071672
Zhu, H., Wang, W., & Li, Y. (2024). The interplay between microbiota and brain-gut axis in epilepsy treatment. In Frontiers in Pharmacology (Vol. 15). https://doi.org/10.3389/fphar.2024.1276551
Funding
Ulster University
Author information
Authors and Affiliations
Contributions
RK was the only author on this article.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kearns, R. The Kynurenine Pathway in Gut Permeability and Inflammation. Inflammation (2024). https://doi.org/10.1007/s10753-024-02135-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10753-024-02135-x