
Abstract
Autoimmune diseases are typically characterized by aberrant activation of immune system that leads to excessive inflammatory reactions and tissue damage. Nevertheless, precise targeted and efficient therapies are limited. Thus, studies into novel therapeutic targets for the management of autoimmune diseases are urgently needed. Radical S-adenosyl methionine domain-containing 2 (RSAD2) is an interferon-stimulated gene (ISG) renowned for the antiviral properties of the protein it encodes, named viperin. An increasing number of studies have underscored the new roles of RSAD2/viperin in immunomodulation and mitochondrial metabolism. Previous studies have shown that there is a complex interplay between RSAD2/vipeirn and mitochondria and that binding of the iron-sulfur (Fe-S) cluster is necessary for the involvement of viperin in mitochondrial metabolism. Viperin influences the proliferation and development of immune cells as well as inflammation via different signaling pathways. However, the function of RSAD2/viperin varies in different studies and a comprehensive overview of this emerging theme is lacking. This review will describe the characteristics of RSAD2/viperin, decipher its function in immunometabolic processes, and clarify the crosstalk between RSAD2/viperin and mitochondria. Furthermore, we emphasize the crucial roles of RSAD2 in autoimmune diseases and its potential application value.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Autoimmune diseases are a complicated group of chronic inflammatory diseases characterized by the inability to distinguish self-antigens from foreign antigens, which ultimately leads to the collapse of immunological tolerance [1]. Currently, uncertainties still exist regarding the underlying mechanisms of autoimmune diseases. Clinical treatment of autoimmune diseases focuses mainly on alleviating the autoimmune response and tissue damage. Nevertheless, the current generation of immunomodulatory drugs exhibits broad-spectrum and non-disease-specific activity, which often results in the occurrence of adverse effects [2]. Consequently, there is an urgent need to develop novel targets for precise intervention and personalized treatment.
Radical S-adenosyl methionine domain-containing 2 (RSAD2), also known as cytomegalovirus-induced gene 5 (cig5), is an interferon (IFN)-induced gene and plays an essential role in innate and adaptive immunity. Viperin, encoded by RSAD2, is a well-known enzyme to inhibit viral replication [3]. Up to date, RSAD2 has been demonstrated to be a potential biomarker in many autoimmune diseases according to the predicted results of bioinformatics, such as rheumatoid arthritis (RA) [4], systemic lupus erythematosus (SLE) [5] and primary Sjögren's syndrome (pSS) [6]. Thus, RSAD2 might possess efficacy in disease prediction and diagnosis as a promising biomarker. However, its performance in the modulation of immunity and mitochondrial metabolism encourages us to further investigate its potential as a therapeutic target.
Accumulating studies have reported a close relationship between RSAD2/viperin and various signaling pathways. For instance, viperin affects Toll-like receptor-7 and -9 (TLR7/9) signaling pathways in plasmacytoid dendritic cells (pDCs), which in turn triggers polyubiquitination of interleukin-1 receptor-associated kinase (IRAK1) and interferon regulatory factor 7 (IRF7)-mediated transcriptional activation of type I IFN (IFN-I) [7]. Similarly, viperin stimulates the production of IFN-I by interacting with tank-binding kinase 1 (TBK1) and stimulator of interferon genes (STING) [8]. The continuous generation of IFN-I may be associated with aberrant immune activation, which contributes to the inflammatory response and tissue destruction observed in autoimmune diseases [9]. Furthermore, there is an interaction between viperin and peroxisomal biogenesis factor 19 (Pex19), which ultimately strengthens the retinoic acid-inducible gene I (RIG-I)-mitochondrial antiviral signaling protein (MAVS) signaling pathway and promotes the antiviral response [10]. Interestingly, studies in this area have revealed that the peripheral blood lymphocytes from patients with SLE exhibit spontaneous MAVS oligomerization, which was associated with the elevated secretion of IFN and mitochondrial oxidative stress [11]. Accordingly, it is reasonable to suspect that viperin may modulate MAVS signaling and downstream immune responses in SLE, influencing the production of IFN-I and other inflammatory mediators. Therefore, deeper insights into the intricate relationship between viperin and the associated signaling pathways may open new avenues for targeted treatment of diseases.
Notably, during human cytomegalovirus (HCMV) infection, viperin affects mitochondrial metabolism when it is translocated from the endoplasmic reticulum (ER) to the matrix of the mitochondrion by binding the viral mitochondrion-localized inhibitor of apoptosis (vMIA) [12]. Viperin affects mitochondrial activity by targeting mitochondria via its N-terminal region, and it is involved in lipid and glucose metabolism [12, 13]. In addition, Fe-S clusters are believed to be essential for the proper functioning of viperin. Three conserved cysteine residues in the structure of viperin are responsible for the formation of a redox-active [4Fe-4S] cluster [14]. By binding to viperin, the [4Fe-4S] cluster influences immunological response, antiviral effects, and mitochondrial function [3, 15]. The multifaceted properties of RSAD2/viperin in signaling pathways point to its significant regulatory role in maintaining the balance of the immune system and mitochondrial metabolism.
While extensive research has described the function of RSAD2/viperin in infectious diseases, the crucial role that RSAD2 plays in autoimmune diseases remains poorly understood. Therefore, this review endeavors to outline the characteristics of RSAD2/viperin, including its structure and signaling pathways. Furthermore, the roles and mechanisms of RSAD2/viperin in immunomodulation and mitochondrial metabolism are highlighted. To provide the overall landscape of the relationship between RSAD2/viperin and autoimmune diseases, we discuss its function and the possible mechanisms by which RSAD2/viperin influences the etiology and progression of autoimmune diseases, thereby providing a novel target for the treatment of autoimmune diseases.
Characteristics of RSAD2/viperin
Structural Features of RSAD2/viperin
RSAD2, situated on human chromosome 2p25.2, consists of a full-length 2858 bp DNA sequence. It contains an open reading frame of 1083 nucleotides, encoding 361 amino acids with a molecular weight of 42.17 kDa (Fig. 1). With slight variations between species but overall high conservation, viperin encompasses three distinct domains: the N-terminal alpha-helix domain, the central S-adenosyl methionine (SAM) domain, and the conserved C-terminal domain. The N-terminal domain features an amphipathic alpha-helix, which varies greatly depending on the species. This alpha-helix, anchored to the lipid layer, facilitates the distribution of viperin to various cellular sites such as the ER membrane, Golgi apparatus, and lipid droplets [16, 17]. Viperin also exerts its antiviral effect via lipid droplet-related mechanisms. It binds to and inhibits farnesyl diphosphate synthase, thereby disrupting lipid rafts and suppressing the budding process of enveloped viruses such as influenza virus [18]. The cardinal feature of the central domain is the CX3CX2C motif, where the three conserved cysteine residues bind iron sites in the [4Fe-4S] cluster. The remaining iron site coordinates with SAM to form a coordination complex, making it easier for the reduced state [4Fe-4S] to transfer electrons to SAM. Subsequently, SAM experiences reductive cleavage to generate the 5′-deoxyadenosyl (5′-dAdo•) radical intermediate [19], which is responsible for extracting a hydrogen atom from cytidine triphosphate (CTP), leading to the production of 3'-deoxy-3',4'-didehydro-CTP (ddhCTP) [20, 21]. Notably, ddhCTP lacks the 3'-hydroxyl group on the ribose portion. Its incorporation into the nascent viral RNA strand halts the strand and consequently stops viral RNA synthesis, effectively impeding viral replication [22]. Despite its significance, limited research has focused on the C-terminal domain, a feature that is predominantly conserved between species. The interaction of the C-terminus with the cytosolic iron-sulfur protein assembly protein is essential for Fe-S cluster formation and viperin stability [15]. In addition, it has been demonstrated that the very last C-terminal amino-acid residues are required for antiviral activity against some viruses [23, 24].

Schematic representation of the gene composition and protein structure of RSAD2 and viperin. The RSAD2 gene is located on chromosome 2, contains 5 exons and 4 introns. The structure of human viperin is composed of 361 amino acids, including the radical SAM core, in which the three conserved cysteines at sites 83, 87 and 90 bind 3 iron atoms of the [4Fe-4S] cluster.
RSAD2/viperin Signaling Pathway
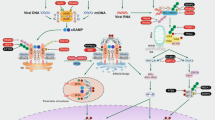
Viperin is a multifunctional regulator of various signaling pathways, not just as an antiviral protein. Understanding the intricate signaling pathways involved in viperin-mediated immune responses is essential for elucidating its biological functions and therapeutic implications. This section comprehensively reviews the current status of knowledge of viperin signaling pathways and highlights its interactions with key immune molecules. Insights into the signaling cascades associated with viperin offer valuable perspectives for the development of new therapeutic strategies to combat immune-related diseases (Fig. 2).

The proposed model for RSAD2 induction and the role of RSAD2/viperin in signaling pathways. The expression of viperin is induced by both IFNs via the JAK-STAT pathway. While phosphorylated STAT levels decrease, unphosphorylated complexes can maintain RSAD2 synthesis upon prolonged IFN exposure. Sensing of viral factors by pattern recognition receptors transduces the immune stimuli into intracellular signals, activating downstream signaling pathways, which triggers the generation of IFN-I, other pro-inflammatory cytokines, and ISGs including RSAD2. 1) RSAD2 can be directly induced by peroxisomal MAVS and downstream molecules IRF1 or IRF3 activation to produce the viperin protein. Viperin facilitates the localization of MAVS in mitochondria and peroxisomes in the RLR-MAVS pathway. 2) Viperin interacts with STING to promote the activation of TBK1 in the cGAS-STING pathway. 3) Viperin triggers TRAF6 self-ubiquitination and IRAK1 ubiquitination in the TLR7/9 pathway. These boost the expression of IFN, proinflammatory cytokinesis, and viperin, forming a complicated positive feedback loop. Abbreviations: IFN, interferon; JAK, janus kinase; TYK2, tyrosine kinase2; STAT, signal transducer and activator of transcription; IRF, interferon regulatory factor; ISGF3, interferon-stimulated gene factor 3; GAF, γ-activated factor; GAS, γ-activated sequence; ISRE, interferon-stimulated response element; ISGs, interferon-stimulated genes; RSAD2, radical S-adenosyl methionine domain-containing 2; RLR, retinoic acid-inducible gene I (RIG-I)-like receptor; MAVS, mitochondrial antiviral signaling; IKK, inhibitor of NF-κB kinase; NF-κB, nuclear factor kappa B; TBK1, tank-binding kinase 1; cGAS, cyclic guanosine monophosphate–adenosine monophosphate synthase; cGAMP, cyclic GMP-AMP; STING, stimulator of interferon genes; TLR, Toll-like receptor; MyD88, myeloid differentiation factor88; TRAF6, tumor necrosis factor receptor-associated factor 6; IRAK1, interleukin-1 receptor-associated kinase 1; Ub, ubiquitinated; P, phosphorylated; U, unphosphorylated form.
JAK-STAT Signaling Pathway
Initially recognized as an IFN-induced gene, RSAD2 was thought to be regulated by IFN-dependent signaling pathways. IFNs primarily signal through the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway [25]. IFN-γ binds to its receptor (composed of two subunits, IFNGR1 and IFNGR2) to induce receptor oligomerization and conformational changes, which activates the JAK1 and JAK2 by transphosphorylation [26]. STAT1 undergoes dimerization after being phosphorylated by JAKs and translocates to the nucleus where it binds to γ-activated sequence (GAS) elements for regulating ISG expression. Additionally, IFN-I (α and β) or IFN-III (λ) can also induce viperin expression, they bind to their specific heterodimeric receptor IFNAR or IFNLR, respectively [27, 28]. The dimerization of IFNAR or IFNLR activates the JAK-STAT signaling pathway and ultimately induces the formation of the heterotrimeric ISG factor 3 complex that directly binds to interferon-stimulated response elements (ISREs) to drive ISG transcription. While phosphorylated STAT levels decrease, unphosphorylated complexes can maintain basal expression of ISGs in response to long-term IFN stimulation [29]. Direct induction of viperin can occur through peroxisomal MAVS and downstream activation of IRF1 or IRF3 (discussed later) [30, 31].
TLR Signaling Pathway
TLRs, a type of pattern recognition receptors, are important mediators between innate and adaptive immunity [32]. Activation of TLR triggers downstream signaling cascades, leading to the production of pro-inflammatory cytokines, and other mediators of immune responses [33]. The myeloid differentiation primary response gene 88 (MyD88)-dependent pathway is responsible for proinflammatory cytokine expression. MyD88 recruits IRAK4, which in turn activates IRAK1. Activated IRAK1 then recruits and binds to TNF receptor-associated factor 6 (TRAF6). Subsequently, TRAF6 forms complexes with transforming growth factor-β-activating kinase (TAK1) and TAK-binding proteins. This complex plays several critical roles, including the activation of the nuclear factor kappa B kinase (IKK)/ nuclear factor kappa B (NF-κB) pathway [34]. Another MyD88-independent pathway mediates the induction of IFN-I and ISGs, where adaptor Toll/IL-1 receptor (TIR) domain-containing adaptor (TRIF) is implicated in. TRIF acts on two proteins, TBK1 and IKKε. IKKε-TBK1 activates IRF3, which enters the nucleus to stimulate IFN expression [35].
Evidence has shown that reciprocal regulation of viperin and TLR pathways upon exposure to stress and infection. TLR activation induces the expression of viperin, which in turn modulates TLR signaling cascades to fine-tune the immune response [7]. In the TLR7/9-IFN-I signaling pathway, viperin activates the E3 ligase activity of TRAF6 [36]. It has also been suggested that viperin can specifically stimulate ubiquitination of IRAK1 by TRAF6, rather than causing general ubiquitination of cellular proteins [37]. And ubiquitination of IRAK1 is a crucial component of the IRF7-mediated IFN-I production pathway. Additionally, viperin targets viral proteins for proteasomal degradation by recruiting the protein ubiquitination [36]. Notably, dysregulated TLR-viperin interactions may cause the overproduction of pro-inflammatory cytokines. It was demonstrated that knockdown of RSAD2 inhibits the expression of IL-1β, IL-6, and TNF-α by suppressing the TLR2/MyD88/NF-κB pathway [38]. It has also been suggested that viperin can act as a negative regulatory factor in the inflammatory response. By suppressing the formation of the IRAK1/TRAF6/TAK1 complex of the TLR4 pathway, viperin inhibits IKK, thereby suppressing the production of nitric oxide and proinflammatory cytokines [39].
RIG-I-MAVS Signaling Pathway
Three members constitute the cytoplasmic receptor family known as RIG-I-like receptors (RLRs): laboratory of genetics and physiology 2 (LGP2), melanoma differentiation-associated protein 5 (MDA5), and retinoic acid-inducible gene I (RIG-1) [40]. Initially, RLR plays an important role in RNA sensing, and its dysregulation is involved in the development of autoimmune diseases [41]. As a cytoplasmic receptor family, RLR initiates the oligomerization of MAVS by conducting a caspase activation and recruitment domain (CARD)-CARD connection upon detection of abnormal double-stranded RNA [42]. Oligomerized MAVS propel a signaling cascade, subsequently activating TBK1 and the cytosolic kinases IKK, which leads to activation of IRF3, IRF7, and NF-κB, turning on the expression of IFNs and other pro-inflammatory cytokines [30, 43].
Stimulation of the key adapter molecule MAVS within the RLR pathway ultimately results in the upregulation of numerous ISGs, including RSAD2 [44]. Moreover, to induce optimal IFN-β in this pathway, it is necessary for MAVS to colocalize on mitochondria and peroxisome membranes [10]. It is noteworthy that viperin facilitates this reaction by forming a close association with MAVS located in mitochondria and mitochondria-associated ER membrane (MAM) [10]. However, the underlying mechanisms and extent of this association are still largely unclear. Moreover, viperin is among the group of IRF1-regulated genes. Previous evidence suggested that IRF1, one of the downstream molecules of MAVS, can bind to the murine viperin promoter to the two proximal IRF elements and thereby directly induce viperin expression, independently of IFNs [45].
STING Signaling Pathway
STING, an ER adaptor, senses abnormal DNA in cells, triggering the production of IFN-I. It responds predominantly to cytosolic DNA, viral or bacterial infection, and cellular damage [46]. Various cytosolic DNA sensors, such as cyclic GMP-AMP synthase (cGAS), recognize DNA and catalyze the production of cyclic GMP-AMP (cGAMP), a second messenger [47]. cGAMP then binds to STING in the ER and leads to a conformational change of STING that activates its signaling function [46]. STING then translocates from the ER to the perinuclear region and forms a complex with TBK1, which is necessary for TBK1 activation [48]. TBK1 phosphorylates multiple transcription factors, including IRF3 and NF-κB [49]. This initiates the transcription of innate immunity-related genes, including IFN-I [50]. In a previous experiment, viperin was shown to bind to STING and promote enhanced polyubiquitination of TBK1, thereby triggering an elevated IFN-I response [8]. Despite the evidence linking viperin to STING pathway enhancement, there is limited data obtained to validate the underlying mechanisms. Targeting STING signaling pathways or modulating viperin expression may offer novel approaches for understanding autoimmune diseases characterized by dysregulated IFN-I responses. Therefore, further experimental analysis and discussion of the data are needed to determine the specific mechanism by which viperin enhances STING signaling activation.
Roles and Mechanisms of RSAD2/viperin in Immunomodulation and Mitochondrial Metabolism
Effects of RSAD2/viperin on Immune Cells
RSAD2 is typically expressed at low levels under physiological conditions, but its expression is notably elevated in several pathological situations, including cancer [51], infectious diseases [52], and autoimmune diseases [53]. DCs, macrophages, neutrophils, T lymphocytes, and B lymphocytes are the major cell types that express RSAD2. The expression level of RSAD2 significantly affects the maturation, differentiation and functional status of immune cells, which can lead to immune dysfunction and persistent inflammation. In this section, we will explore the link between RSAD2/viperin and immune cells and provide perspectives on its function in the immune system.
DCs
DCs are the most potent professional antigen-presenting cells that are central to both innate and adaptive immunity. They can be broadly classified into classical DCs, pDCs, Langerhans cells, and monocyte-derived DCs [54]. RSAD2 simulates IFN-I secretion by regulating the TLR7 and TLR9-IRAK1 signaling axis in pDCs. IRAK1 and TRAF6, interact with viperin and are recruited to liposomes. This process promotes the K63-linked ubiquitination of IRAK1, inducing the nuclear translocation of IRF7/9 thereby promoting IFN-β production [7]. Another study co-transfected HEK 293 T cells with viperin, IRAK1 and TRAF6, quantified viperin by western blotting, and measured the formation of 5'-dA. It was found that when the [4Fe-4S] cluster is removed during this process, the stability of viperin is destroyed and the polyubiquitination of IRAK1 cannot be stimulated by TRAF6 [37]. Furthermore, RSAD2 has been demonstrated to be a crucial factor in DC maturation as well as in T cell activation, particularly via the IRF7-mediated signaling pathway. Knockdown of RSAD2 in mature DCs resulted in a remarkable reduction in the secretion of proinflammatory cytokines and suppressed their ability to stimulate T cells [55].
Macrophages
Macrophages play a pivotal role in immunity, participating in both specific and non-specific immunity in vivo [56]. Macrophages can be polarized into classically activated macrophages (M1) and alternatively activated macrophages (M2), which play pro-inflammatory and anti-inflammatory roles, respectively [57]. Whether viperin is associated with the polarization and function of macrophages varies depending on the specific circumstances. A previous study indicated that RSAD2 was significantly elevated in the M1 phenotype compared with the M0 and M2 phenotypes [58]. Overactivation of M1 macrophages has been linked to various mechanisms of chronic inflammatory diseases, including autoimmune diseases [59, 60]. Nevertheless, another study demonstrated that viperin did not affect macrophage polarization, but did affect the production of inflammatory cytokines following mycobacterial infection [39]. In addition, a recent study highlighted that viperin is capable of inhibiting the production of IFN-γ in macrophages and the lungs of mice, thereby promoting the survival of Mtb [61]. Therefore, the development of inflammation caused by the activation of RSAD2 is a topic worthy of further investigation.
Neutrophils
Neutrophils develop and differentiate in the bone marrow before being released into the bloodstream. Neutrophil extracellular traps (NETs) are typically produced and released through a unique process of programmed inflammatory cell death, a specific way in which neutrophils die [62]. SLE NETs activate pDCs and induce IFN-I secretion, which also triggers NETosis in healthy neutrophils [63]. RSAD2 has been demonstrated to be highly expressed and induced in neutrophils in RA [64], acute lymphocytic choriomeningitis virus [65] and asthma [66], and this process is also associated with IFN. Therefore, it is worth exploring whether RSAD2, as an IFN-induced gene, is involved in the specific inflammatory cell death mode of NETs.
Lymphocytes
B lymphocytes are antigen-presenting cells that, in addition to specialized humoral immune functions, contribute to immune regulation by producing antibodies. CD19, existing on the surface of B cells, acts as a component of B cell activation co-receptor, which enhances the sensitivity of B cells to antigen stimulation and participates in B cell activation [67]. A previous experiment demonstrated that the expression of c-Rel, p50, and p-p65/t-p65 decreased in CD40L-induced CD19+ B cells after knockdown of RSAD2, indicating that knockdown of RSAD2 deactivates the NF‑κB signaling pathway of CD19+ B cells [68]. It has been shown that the overexpression plasmids of p65 partially alleviate the inhibitory effects of si-RSAD2-2 on proliferation, immunoglobulin production, and IL-10 expression in CD40L-induced CD19+ B cells. This indicates that RSAD2 controls B cell activation through the NF-κB signaling pathway [68].
T cells are divided into two functionally distinct subgroups, the CD4+ T cells and the CD8+ cytotoxic T cells. It is essential that CD4+ and CD8+ T cells are adequately activated to proliferate, clonally expand, and provide effector functions in order to ensure the efficient clearance of infection by pathogens [69]. According to a recent study, RSAD2 is expressed at increased levels in all T cell subsets, particularly CD4+ T cells, in the context of immune disorders [70]. It was discovered that different IFNs have different stimulatory effects on RSAD2 at different periods. Consequently, it was postulated that RSAD2 can influence the differentiation of CD4+ naïve T cells into Th17 and Tfh cells and is regulated by IFN-I. Furthermore, the study demonstrated that the absence of RSAD2 can lead to a decrease in Th17 and Tfh cells, whereas the presence of RSAD2 can promote the differentiation of Th17 and Tfh cells in individuals with SLE [70]. Guillaume Carissimo et al. showed that viperin controls the stimulation of murine chikungunya virus-specific pathogenic T cells: IFN-γ producing Th1. They demonstrated that the observed increase in IFN-γ producing T-cell stimulation may be attributed to an elevation in activating and polarizing soluble mediators released by APCs during viral stimulation [71]. Additionally, another experiment demonstrated that viperin is required for optimal Th2 responses and T cell receptor-mediated activation of NF-κB and AP-1 [72].
Crucial Roles and Mechanisms of RSAD2/viperin in Mitochondrial Metabolisms
Mitochondria play multifaceted biological functions within cells, acting as the primary site of material and energy metabolism. It was found that mitochondria are involved in specific signaling pathways by interacting with certain proteins and affecting the expression of RSAD2/viperin. Viperin could also target mitochondria via a variety of transport mechanisms and influence mitochondrial metabolism. The [4Fe-4S] cluster is a very important component of viperin in this process. In this section, we aimed to clarify the relationship between RSAD2/viperin and mitochondria, as well as the essential role of the [4Fe-4S] cluster for viperin to acquire a more profound comprehension of the underlying mechanisms (Fig. 3).

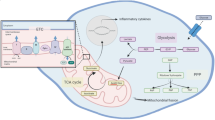
Mechanisms of RSAD2/viperin involving mitochondrial and cellular metabolisms. 1) Pex19 is a cytoplasmic chaperone protein that, in conjunction with Pex3, is responsible for the transport of peroxisomal membrane proteins to the organelle. Pex19 interacts with vipeirn to position the peroxisome at the mitochondrial/MAM MAVS signaling synapse, thereby strengthening the RIG-I-MAV signaling pathway and promoting the expression of IFN-I and RSAD2 in the microenvironment of viral infection or immune stimulation (purple arrows illustrate the process). 2) The exact mechanism by which viperin transfers from the ER to mitochondria is unknown. Under the infection of HCMV, viperin can target mitochondria by binding to Cys44 of vMIA. Additionally, the localization of vMIA and viperin in the MAM also reveals a potential new mechanism for the transfer of viperin through the MAM (pink arrows illustrate the process). 3) When viperin enters into mitochondria, it binds to the β-subunit of the mitochondrial trifunctional protein (TFP, HADHB), thus the β-oxidation of fatty acids was blocked. This sets off a sequence of ongoing processes that boost viral replication while encouraging glycolysis and lipogenesis (blue arrows illustrate the process). Abbreviations: Pex19, peroxisomal biogenesis factor 19; MAM, mitochondria-associated ER membrane; MAVS, mitochondrial antiviral-signaling protein; RIG-I, retinoic acid-induced gene I; ER, endoplasmic reticulum; TRAF3, tumor necrosis factor receptor-associated factor 3; TBK1, tank-binding kinase 1; TFG, TRK-fused gene; mTOR, mechanistic target of rapamycin; IRF3, interferon regulatory factor 3; IFN-I, type I interferons; RSAD2, radical S-adenosyl methionine domain-containing 2; vMIA, viral mitochondrion-localized inhibitor of apoptosis; TFP, mitochondrial trifunctional protein; AMP, adenosine monophosphate; AMPK, AMP-activated protein kinase; GLUT4, glucose transporters 4; ChREBP, carbohydrate response element binding protein; ChoRE, carbohydrate response element; SREBP1, sterol regulatory element-binding protein 1; LDs, lipid droplets.
The Crosstalk Between RSAD2/vipeirn and Mitochondria
A multitude of investigations have demonstrated the intimate relationship between mitochondria and viperin. MAVS located on mitochondria are implicated in the induction of RSAD2/viperin. Viperin was discovered to interact with Pex19 to anchor the peroxisome to the mitochondrial/MAM MAVS signaling synapse, which strengthens the RIG-I-MAVS signaling pathway and encourages the antiviral response [10]. The TRK-fused gene (TFG), a protein that interacts with TRAF3 and potentially activates mechanistic target of rapamycin (mTOR) after Sendai virus infection, has been identified in a separate study [44]. Activation of mTOR enabled TBK1 to phosphorylate mTOR on serine 2159, which in turn stimulated the involvement of mTOR in the RIG-I-MAV signaling pathway [44]. As we have already mentioned, the expression of IFN and RSAD2 is induced by the activation of the RIG-I-MAVS signaling pathway.
Additionally, several studies are exploring the relationship between STING and mitochondria. It is noteworthy that Maekawa et al. have demonstrated that mitochondrial damage in tubular cells results in the leakage of mitochondrial DNA (mtDNA) into the cytosol, most likely through BAX pores on the mitochondria. This process initiates cGAS-STING signaling, which in turn triggers tubular inflammatory responses in cisplatin-induced acute kidney injury [73]. One review explored the interactions between ER stress, mitochondrial dysfunction, and STING activation in various physiological and pathological conditions [74]. The stimulation of IFN-treated neutrophils or neutrophils from lupus patients with anti-RNP immune complexes can result in the release of oxidized mtDNA through multiple mechanisms, which has been extensively reviewed in the article [74]. For instance, extracellular oxidized mtDNA in lupus is recognized by monocytes in a way that is reliant on STING, and pDC internalizes it through the receptor for advanced glycation end products [75]. Collectively, ER stress can activate STING through calcium/ROS-mediated mitochondrial damage and release of mtDNA into the cytosol. This activation of STING leads to the production of IFN-I and immune responses. As previously stated, viperin has been shown to bind to STING and TBK1 in the context of viral infection, thereby enhancing the IFN-I response. This leads us to hypothesize that viperin may also be involved in the process by which mitochondrial damage induces inflammation via cGAS-STING signaling in a broader context, such as oxidative stress, inflammatory stimuli, and autoimmunity, not just viral infection.
Viperin Affects Metabolic Processes by Targeting Mitochondria
Viperin can enter the mitochondria via a variety of transport mechanisms. Following infection with HCMV, viperin binds to the vMIA and targets the mitochondria through N-terminal mitochondrial localization signals to achieve transfer from the ER to the mitochondria [12]. The N-terminal domain of viperin interacts with cysteine residue 44 (Cys44) of vMIA which is necessary for their interaction. Furthermore, Cys44 of vMIA is essential for the transport of viperin and its antiviral activity [76]. It is worth noting that vMIA can cross from the ER to the outer mitochondrial membrane via MAM [77]. This suggests that viperin may bind to vMIA and be localized to the mitochondria via MAM. Indeed, it has been proposed that viperin is localized in the MAM and interacts with MAVS to prevent excessive immune responses as a regulator of the IFN response, which will deepen our understanding of the functions and transit mechanisms of viperin [78]. In addition, rotaviral non-structural protein 4 (RV-NSP4) has been shown to trigger the translocation of viperin from ER to mitochondria during rotavirus infection. Viperin can penetrate the mitochondrial membrane via the N-terminal domain, bind to NSP4 via the free radical SAM region and the C-terminal region, and prevent rotavirus release by inhibiting apoptosis [79].
In mitochondria, HCMV-induced viperin binds to the β-subunit of the mitochondrial trifunctional protein (HADHB), which can catalyze the final three steps of the β-oxidation pathway of fatty acids. Protein–protein interactions facilitate the activation of viperin by HADHB, which in turn leads to the synthesis of the chain termination inhibitor of RNA polymerase (ddhCTP) [12]. The function of ddhCTP in mitochondria remains to be fully elucidated. Given that it was previously identified as a chain terminator for RNA-dependent RNA polymerases, it may have a hitherto unappreciated role in influencing mitochondrial transcription [21]. Additionally, the localization of viperin in mitochondria can inhibit the thiolase activity of HADHB and simultaneously promote the degradation of HADHB via the proteasomal route upon retrotranslocation to the outer mitochondrial membrane [12, 80]. Inhibition of HADHB by viperin can reduce the thiolysis of β-ketoacyl-CoA, thus blocking the β-oxidation of fatty acids [80]. When viperin targets mitochondria, there is a reduction in ATP and NADH levels, which leads to the destruction of the actin cytoskeleton and the facilitation of viral replication [80]. It is often observed that the accumulation of AMP concomitantly with the depletion of ATP. AMP can activate AMP-activated protein kinase (AMPK), which in turn triggers the upregulation of glucose transporters 4 (GLUT4), increasing the amount of cytoplasmic glucose available for glycolysis and lipid synthesis, as well as the translocation of carbohydrate response element binding protein (ChREBP) to the nucleus. This in turn causes an increase in de novo lipogenesis, lipid droplet accumulation, and viral envelope formation due to the increased transcription of genes encoding lipogenic enzymes [13, 81].
A recent study has indicated that viperin plays a role in metabolic alteration and accelerates the progression of cancer. In a manner analogous to the aforementioned process, viperin facilitates lipogenesis and glycolysis by blocking fatty acid β-oxidation [51]. Research has also shown that the PI3K/AKT/mTOR/HIF-1 and JAK/STAT signaling pathways enhance the expression of viperin in the tumor microenvironment due to insufficient fatty acids, oxygen, and the production of IFNs [51]. An interesting phenomenon is that viperin stimulates sterol regulatory element binding protein 1 (SREBP1) and ChREBP via increased glucose uptake as cancer progresses. Both SREBP1 and ChREBP work in concert to stimulate lipogenesis [51]. Although it has been demonstrated that HCMV infection may cause the cleavage of SREBP1, this effect was not found to be related to viperin expression [82].
Regardless of the method used, viperin targeting mitochondria can replicate all subsequent metabolic outcomes, which leads us to hypothesize that there is a link with autoimmune diseases, as dysregulated lipid metabolism has already been strongly associated with the pathogenesis and progression of autoimmune diseases [83, 84]. A review of the direct interaction between RSAD2 and mitochondria will provide new insights into the pathophysiology of autoimmune diseases and potential future treatment options.
The Involvement of Viperin in Metabolic Processes Requires the Binding of Fe-S Clusters
Viperin is a radical SAM enzyme and contains a redox-active [4Fe-4S] cluster coordinated by the three conserved cysteines [85]. The [4Fe-4S] cluster serves to anchor the SAM cofactor, thereby facilitating the formation of the highly reactive 5′-deoxy-5′-adenosyl radical [86]. A research team employed multi-template homology modeling and molecular dynamics simulations to demonstrate that the removal of the [4Fe-4S] cluster resulted in the collapse of the tertiary structure of viperin [87]. Furthermore, conformational analysis using circular dichroism and steady-state fluorescence spectroscopy was performed on the four purified mutant proteins, which showed that they were partially unfolded, conformationally unstable, and prone to aggregation. The researchers proposed that the lack of antiviral activity exhibited by the mutant protein could be attributed to its reduced conformational stability [87].
Cytosolic Fe-S assembly component 1 (CIAO1/CIA1), an interaction factor of viperin, contributes to the antiviral effect of viperin and is thought to facilitate the capacity of viperin to bind iron. In one study, the presence of an Fe-S cluster was indicated by the incorporation of 55Fe, which was detected by radiolabelling in vivo. Silencing of CIAO1 by siRNA resulted in a reduction in the levels of viperin and 55Fe binding, as well as its antiviral efficacy against TBEV [15]. It suggests that viperin requires the assembly of Fe-S clusters and activation of its function depends on the interaction with CIAO1.
The involvement of viperin in metabolic reprogramming necessitates mitochondrial localization and the binding of Fe-S clusters [88]. Meanwhile, Fe-S clusters have a significant impact on mitochondrial energy production and substance metabolism through their binding to viperin. Viperin can directly target mitochondria to replicate the results of lipid metabolism in the absence of HCMV infection [13]. Viperin binding to the Fe-S cluster targeting mitochondria contributes to ATP depletion. This conclusion was corroborated by measuring the intracellular ATP level of murine embryonic fibroblasts with viperin knockout expressing vMIA-Myc (which induces endogenous viperin), WT toxic protein, and finding that ATP levels decreased by approximately 50% solely in cells expressing the mitochondria localization sequence-viperin [80]. Indeed, in most eukaryotes, mitochondria play a crucial role in the biogenesis of all cellular Fe-S proteins [89]. The Fe-S cluster assembly mechanism in mitochondria, which consists of up to 18 distinct proteins, is responsible for synthesizing Fe-S clusters from scratch and inserting them into target apoproteins [90, 91].
The association between RSAD2/vipeirn and the Fe-S cluster suggests that it may be relevant to iron metabolism. It has been demonstrated that iron metabolism is intricately linked to the pathogenesis of autoimmune diseases [92]. Moreover, ferroptosis is a new mode of cell death that is driven by iron-dependent lipid peroxidation. A consensus clustering analysis has demonstrated that RSAD2 is associated with both lipid metabolism and ferroptosis [93]. Therefore, elucidating the function of RSAD2 in iron metabolism is anticipated to represent a wholly novel area of investigation for future research on RSAD2.
Crucial Roles of RSAD2/viperin in Autoimmune Diseases
As summarized in the former text, RSAD2/viperin is closely associated with immune function and mitochondrial metabolism at the cellular and molecular levels, potentially leading to the onset and progression of autoimmune diseases. Thus, our goal in this part is to give an overview of the critical functions that RSAD2/viperin plays in autoimmune diseases (Table 1).
AGS
Aicardi-Goutières syndrome (AGS) is defined as a genetic neuroinflammatory disease characterized by increased IFN-α levels with ensuing ISG expression [94, 95]. Considered the prototype of type I interferonopathies, AGS is an enormous yet poorly understood problem in the field of neuroscience, necessitating further research into effective treatments using inhibitors to block IFN activation in patients [95].
The upregulation of IFN-I signaling appears to be a lifelong phenotype so that AGS patients benefit from IFN-related therapy at any age [96]. Patients with AGS often onset in early infancy, concurrent with a history of severe manifestations during this period [97]. Notably, under the circumstances of increased ISG expression, several AGS patients are susceptible to developing SLE in early childhood [98,99,100]. In a study of clinical AGS samples, persistently elevated expression of ISGs (including RSAD2) in peripheral blood was demonstrated to lead to a disease exacerbation through an increase in pro-inflammatory and immunomodulatory proteins [101]. They pointed out that RSAD2, as an AGS indicator, showed a more sensitive and specific ability to identify IFN signaling compared to standard inflammatory markers [101]. In addition, recent studies have indicated that RSAD2, one of the most hypomethylated ISGs in AGS, correlates with phenotype, inflammation, and disease stage by specifically expressing hypomethylated cytosine-phosphate-guanine (CpG) dinucleotides in peripheral blood mononuclear cells (PBMCs) of "severe" patients [102]. It turns out that the upregulated expression of RSAD2 in patients with severe phenotype according to a neurologic severity score is correlated with this differential methylation pattern [102]. Through our understanding of these studies, we emphasize the significance of RSAD2 in IFN-dependent autoimmunity and autoinflammation and the possibility of RSAD2-targeted therapy.
DM
Dermatomyositis (DM) is an acquired autoimmune disease that affects the skin and muscles, however, the underlying mechanisms were unclear [103]. Owing to its clinical heterogeneity, DM is a hard-to-diagnose disorder, and its cutaneous manifestations have different characteristics in terms of time course and severity [104]. As one of the major subsets of idiopathic inflammatory myopathy, DM has been proven to be pertinent in pDCs which are the professional producers of IFN-I, and its ensuing co-regulation of IFN-driven chemokines [105, 106]. Remarkably, RSAD2 takes active parts in the IFN-I signaling pathway and the cellular response to IFN-I that is linked to immune-related processes in DM [107]. Apart from IFN-related factors, RSAD2 is also thought to be implicated in virus response, negative regulation of viral genome replication, and other processes [108].
RSAD2, the DM-related gene, is found to be significant in the development of myocarditis in DM. Recent advances in DM research focusing on myocardial damage have shown that upregulation of RSAD2 in the myocardium of patients is positively associated with M2 macrophage proliferation, which has been implicated in the pathogenesis of DM [107, 109]. In serum exosomes from DM patients with myocarditis, some RSAD2 target miRNAs were also reported to be upregulated compared to normal controls [107]. Despite the achievements in elucidating the function of RSAD2 in the pathogenesis of DM, it should not be taken for granted that RSAD2 forms the cornerstone of DM, as its role in this field remains unclear. It is expected that RSAD2 could be a potential intervention in DM owing to its significant mechanism.
RA
The hallmark of RA is chronic inflammation, of which, synovial joint inflammation is a typified disease manifestation, indicating that the overproduction of proinflammatory cytokines and chemokines is at the core of the disease [110]. Notwithstanding the etiology remains unclear, a growing body of research suggests that RSAD2 is involved in the pathogenesis of RA.
Expression of RSAD2 mRNA in the peripheral blood of RA patients was shown to exacerbate the RA disease progression by modulating the IFN-I response [64]. In addition, several investigators have reported differential expression of interferon response genes (IRGs), including RSAD2, in all peripheral blood cell types, correspondingly equipped with IFN-I signaling ability [111]. PBMCs are considered a major ingredient that plays a critical role in RA by secreting IFN-I. Clinical studies have shown that decreased RSAD2 expressions in PBMCs are closely related to better clinical responses, and could prospectively discriminate rituximab (RTX)-unresponsive RA patients [112]. Patients with RA are typically administered immunosuppressive treatments, including disease-modifying anti-rheumatic drugs and glucocorticoids. In the event of treatment failure, biologics such as TNF blocking agents and RTX, a B-cell depletion therapy, are typically employed [113]. The clinical response of RTX therapy is related to the activation of the IFN system. The sole distinction between RTX responders and non-responders is pharmacodynamic changes in the expression of a selective group of genes all regulated by IFN-I [114]. Based on the premise that only IRGs have clinical relevance to RTX in all the genes in the human genome, a study demonstrated that the IFN-score of IRGs (EPSTI1, MX1 and RSAD2) performed best as a predictor of RTX response (AUC = 0.87) [112]. Whereas 5 or 8 gene sets of IRGs (both including RSAD2) manifest lower predictive power to RTX response, which may indicate the significance of selecting the appropriate combination of biomarkers to predict disease [112]. Abundant RSAD2 expression was observed in RA patient polymorphonuclear granulocytes, which are more potent than PBMCs in inducing IRGs [64]. Moreover, RSAD2 is abundant in synovial macrophages, a central driver of cartilage destruction as well as inflammation and autoimmunity, providing insight into the pathogenesis of RA [115, 116]. Therefore, RSAD2 may be considered a potential treatment target with clinical value for monitoring RA progression.
SLE
SLE is a prototypical chronic autoimmune disease involving multiple organ damage [117]. The complex mechanisms of SLE include the dysregulations of IFN-I and several cellular components of innate and immune responses [118, 119]. Although myriad SLE treatments such as immunomodulators and immunosuppressants are available, current options may not address the complexity and heterogeneity of SLE [120, 121]. Therefore, better therapeutic strategies are urgently needed to conquer the disease.
RSAD2 plays an important role in the onset and progression of SLE by inducing increased levels of IFN-I, particularly IFN-α, which is considered to be the principal pathogenic mediator in SLE [122]. The positive feedback loop between IFN-I synthesis and RSAD2 expression during SLE onset is confirmed and well-described. Activation of the IFN-I signal pathway upregulated RSAD2 expression, which in turn triggers an increase in IFN levels and ultimately exacerbates the disease severity [123, 124]. Consistent with RSAD2, the lncRNA negative regulator of the IFN response (NRIR), which is induced by IFN-α, exhibits a close association with the IFN-I pathway and is located adjacent to RSAD2 in the genome [5, 125]. The NRIR-RSAD2 interaction pairs are critical for the pathogenesis of SLE, and together they are involved in both viral infection and the innate immune response [125]. The expression levels of several ISGs, including RSAD2 and NRIR, are intimately related to the typical symptom of thrombocytopenia which regularly indicates the severity of the disease. SLE patients with thrombocytopenia showed lower expression levels of RSAD2 and NRIR than those with normal platelet counts. Further, compared to normal SLE patients, SLE patients with anemia are also found to have lower levels of RSAD2 instead of NRIR [125].
It is well-established that pDCs are referred to as the main culprit for SLE due to the amplification of IFN-I it produces [120, 126]. RSAD2 is abundantly produced in concentrated suspensions of pDCs and serves as an intermediary in the induction of pDCs to produce IFN-I via TLR7 and TLR9, and its excessive activation then further affects SLE progression [7, 127].
RSAD2 activities in T cells are implicated in the development of SLE. As critical components of the immune system, T lymphocytes have gained increasing recognition as key players in the pathogenesis of SLE, exhibiting profound multifaceted aberrations defects that are intertwined with SLE-associated symptom severity and disease phenotype [128,129,130]. IFN-α is also involved in the regulation of RSAD2 expression in CD4+ T cells [70]. Additionally, the enhancement of TRIM5 transcriptional activity can be achieved by repeated viral infection, further resulting in the upregulation of RSAD2 that drives SLE autoimmunity. Concertedly, TRIM5 transcriptional activity is inversely associated with naïve CD4+ T cells [131]. Recent advances in bioinformatics research show that gene signatures in CD4+ T cells reveal the therapeutic potential of RSAD2 as an IFN signature gene that is shared across multiple autoimmune diseases [132]. In addition, studies have unveiled a fundamental role for RSAD2 in Th2 cell development by modulating the activities of NF-κB and AP-1 and indirectly promoting T cell receptor-mediated activation of GATA binding protein 3 (GATA3) which can inhibit viral replication in SLE [72, 132].
Epigenetic dysregulation of B cell differentiation is thought to be a crucial factor in SLE pathogenesis [133]. One study revealed that under IFN-α regulation, RSAD2 facilitates the differentiation of Th17 and Tfh cells, which in turn promotes B cell activation [70]. In light of these findings, novel therapies aimed at inducing B-cell insensitivity/tolerance are worth endeavoring. The above findings emphasize the importance of RSAD2 expression and the different roles it plays in different cell types and individuals. At present, SLE diagnosis and assessment may focus on clinical and immunologic biomarkers [119]. To our knowledge, RSAD2 has the potential to be a biomarker that distinguishes SLE patients from those with other immune system disorders and healthy individuals as RSAD2 levels in patients are related to the disease severity of SLE [125].
Despite the heterogeneity of SLE, over 80% of patients have an IFN signature and show associated transcriptional signatures in the blood [134, 135]. The elevated expression level of ISGs in SLE is directly related to disease activity, and ISGs can also serve as laboratory biomarkers to help manage clinical symptoms [125, 136]. Compared with healthy controls (HCs), RSAD2 demonstrates high expression, persistent hypomethylation and concomitant presence of a nine-fold transcription activity in B cells. These distinctive characteristics enable RSAD2 to differentiate itself from the other four-fold upregulated genes, establishing it as an epigenetic biomarker with the potential to inform future diagnosis [137, 138]. Notwithstanding its importance as a biomarker, there seems no obvious relationship with common inflammation-related indicators [125]. Existing research confirms phenotypic differences between ethnicities regarding DNA methylation [139]. There is a negative correlation between patient age and RSAD2 expression levels, as reflected by younger patients typically exhibiting a higher IFN signature [125]. It is interesting to note that though SLE is a female-biased disease, RSAD2 gene expression is higher in SLE males than in SLE females [140]. Therefore, individual-dependent differences in SLE patients, such as ethnicity, age, gender, etc., also need to be taken into consideration in the diagnosis of SLE [141, 142].
Lupus nephritis (LN) is defined as a major cause of overall morbidity and mortality in SLE patients [135, 143]. Receiver operating characteristic analysis showed the good diagnostic performance of MX dynamin-like GTPase 1 (MX1)-RSAD2 pair for LN (area under the curve (AUC) > 0.6), and their upregulation may play a molecular regulatory role in LN progression through co-regulation and positive correlativity with cell infiltration [144, 145]. Moreover, rash-related symptoms are more common in the majority of patients with high ISG expression [146]. Immunohistochemistry results confirmed that viperin is highly expressed in SLE and showed differential expression in the kidneys, blood, and skin lesions of HCs and SLE patients [147]. In summary, these results demonstrate the promising utility of RSAD2/viperin as a new diagnostic biomarker in SLE.
SSc
Systemic sclerosis (SSc) is a rare fibrosing disease caused by an autoimmune disorder. Previous studies have unequivocally demonstrated that alterations in the equilibrium between the innate and acquired immune system, shaped by a predisposing genetic background are crucial for the initial disease process along with extrinsic variables [148, 149]. Combinations of immunosuppressive drugs are considered the traditional treatment standards of care, and there is growing evidence for the use of immunosuppression to treat specific complications [150, 151]. Nevertheless, the pressing need to determine the immune-related pathogenesis remains, given that current therapies are not curative [150].
Hypomethylation is common at the CpG site of RSAD2 in blood cells [152]. Different RSAD2 methylation in a single blood cell type in SSc results in further cellular dysregulation [152, 153]. The methylation and overexpression of RSAD2 in the blood of patients exert its function in the immune response via the IFN-I signaling pathway, suggesting its potential role as a sensitive or epigenetic blood biomarker [138, 152].
SSc with pulmonary arterial hypertension (SSc-PAH) is a complication with high mortality and few available evaluation indicators [154, 155]. A recent study has revealed that SSc-PAH patients exhibit higher expression of RSAD2 in PBMCs compared to patients with SSc alone [153]. The upregulation of RSAD2 is involved in the IFN-I signaling pathway and response to viruses. The high relevance of RSAD2 and SSc-PAH has been well studied and RSAD2 has emerged as a precise therapeutic target for SSc-PAH [153].
SS
Sjögren's syndrome (SS) is an incurable multisystem autoimmune disease that causes hypofunction of the salivary and lacrimal glands [156, 157]. In SS patients with autoantibodies to Ro/SSA (SSRo+), a differentially expressed protein-coding RNA RSAD2, is upregulated and involved in cell metabolism and protein processing [158]. Researchers delved into the intricate correlations between the IFN-upregulated gene RSAD2 and differentially expressed lncRNAs in Kasumi-3 cells from 27 SSRo+ subphenotypes [158]. Although these findings point to the significance of RSAD2+ Kasumi-3 cells in the pathogenesis of SS, further work is required to interpret the diagnostic and therapeutic value of RSAD2 in SS.
In fact, there are two distinct types of SS: pSS and secondary SS. pSS is a chronic inflammatory rheumatologic disease that primarily affects exocrine glands, resulting in lymphocyte infiltration and subsequent oral and ocular dryness [159]. In contrast to SS, no additional connective tissue diseases occur in pSS [160]. The cardinal pathogenetic features of pSS lie in the abnormal lymphocyte-mediated, immunoglobulin-mediated and T cell-mediated hyperactivation of B cells [161, 162].
RSAD2 plays a unique role in the pathogenesis of pSS [163]. Mechanistically, endosomal TLR7 and the downstream signaling molecule RSAD2 are upregulated, and the latter conversely promotes TLR7-mediated pathogenic IFN-I production in pDCs [164]. In addition, the expression of RSAD2 and IFIH1 showed a strong correlation in patient monocytes, indicating the potential involvement of RSAD2 in the RLR pathway [164].
RSAD2 is overexpressed in patient CD19+ B cells and may play a role in pSS by modulating B cell hyperactivity [68]. CD40-CD40L is a crucial co-stimulatory pathway for B cell activation. Upon CD40L stimulation, RSAD2 is upregulated in patient CD19+ B cells and activated in antiviral and antitumor immune responses [68, 165]. Whilst it is worth noting that RSAD2 appears to have a greater effect on CD19+ B cells, even after CD40L stimulation. Silence of RSAD2 could downregulate the cellular activity by inhibiting the NF-κB pathway [60]. Analogously, a reduction in NF-κB DNA-binding complex activity was observed in CD4+ T cells lacking RSAD2 [68]. Taken together, RSAD2 might represent a novel therapeutic target in pSS.
MS
The most prevalent chronic inflammatory disease affecting the central nervous system is multiple sclerosis (MS), which involves several susceptible genes in addition to exposure to environmental factors of elusive etiology [166, 167]. However, the prognosis of MS is a multifaceted issue varying considerably across individuals. Herein, biomarkers emerge as invaluable instruments, offering advantages in clinical prediction and optimization of therapy [168].
IFN-β therapy (IFN-β-1a or IFN-β-1b) is one of the significant therapeutic strategies created specifically for the treatment of MS, and its efficacy and safety have been confirmed [169]. Since the advent of IFN-β therapy, patients with relapsing MS have been offered a better approach to alleviating relapse rates and irreversible neurological lesions [170]. Gene expression of RSAD2 and MX1 has been shown to increase in parallel following IFN-β treatment, as evidenced by a study of neural precursor cells expressing the IFN-α receptor in mouse embryos [171, 172]. Apparent differential expression of RSAD2 occurs at different times during the process with IFN-β treatment, and the changes are detectable early and long-lasting [173]. IFN-β exerts its therapeutic effects in therapy by influencing the bioactivity of neutralizing antibodies (NAbs) through binding to IFN-α receptor-induced viperin [172]. In contrast, NAbs significantly reduce the expression of viperin and myxovirus resistance protein A (MxA) and hinder the interaction between IFN-β and IFN-α receptors, thereby compromising the ability to interact and can reduce the therapeutic efficacy of IFN-β [174]. Evidence from both clinical and basic research demonstrated a relationship between RSAD2 and MxA with disease activity and poorer prognosis after IFN-β treatment. And the potency to upregulate RSAD2 of IFN-β at various time points is stronger than MX1, RSAD2 has thus become a novel in vivo biomarker for immune cell responsiveness to IFN-β [175, 176]. As mentioned above, viperin could function as an early clinical biomarker for IFN-β to predict its bioavailability and therapeutic response, which is consistent with the long-term need for IFN-β treatment [174].
Conclusion and Perspectives
The investigation of autoimmune diseases has undergone a transition from a focus on macroscopic clinical manifestations to a concentration on microscopic molecular mechanisms. To better understand the pathophysiology of autoimmune diseases and to identify potential diagnostic biomarkers and therapeutic targets, in-depth clinical and molecular insights are required. The potential role of RSAD2 as a diagnostic biomarker has been demonstrated in numerous autoimmune diseases. However, the challenge is that further research is necessary to ascertain the potential clinical application value of RSAD2 in autoimmune diseases.
Previous experimental studies have investigated the antiviral effect of viperin, and more recently, a correlation between RSAD2, lipid metabolism, and ferroptosis was demonstrated [93, 177]. However, a comprehensive immune-centered perspective is lacking to fully comprehend the function of RSAD2/viperin. Several studies indicated that RSAD2/viperin can regulate mitochondrial metabolism [76, 178]. Nevertheless, it is still unclear how exactly viperin is transported from the ER to the mitochondria and how it contributes to the immune metabolism process. The intricate connection between RSAD2/viperin, mitochondrial metabolism, and autoimmune diseases is worthy of further investigation.
Research has demonstrated that RSAD2 is strongly linked to inflammatory signaling pathways and has an impact on cellular immune metabolism. The suppression of RSAD2 by the use of RSAD2 siRNA resulted in the attenuation of the NF‑κB signaling pathway of CD19+ B cells in pSS [68]. Moreover, following IFN-α stimulation, the production of RSAD2 is enhanced in CD4+ T cells, which in turn affects the differentiation of Th17 and Tfh cells in SLE patients [70]. A growing body of evidence indicates that downregulation of RSAD2 and inhibition of associated signaling pathways may contribute to the alleviation of autoimmune response. For instance, a recent study demonstrated that CSNK1A1 (a serine/threonine protein kinase) can promote the autophagic degradation of STING [179]. This suggests that SSTC3, a selective CSNK1A1 agonist, may indirectly reduce the expression of ISGs such as RSAD2 by inhibiting STING-related signaling pathways, thereby promoting autoimmune homeostasis [179]. Nevertheless, it is vital to recognize that the precise mechanism of RSAD2 in autoimmune diseases, and the applications of downregulation of RSAD2 in autoimmune diseases still require further substantiation.
In a study investigating the potential of mesenchymal stem cells-derived exosomes for neuropathic pain relief, RSAD2 was demonstrated to be downregulated by these exosomes [38]. This suggests that in addition to the targeted inhibition of the high expression of RSAD2 in immune cells as mentioned above, exosomes have the potential for RSAD2-related therapy as a cell-free therapeutic approach, offering the advantages of a targeted effect, minimal toxicity, and lower immunogenicity. In conclusion, RSAD2 offers a perspective on the regulation of immune homeostasis and autoimmune reactivity. The integration of increasingly innovative targets and therapeutic strategies will bring new hope for the treatment of autoimmune diseases.
Data Availability
No datasets were generated or analysed during the current study.
Abbreviations
- RSAD2:
-
Radical S-adenosyl methionine domain-containing 2
- ISG:
-
Interferon-stimulated gene
- Fe-S:
-
Iron-sulfur
- IFN:
-
Interferon
- RA:
-
Rheumatoid arthritis
- SLE:
-
Systemic lupus erythematosus
- pSS:
-
Primary Sjögren's syndrome
- TLR7/9:
-
Toll-like receptor-7 and -9
- pDCs:
-
Plasmacytoid dendritic cells
- IRAK:
-
Interleukin-1 receptor-associated kinase;
- IRF7:
-
Interferon regulatory factor 7
- IFN-I:
-
Type I IFN
- TBK1:
-
Tank-binding kinase 1
- STING:
-
Stimulator of interferon genes
- RIG-I:
-
Retinoic acid-induced gene I
- MAVS:
-
Mitochondrial antiviral signaling protein
- HCMV:
-
Human cytomegalovirus
- ER:
-
Endoplasmic reticulum
- vMIA:
-
Viral mitochondrion-localized inhibitor of apoptosis
- SAM:
-
Central S-adenosyl methionine
- TRAF6:
-
Tumor necrosis factor receptor-associated factor 6
- NF-κB:
-
Nuclear factor kappa B
- MAM:
-
Mitochondria-associated ER membrane
- cGAS:
-
Cyclic GMP-AMP synthase
- HADHB:
-
β-Subunit of the mitochondrial trifunctional protein
- CIAO1/CIA1:
-
Cytosolic Fe-S assembly component 1
- AGS:
-
Aicardi-Goutières syndrome
- DM:
-
Dermatomyositis
- SSc:
-
Systemic sclerosis
- SS:
-
Sjögren's syndrome
- MS:
-
Multiple sclerosis
References
Pisetsky, D.S. 2023. Pathogenesis of autoimmune disease. Nature Reviews Nephrology 19 (8): 509–524. https://doi.org/10.1038/s41581-023-00720-1.
Fugger, L., L.T. Jensen, and J. Rossjohn. 2020. Challenges, progress, and prospects of developing therapies to treat autoimmune diseases. Cell 181 (1): 63–80. https://doi.org/10.1016/j.cell.2020.03.007.
Seo, J.Y., R. Yaneva, and P. Cresswell. 2011. Viperin: A multifunctional, interferon-inducible protein that regulates virus replication. Cell Host & Microbe 10 (6): 534–539. https://doi.org/10.1016/j.chom.2011.11.004.
Fang, Q., T. Li, P. Chen, Y. Wu, T. Wang, L. Mo, et al. 2021. Comparative analysis on abnormal methylome of differentially expressed genes and disease pathways in the immune cells of RA and SLE. Frontiers in Immunology 12: 668007. https://doi.org/10.3389/fimmu.2021.668007.
Feng, X., J. Huang, Y. Liu, L. Xiao, D. Wang, B. Hua, et al. 2015. Identification of interferon-inducible genes as diagnostic biomarker for systemic lupus erythematosus. Clinical Rheumatology 34 (1): 71–79. https://doi.org/10.1007/s10067-014-2799-4.
Imgenberg-Kreuz, J., J.K. Sandling, K.B. Norheim, S.J.A. Johnsen, R. Omdal, A.C. Syvänen, et al. 2021. DNA Methylation-based interferon scores associate with sub-phenotypes in Primary Sjögren’s Syndrome. Frontiers in Immunology 12: 702037. https://doi.org/10.3389/fimmu.2021.702037.
Saitoh, T., T. Satoh, N. Yamamoto, S. Uematsu, O. Takeuchi, T. Kawai, et al. 2011. Antiviral protein Viperin promotes Toll-like receptor 7- and Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Immunity 34 (3): 352–363. https://doi.org/10.1016/j.immuni.2011.03.010.
Crosse, K.M., E.A. Monson, A.B. Dumbrepatil, M. Smith, Y.Y. Tseng, K.H. Van der Hoek, et al. 2021. Viperin binds STING and enhances the type-I interferon response following dsDNA detection. Immunology and Cell Biology 99 (4): 373–391. https://doi.org/10.1111/imcb.12420.
Crow, M.K., M. Olferiev, and K.A. Kirou. 2019. Type I interferons in Autoimmune Disease. Annual Review of Pathology 14: 369–393. https://doi.org/10.1146/annurev-pathol-020117-043952.
Khantisitthiporn, O., B. Shue, N.S. Eyre, C.W. Nash, L. Turnbull, C.B. Whitchurch, et al. 2021. Viperin interacts with PEX19 to mediate peroxisomal augmentation of the innate antiviral response. Life Science Alliance 4 (7). https://doi.org/10.26508/lsa.202000915.
Buskiewicz, I.A., T. Montgomery, E.C. Yasewicz, S.A. Huber, M.P. Murphy, R.C. Hartley, et al. 2016. Reactive oxygen species induce virus-independent MAVS oligomerization in systemic lupus erythematosus. Science Signaling 9 (456): ra115. https://doi.org/10.1126/scisignal.aaf1933.
Dumbrepatil, A.B., K.A. Zegalia, K. Sajja, R.T. Kennedy, and E.N.G. Marsh. 2020. Targeting viperin to the mitochondrion inhibits the thiolase activity of the trifunctional enzyme complex. Journal of Biological Chemistry 295 (9): 2839–2849. https://doi.org/10.1074/jbc.RA119.011526.
Seo, J.Y., and P. Cresswell. 2013. Viperin regulates cellular lipid metabolism during human cytomegalovirus infection. PLoS Pathogens 9 (8): e1003497. https://doi.org/10.1371/journal.ppat.1003497.
Rivera-Serrano, E.E., A.S. Gizzi, J.J. Arnold, T.L. Grove, S.C. Almo, and C.E. Cameron. 2020. Viperin reveals its true function. Annual Review of Virology 7 (1): 421–446. https://doi.org/10.1146/annurev-virology-011720-095930.
Upadhyay, A.S., K. Vonderstein, A. Pichlmair, O. Stehling, K.L. Bennett, G. Dobler, et al. 2014. Viperin is an iron-sulfur protein that inhibits genome synthesis of tick-borne encephalitis virus via radical SAM domain activity. Cellular Microbiology 16 (6): 834–848. https://doi.org/10.1111/cmi.12241.
Hinson, E.R., and P. Cresswell. 2009. The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic alpha-helix. Proceedings of the National Academy of Sciences of the United States of America. 106 (48): 20452–20457. https://doi.org/10.1073/pnas.0911679106.
Hinson, E.R., and P. Cresswell. 2009. The N-terminal amphipathic alpha-helix of viperin mediates localization to the cytosolic face of the endoplasmic reticulum and inhibits protein secretion. Journal of Biological Chemistry 284 (7): 4705–4712. https://doi.org/10.1074/jbc.M807261200.
Wang, X., E.R. Hinson, and P. Cresswell. 2007. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host & Microbe 2 (2): 96–105. https://doi.org/10.1016/j.chom.2007.06.009.
Krebs, C., W.E. Broderick, T.F. Henshaw, J.B. Broderick, and B.H. Huynh. 2002. Coordination of adenosylmethionine to a unique iron site of the [4Fe-4S] of pyruvate formate-lyase activating enzyme: A Mössbauer spectroscopic study. Journal of the American Chemical Society 124 (6): 912–913. https://doi.org/10.1021/ja017562i.
Broderick, W.E., B.M. Hoffman, and J.B. Broderick. 2018. Mechanism of radical initiation in the radical S-Adenosyl-l-methionine superfamily. Accounts of Chemical Research. 51 (11): 2611–2619. https://doi.org/10.1021/acs.accounts.8b00356.
Gizzi, A.S., T.L. Grove, J.J. Arnold, J. Jose, R.K. Jangra, S.J. Garforth, et al. 2018. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 558 (7711): 610–614. https://doi.org/10.1038/s41586-018-0238-4.
Wein, T., and R. Sorek. 2022. Bacterial origins of human cell-autonomous innate immune mechanisms. Nature Reviews Immunology. 22 (10): 629–638. https://doi.org/10.1038/s41577-022-00705-4.
Helbig, K.J., J.M. Carr, J.K. Calvert, S. Wati, J.N. Clarke, N.S. Eyre, et al. 2013. Viperin is induced following dengue virus type-2 (DENV-2) infection and has anti-viral actions requiring the C-terminal end of viperin. PLoS Neglected Tropical Diseases 7 (4): e2178. https://doi.org/10.1371/journal.pntd.0002178.
Vanwalscappel, B., G. Gadea, and P. Desprès. 2019. A Viperin Mutant Bearing the K358R Substitution Lost its Anti-ZIKA Virus Activity. International Journal of Molecular Sciences. 20 (7). https://doi.org/10.3390/ijms20071574.
Subramaniam, P.S., B.A. Torres, and H.M. Johnson. 2001. So many ligands, so few transcription factors: A new paradigm for signaling through the STAT transcription factors. Cytokine 15 (4): 175–187. https://doi.org/10.1006/cyto.2001.0905.
Bach, E.A., M. Aguet, and R.D. Schreiber. 1997. The IFN gamma receptor: A paradigm for cytokine receptor signaling. Annual Review of Immunology 15: 563–591. https://doi.org/10.1146/annurev.immunol.15.1.563.
Pervolaraki, K., S. Rastgou Talemi, D. Albrecht, F. Bormann, C. Bamford, J.L. Mendoza, et al. 2018. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PLoS Pathogens. 14 (11): e1007420. https://doi.org/10.1371/journal.ppat.1007420.
Zhou, Z., O.J. Hamming, N. Ank, S.R. Paludan, A.L. Nielsen, and R. Hartmann. 2007. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. Journal of Virology 81 (14): 7749–7758. https://doi.org/10.1128/jvi.02438-06.
Michalska, A., K. Blaszczyk, J. Wesoly, and H.A.R. Bluyssen. 2018. A Positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type I and type II IFN responses. Frontiers in Immunology 9: 1135. https://doi.org/10.3389/fimmu.2018.01135.
Seth, R.B., L. Sun, C.K. Ea, and Z.J. Chen. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122 (5): 669–682. https://doi.org/10.1016/j.cell.2005.08.012.
Grandvaux, N., M.J. Servant, B. tenOever, G.C. Sen, S. Balachandran, G.N. Barber, et al. 2002. Transcriptional profiling of interferon regulatory factor 3 target genes: Direct involvement in the regulation of interferon-stimulated genes. Journal of Virology 76 (11): 5532–5539. https://doi.org/10.1128/jvi.76.11.5532-5539.2002.
Kaisho, T., and S. Akira. 2006. Toll-like receptor function and signaling. Journal of Allergy and Clinical Immunology. 117 (5): 979–987; quiz 988. https://doi.org/10.1016/j.jaci.2006.02.023.
Fitzgerald, K.A., and J.C. Kagan. 2020. Toll-like receptors and the control of immunity. Cell 180 (6): 1044–1066. https://doi.org/10.1016/j.cell.2020.02.041.
Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11 (1): 115–122. https://doi.org/10.1016/s1074-7613(00)80086-2.
Yamamoto, M., S. Sato, H. Hemmi, K. Hoshino, T. Kaisho, H. Sanjo, et al. 2003. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301 (5633): 640–643. https://doi.org/10.1126/science.1087262.
Patel, A.M., and E.N.G. Marsh. 2021. The antiviral enzyme, viperin, activates protein ubiquitination by the E3 ubiquitin ligase, TRAF6. Journal of the American Chemical Society 143 (13): 4910–4914. https://doi.org/10.1021/jacs.1c01045.
Dumbrepatil, A.B., S. Ghosh, K.A. Zegalia, P.A. Malec, J.D. Hoff, R.T. Kennedy, et al. 2019. Viperin interacts with the kinase IRAK1 and the E3 ubiquitin ligase TRAF6, coupling innate immune signaling to antiviral ribonucleotide synthesis. Journal of Biological Chemistry. 294 (17): 6888–6898. https://doi.org/10.1074/jbc.RA119.007719.
Gao, X., L.F. Gao, Y.N. Zhang, X.Q. Kong, S. Jia, and C.Y. Meng. 2023. Huc-MSCs-derived exosomes attenuate neuropathic pain by inhibiting activation of the TLR2/MyD88/NF-κB signaling pathway in the spinal microglia by targeting Rsad2. International Immunopharmacology. 114: 109505. https://doi.org/10.1016/j.intimp.2022.109505.
Zhou, X., Z. Zhang, H. Xu, B. Zhu, L. Zhang, L. Lie, et al. 2022. Viperin impairs the innate immune response through the IRAK1-TRAF6-TAK1 axis to promote Mtb infection. Science Signaling. 15 (754): eabe1621. https://doi.org/10.1126/scisignal.abe1621.
Loo, Y.M., and M. Gale Jr. 2011. Immune signaling by RIG-I-like receptors. Immunity 34 (5): 680–692. https://doi.org/10.1016/j.immuni.2011.05.003.
Song, J., M. Li, C. Li, K. Liu, Y. Zhu, and H. Zhang. 2022. Friend or foe: RIG- I like receptors and diseases. Autoimmunity Reviews 21 (10): 103161. https://doi.org/10.1016/j.autrev.2022.103161.
Yoneyama, M., M. Kikuchi, T. Natsukawa, N. Shinobu, T. Imaizumi, M. Miyagishi, et al. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nature Immunology 5 (7): 730–737. https://doi.org/10.1038/ni1087.
Hou, F., L. Sun, H. Zheng, B. Skaug, Q.X. Jiang, and Z.J. Chen. 2011. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146 (3): 448–461. https://doi.org/10.1016/j.cell.2011.06.041.
Khan, K.A., A. Marineau, P. Doyon, M. Acevedo, É. Durette, A.C. Gingras, et al. 2021. TRK-Fused Gene (TFG), a protein involved in protein secretion pathways, is an essential component of the antiviral innate immune response. PLoS Pathogens 17 (1): e1009111. https://doi.org/10.1371/journal.ppat.1009111.
Stirnweiss, A., A. Ksienzyk, K. Klages, U. Rand, M. Grashoff, H. Hauser, et al. 2010. IFN regulatory factor-1 bypasses IFN-mediated antiviral effects through viperin gene induction. Journal of Immunology 184 (9): 5179–5185. https://doi.org/10.4049/jimmunol.0902264.
Ishikawa, H., and G.N. Barber. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455 (7213): 674–678. https://doi.org/10.1038/nature07317.
Cai, X., Y.H. Chiu, and Z.J. Chen. 2014. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Molecular Cell 54 (2): 289–296. https://doi.org/10.1016/j.molcel.2014.03.040.
Mukai, K., H. Konno, T. Akiba, T. Uemura, S. Waguri, T. Kobayashi, et al. 2016. Activation of STING requires palmitoylation at the Golgi. Nature Communications 7: 11932. https://doi.org/10.1038/ncomms11932.
Wei, J., Z. Liu, H. Sun, and L. Xu. 2024. Perillaldehyde ameliorates lipopolysaccharide-induced acute lung injury via suppressing the cGAS/STING signaling pathway. International Immunopharmacology 130: 111641. https://doi.org/10.1016/j.intimp.2024.111641.
Ishikawa, H., Z. Ma, and G.N. Barber. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461 (7265): 788–792. https://doi.org/10.1038/nature08476.
Choi, K.M., J.J. Kim, J. Yoo, K.S. Kim, Y. Gu, J. Eom, et al. 2022. The interferon-inducible protein viperin controls cancer metabolic reprogramming to enhance cancer progression. Journal of Clinical Investigation. 132 (24). https://doi.org/10.1172/jci157302.
Giugliano, S., M. Kriss, L. Golden-Mason, E. Dobrinskikh, A.E. Stone, A. Soto-Gutierrez, et al. 2015. Hepatitis C virus infection induces autocrine interferon signaling by human liver endothelial cells and release of exosomes, which inhibits viral replication. Gastroenterology 148 (2): 392-402.e313. https://doi.org/10.1053/j.gastro.2014.10.040.
Pan, M., Y. Yin, T. Hu, X. Wang, T. Jia, J. Sun, et al. 2023. UXT attenuates the CGAS-STING1 signaling by targeting STING1 for autophagic degradation. Autophagy 19 (2): 440–456. https://doi.org/10.1080/15548627.2022.2076192.
Murphy, T.L., G.E. Grajales-Reyes, X. Wu, R. Tussiwand, C.G. Briseño, A. Iwata, et al. 2016. Transcriptional control of dendritic cell development. Annual Review of Immunology 34: 93–119. https://doi.org/10.1146/annurev-immunol-032713-120204.
Jang, J.S., J.H. Lee, N.C. Jung, S.Y. Choi, S.Y. Park, J.Y. Yoo, et al. 2018. Rsad2 is necessary for mouse dendritic cell maturation via the IRF7-mediated signaling pathway. Cell Death & Disease 9 (8): 823. https://doi.org/10.1038/s41419-018-0889-y.
Navegantes, K.C., R. de Souza Gomes, P.A.T. Pereira, P.G. Czaikoski, C.H.M. Azevedo, and M.C. Monteiro. 2017. Immune modulation of some autoimmune diseases: The critical role of macrophages and neutrophils in the innate and adaptive immunity. Journal of Translational Medicine 15 (1): 36. https://doi.org/10.1186/s12967-017-1141-8.
Sica, A., and A. Mantovani. 2012. Macrophage plasticity and polarization: In vivo veritas. Journal of Clinical Investigation 122 (3): 787–795. https://doi.org/10.1172/jci59643.
Li, P., Z. Hao, J. Wu, C. Ma, Y. Xu, J. Li, et al. 2021. Comparative proteomic analysis of polarized human THP-1 and mouse RAW264.7 macrophages. Frontiers in Immunology. 12 700009. https://doi.org/10.3389/fimmu.2021.700009.
Liu, Y.C., X.B. Zou, Y.F. Chai, and Y.M. Yao. 2014. Macrophage polarization in inflammatory diseases. International Journal of Biological Sciences. 10 (5): 520–529. https://doi.org/10.7150/ijbs.8879.
McInnes, I.B., and G. Schett. 2017. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 389 (10086): 2328–2337. https://doi.org/10.1016/s0140-6736(17)31472-1.
Liang, Y., Y. Liang, Q. Wang, Q. Li, Y. Huang, R. Li, et al. 2024. Viperin inhibits interferon-γ production to promote Mycobacterium tuberculosis survival by disrupting TBK1-IKKε-IRF3-axis and JAK-STAT signaling. Inflammation Research. https://doi.org/10.1007/s00011-024-01873-w.
Gupta, S., and M.J. Kaplan. 2021. Bite of the wolf: innate immune responses propagate autoimmunity in lupus. Journal of Clinical Investigation. 131 (3). https://doi.org/10.1172/jci144918.
Garcia-Romo, G.S., S. Caielli, B. Vega, J. Connolly, F. Allantaz, Z. Xu, et al. 2011. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Science Translational Medicine. 3 (73): 73ra20. https://doi.org/10.1126/scitranslmed.3001201.
de Jong, T.D., J. Lübbers, S. Turk, S. Vosslamber, E. Mantel, H.J. Bontkes, et al. 2016. The type I interferon signature in leukocyte subsets from peripheral blood of patients with early arthritis: A major contribution by granulocytes. Arthritis Research & Therapy 18: 165. https://doi.org/10.1186/s13075-016-1065-3.
Hinson, E.R., N.S. Joshi, J.H. Chen, C. Rahner, Y.W. Jung, X. Wang, et al. 2010. Viperin is highly induced in neutrophils and macrophages during acute and chronic lymphocytic choriomeningitis virus infection. Journal of Immunology 184 (10): 5723–5731. https://doi.org/10.4049/jimmunol.0903752.
da Silva, J., C. Hilzendeger, C. Moermans, F. Schleich, M. Henket, T. Kebadze, et al. 2017. Raised interferon-β, type 3 interferon and interferon-stimulated genes - evidence of innate immune activation in neutrophilic asthma. Clinical and Experimental Allergy. 47 (3): 313–323. https://doi.org/10.1111/cea.12809.
Pieper, K., B. Grimbacher, and H. Eibel. 2013. B-cell biology and development. Journal of Allergy and Clinical Immunology. 131 (4): 959–971. https://doi.org/10.1016/j.jaci.2013.01.046.
Zhu, H., J. Zheng, Y. Zhou, T. Wu, and T. Zhu. 2021. Knockdown of RSAD2 attenuates B cell hyperactivity in patients with primary Sjögren’s syndrome (pSS) via suppressing NF-κb signaling pathway. Molecular and Cellular Biochemistry 476 (5): 2029–2037. https://doi.org/10.1007/s11010-021-04070-z.
Wik, J.A., and B.S. Skålhegg. 2022. T cell metabolism in infection. Frontiers in Immunology 13: 840610. https://doi.org/10.3389/fimmu.2022.840610.
Xin, Y., Z. He, Y. Mei, X. Li, Z. Zhao, M. Zhao, et al. 2023. Interferon-α regulates abnormally increased expression of RSAD2 in Th17 and Tfh cells in systemic lupus erythematosus patients. European Journal of Immunology. 53 (8): e2350420. https://doi.org/10.1002/eji.202350420.
Carissimo, G., T.H. Teo, Y.H. Chan, C.Y. Lee, B. Lee, A. Torres-Ruesta, et al. 2019. Viperin controls chikungunya virus-specific pathogenic T cell IFNγ Th1 stimulation in mice. Life Science Alliance. 2 (1). https://doi.org/10.26508/lsa.201900298.
Qiu, L.Q., P. Cresswell, and K.C. Chin. 2009. Viperin is required for optimal Th2 responses and T-cell receptor-mediated activation of NF-kappaB and AP-1. Blood 113 (15): 3520–3529. https://doi.org/10.1182/blood-2008-07-171942.
Maekawa, H., T. Inoue, H. Ouchi, T.M. Jao, R. Inoue, H. Nishi, et al. 2019. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Reports 29 (5): 1261-1273.e1266. https://doi.org/10.1016/j.celrep.2019.09.050.
Smith, J.A. 2020. STING, the endoplasmic reticulum, and mitochondria: Is three a crowd or a conversation? Frontiers in Immunology. 11: 611347. https://doi.org/10.3389/fimmu.2020.611347.
Caielli, S., S. Athale, B. Domic, E. Murat, M. Chandra, R. Banchereau, et al. 2016. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. Journal of Experimental Medicine 213 (5): 697–713. https://doi.org/10.1084/jem.20151876.
Kim, J.J., S. Hong, and J.Y. Seo. 2023. A cysteine residue of human cytomegalovirus vMIA protein plays a crucial role in viperin trafficking to control viral infectivity. Journal of Virology 97 (6): e0187422. https://doi.org/10.1128/jvi.01874-22.
Colberg-Poley, A.M., G.H. Patterson, K. Salka, S. Bhuvanendran, D. Yang, and J.K. Jaiswal. 2015. Superresolution imaging of viral protein trafficking. Medical Microbiology and Immunology 204 (3): 449–460. https://doi.org/10.1007/s00430-015-0395-0.
Hee, J.S., and P. Cresswell. 2017. Viperin interaction with mitochondrial antiviral signaling protein (MAVS) limits viperin-mediated inhibition of the interferon response in macrophages. PLoS ONE 12 (2): e0172236. https://doi.org/10.1371/journal.pone.0172236.
Sarkar, R., S. Nandi, M. Lo, A. Gope, and M. Chawla-Sarkar. 2021. Viperin, an IFN-stimulated protein, delays rotavirus release by inhibiting Non-Structural Protein 4 (NSP4)-induced intrinsic apoptosis. Viruses. 13 (7). https://doi.org/10.3390/v13071324.
Seo, J.Y., R. Yaneva, E.R. Hinson, and P. Cresswell. 2011. Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science 332 (6033): 1093–1097. https://doi.org/10.1126/science.1202007.
Herman, M.A., O.D. Peroni, J. Villoria, M.R. Schön, N.A. Abumrad, M. Blüher, et al. 2012. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 484 (7394): 333–338. https://doi.org/10.1038/nature10986.
Yu, Y., T.G. Maguire, and J.C. Alwine. 2012. Human cytomegalovirus infection induces adipocyte-like lipogenesis through activation of sterol regulatory element binding protein 1. Journal of Virology 86 (6): 2942–2949. https://doi.org/10.1128/jvi.06467-11.
Robinson, G., I. Pineda-Torra, C. Ciurtin, and E.C. Jury. 2022. Lipid metabolism in autoimmune rheumatic disease: implications for modern and conventional therapies. Journal of Clinical Investigation. 132 (2). https://doi.org/10.1172/jci148552.
Sun, W., P. Li, J. Cai, J. Ma, X. Zhang, Y. Song, et al. 2022. Lipid metabolism: Immune regulation and therapeutic prospectives in systemic lupus erythematosus. Frontiers in Immunology 13: 860586. https://doi.org/10.3389/fimmu.2022.860586.
Shaveta, G., J. Shi, V.T. Chow, and J. Song. 2010. Structural characterization reveals that viperin is a radical S-adenosyl-L-methionine (SAM) enzyme. Biochemical and Biophysical Research Communications 391 (3): 1390–1395. https://doi.org/10.1016/j.bbrc.2009.12.070.
Fenwick, M.K., Y. Li, P. Cresswell, Y. Modis, and S.E. Ealick. 2017. Structural studies of viperin, an antiviral radical SAM enzyme. Proceedings of the National Academy of Sciences of the United States of America 114 (26): 6806–6811. https://doi.org/10.1073/pnas.1705402114.
Haldar, S., S. Paul, N. Joshi, A. Dasgupta, and K. Chattopadhyay. 2012. The presence of the iron-sulfur motif is important for the conformational stability of the antiviral protein, Viperin. PLoS ONE 7 (2): e31797. https://doi.org/10.1371/journal.pone.0031797.
Weinstein, A.G., I. Godet, and D.M. Gilkes. 2022. The rise of viperin: the emerging role of viperin in cancer progression. Journal of Clinical Investigation. 132 (24). https://doi.org/10.1172/jci165907.
Lill, R., and S.A. Freibert. 2020. Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annual Review of Biochemistry 89: 471–499. https://doi.org/10.1146/annurev-biochem-013118-111540.
Zheng, L., V.L. Cash, D.H. Flint, and D.R. Dean. 1998. Assembly of iron-sulfur clusters. Identification of an iscSUA-hscBA-fdx gene cluster from Azotobacter vinelandii. Journal of Biological Chemistry. 273 (21): 13264–13272. https://doi.org/10.1074/jbc.273.21.13264.
Freibert, S.A., A.V. Goldberg, C. Hacker, S. Molik, P. Dean, T.A. Williams, et al. 2017. Evolutionary conservation and in vitro reconstitution of microsporidian iron-sulfur cluster biosynthesis. Nature Communications 8: 13932. https://doi.org/10.1038/ncomms13932.
Nairz, M., and G. Weiss. 2020. Iron in infection and immunity. Molecular Aspects of Medicine 75: 100864. https://doi.org/10.1016/j.mam.2020.100864.
Yang, P., J. Lu, P. Zhang, and S. Zhang. 2023. Comprehensive analysis of prognosis and immune landscapes based on lipid-metabolism- and ferroptosis-associated signature in uterine corpus endometrial carcinoma. Diagnostics (Basel). 13 (5). https://doi.org/10.3390/diagnostics13050870.
Zhang, X., D. Bogunovic, B. Payelle-Brogard, V. Francois-Newton, S.D. Speer, C. Yuan, et al. 2015. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature 517 (7532): 89–93. https://doi.org/10.1038/nature13801.
Tonduti, D., E. Fazzi, R. Badolato, and S. Orcesi. 2020. Novel and emerging treatments for Aicardi-Goutières syndrome. Expert Review of Clinical Immunology 16 (2): 189–198. https://doi.org/10.1080/1744666x.2019.1707663.
Crow, Y.J., J. Shetty, and J.H. Livingston. 2020. Treatments in Aicardi-Goutières syndrome. Developmental Medicine and Child Neurology 62 (1): 42–47. https://doi.org/10.1111/dmcn.14268.
Crow, Y.J., D.S. Chase, J. Lowenstein Schmidt, M. Szynkiewicz, G.M. Forte, H.L. Gornall, et al. 2015. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. American Journal of Medical Genetics - Part A. 167a (2): 296–312. https://doi.org/10.1002/ajmg.a.36887.
Rice, G., T. Patrick, R. Parmar, C.F. Taylor, A. Aeby, J. Aicardi, et al. 2007. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. American Journal of Human Genetics 81 (4): 713–725. https://doi.org/10.1086/521373.
Crow, Y.J., and N. Manel. 2015. Aicardi-Goutières syndrome and the type I interferonopathies. Nature Reviews Immunology 15 (7): 429–440. https://doi.org/10.1038/nri3850.
Rice, G., W.G. Newman, J. Dean, T. Patrick, R. Parmar, K. Flintoff, et al. 2007. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. American Journal of Human Genetics 80 (4): 811–815. https://doi.org/10.1086/513443.
Wang, B.X., S.A. Grover, P. Kannu, G. Yoon, R.M. Laxer, E.A. Yeh, et al. 2017. Interferon-stimulated gene expression as a preferred biomarker for disease activity in Aicardi-Goutières Syndrome. Journal of Interferon and Cytokine Research 37 (4): 147–152. https://doi.org/10.1089/jir.2016.0117.
Garau, J., A. Charras, C. Varesio, S. Orcesi, F. Dragoni, J. Galli, et al. 2023. Altered DNA methylation and gene expression predict disease severity in patients with Aicardi-Goutières syndrome. Clinical Immunology 249: 109299. https://doi.org/10.1016/j.clim.2023.109299.
Dalakas, M.C. 2015. Inflammatory muscle diseases. New England Journal of Medicine 372 (18): 1734–1747. https://doi.org/10.1056/NEJMra1402225.
DeWane, M.E., R. Waldman, and J. Lu. 2020. Dermatomyositis: Clinical features and pathogenesis. Journal of the American Academy of Dermatology. 82 (2): 267–281. https://doi.org/10.1016/j.jaad.2019.06.1309.
Dalakas, M.C., and R. Hohlfeld. 2003. Polymyositis and dermatomyositis. Lancet 362 (9388): 971–982. https://doi.org/10.1016/s0140-6736(03)14368-1.
Oddis, C.V., and R. Aggarwal. 2018. Treatment in myositis. Nature Reviews Rheumatology 14 (5): 279–289. https://doi.org/10.1038/nrrheum.2018.42.
Zhang, Y., L. Shan, D. Li, Y. Tang, W. Qian, J. Dai, et al. 2023. Identification of key biomarkers associated with immune cells infiltration for myocardial injury in dermatomyositis by integrated bioinformatics analysis. Arthritis Research & Therapy 25 (1): 69. https://doi.org/10.1186/s13075-023-03052-4.
Liu, W., W.J. Zhao, and Y.H. Wu. 2020. Study on the differentially expressed genes and signaling pathways in dermatomyositis using integrated bioinformatics method. Medicine (Baltimore) 99 (34): e21863. https://doi.org/10.1097/md.0000000000021863.
Matsuda, S., T. Kotani, T. Ishida, K. Fukui, Y. Fujiki, T. Suzuka, et al. 2020. Exploration of pathomechanism using comprehensive analysis of serum cytokines in polymyositis/dermatomyositis-interstitial lung disease. Rheumatology (Oxford) 59 (2): 310–318. https://doi.org/10.1093/rheumatology/kez301.
Burmester, G.R., E. Feist, and T. Dörner. 2014. Emerging cell and cytokine targets in rheumatoid arthritis. Nature Reviews Rheumatology. 10 (2): 77–88. https://doi.org/10.1038/nrrheum.2013.168.
Zheng, Q., D. Wang, R. Lin, Q. Lv, and W. Wang. 2022. IFI44 is an immune evasion biomarker for SARS-CoV-2 and Staphylococcus aureus infection in patients with RA. Frontiers in Immunology 13: 1013322. https://doi.org/10.3389/fimmu.2022.1013322.
Raterman, H.G., S. Vosslamber, S. de Ridder, M.T. Nurmohamed, W.F. Lems, M. Boers, et al. 2012. The interferon type I signature towards prediction of non-response to rituximab in rheumatoid arthritis patients. Arthritis Research & Therapy 14 (2): R95. https://doi.org/10.1186/ar3819.
Smolen, J.S., R.B.M. Landewé, S.A. Bergstra, A. Kerschbaumer, A. Sepriano, D. Aletaha, et al. 2023. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Annals of the Rheumatic Diseases 82 (1): 3–18. https://doi.org/10.1136/ard-2022-223356.
Vosslamber, S., H.G. Raterman, T.C. van der Pouw Kraan, M.W. Schreurs, B.M. von Blomberg, M.T. Nurmohamed, et al. 2011. Pharmacological induction of interferon type I activity following treatment with rituximab determines clinical response in rheumatoid arthritis. Annals of the Rheumatic Diseases 70 (6): 1153–1159. https://doi.org/10.1136/ard.2010.147199.
Asif Amin, M., D.A. Fox, and J.H. Ruth. 2017. Synovial cellular and molecular markers in rheumatoid arthritis. Seminars in Immunopathology 39 (4): 385–393. https://doi.org/10.1007/s00281-017-0631-3.
He, P., Z. Zhang, W. Liao, D. Xu, M. Fu, and Y. Kang. 2016. Screening of gene signatures for rheumatoid arthritis and osteoarthritis based on bioinformatics analysis. Molecular Medicine Reports 14 (2): 1587–1593. https://doi.org/10.3892/mmr.2016.5423.
Kiriakidou, M., and C.L. Ching. 2020. Systemic Lupus Erythematosus. Annals of Internal Medicine. 172 (11): Itc81-itc96. https://doi.org/10.7326/aitc202006020.
Chiche, L., N. Jourde-Chiche, E. Whalen, S. Presnell, V. Gersuk, K. Dang, et al. 2014. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis & Rheumatology 66 (6): 1583–1595. https://doi.org/10.1002/art.38628.
Yu, H., Y. Nagafuchi, and K. Fujio. 2021. Clinical and Immunological Biomarkers for Systemic Lupus Erythematosus. Biomolecules. 11 (7). https://doi.org/10.3390/biom11070928.
Fava, A., and M. Petri. 2019. Systemic lupus erythematosus: Diagnosis and clinical management. Journal of Autoimmunity 96: 1–13. https://doi.org/10.1016/j.jaut.2018.11.001.
Durcan, L., T. O’Dwyer, and M. Petri. 2019. Management strategies and future directions for systemic lupus erythematosus in adults. Lancet 393 (10188): 2332–2343. https://doi.org/10.1016/s0140-6736(19)30237-5.
Zhao, L., X. Hu, F. Xiao, X. Zhang, L. Zhao, and M. Wang. 2022. Mitochondrial impairment and repair in the pathogenesis of systemic lupus erythematosus. Frontiers in Immunology 13: 929520. https://doi.org/10.3389/fimmu.2022.929520.
Khamashta, M., J.T. Merrill, V.P. Werth, R. Furie, K. Kalunian, G.G. Illei, et al. 2016. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: A randomised, double-blind, placebo-controlled study. Annals of the Rheumatic Diseases 75 (11): 1909–1916. https://doi.org/10.1136/annrheumdis-2015-208562.
Joseph, S., N.I. George, B. Green-Knox, E.L. Treadwell, B. Word, S. Yim, et al. 2019. Epigenome-wide association study of peripheral blood mononuclear cells in systemic lupus erythematosus: Identifying DNA methylation signatures associated with interferon-related genes based on ethnicity and SLEDAI. Journal of Autoimmunity 96: 147–157. https://doi.org/10.1016/j.jaut.2018.09.007.
Shen, M., C. Duan, C. Xie, H. Wang, Z. Li, B. Li, et al. 2022. Identification of key interferon-stimulated genes for indicating the condition of patients with systemic lupus erythematosus. Frontiers in Immunology 13: 962393. https://doi.org/10.3389/fimmu.2022.962393.
Chan, V.S., Y.J. Nie, N. Shen, S. Yan, M.Y. Mok, and C.S. Lau. 2012. Distinct roles of myeloid and plasmacytoid dendritic cells in systemic lupus erythematosus. Autoimmunity Reviews 11 (12): 890–897. https://doi.org/10.1016/j.autrev.2012.03.004.
Liao, A.P., M. Salajegheh, C. Morehouse, R. Nazareno, R.G. Jubin, B. Jallal, et al. 2010. Human plasmacytoid dendritic cell accumulation amplifies their type 1 interferon production. Clinical Immunology 136 (1): 130–138. https://doi.org/10.1016/j.clim.2010.02.014.
Katsuyama, T., G.C. Tsokos, and V.R. Moulton. 2018. Aberrant T cell signaling and subsets in systemic lupus erythematosus. Frontiers in Immunology 9: 1088. https://doi.org/10.3389/fimmu.2018.01088.
Moulton, V.R., A. Suarez-Fueyo, E. Meidan, H. Li, M. Mizui, and G.C. Tsokos. 2017. Pathogenesis of human systemic lupus erythematosus: A cellular perspective. Trends in Molecular Medicine 23 (7): 615–635. https://doi.org/10.1016/j.molmed.2017.05.006.
Tsokos, G.C., M.S. Lo, P. Costa Reis, and K.E. Sullivan. 2016. New insights into the immunopathogenesis of systemic lupus erythematosus. Nature Reviews Rheumatology 12 (12): 716–730. https://doi.org/10.1038/nrrheum.2016.186.
Pan, Z., Q. Yang, X. Zhang, X. Xu, Y. Sun, F. Zhou, et al. 2023. TRIM5 promotes systemic lupus erythematosus through CD4(+) T cells and macrophage. International Journal of General Medicine 16: 3567–3580. https://doi.org/10.2147/ijgm.S416493.
Sezin, T., A. Vorobyev, C.D. Sadik, D. Zillikens, Y. Gupta, and R.J. Ludwig. 2017. Gene expression analysis reveals novel shared gene signatures and candidate molecular mechanisms between pemphigus and systemic lupus erythematosus in CD4(+) T cells. Frontiers in Immunology 8: 1992. https://doi.org/10.3389/fimmu.2017.01992.
Rawlings, D.J., G. Metzler, M. Wray-Dutra, and S.W. Jackson. 2017. Altered B cell signalling in autoimmunity. Nature Reviews Immunology 17 (7): 421–436. https://doi.org/10.1038/nri.2017.24.
Banchereau, R., S. Hong, B. Cantarel, N. Baldwin, J. Baisch, M. Edens, et al. 2016. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 165 (6): 1548–1550. https://doi.org/10.1016/j.cell.2016.05.057.
Baechler, E.C., F.M. Batliwalla, G. Karypis, P.M. Gaffney, W.A. Ortmann, K.J. Espe, et al. 2003. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proceedings of the National Academy of Sciences of the United States of America. 100 (5): 2610–2615. https://doi.org/10.1073/pnas.0337679100.
Kato, Y., J. Park, H. Takamatsu, H. Konaka, W. Aoki, S. Aburaya, et al. 2018. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Annals of the Rheumatic Diseases 77 (10): 1507–1515. https://doi.org/10.1136/annrheumdis-2018-212988.
Becker, A.M., K.H. Dao, B.K. Han, R. Kornu, S. Lakhanpal, A.B. Mobley, et al. 2013. SLE peripheral blood B cell, T cell and myeloid cell transcriptomes display unique profiles and each subset contributes to the interferon signature. PLoS ONE 8 (6): e67003. https://doi.org/10.1371/journal.pone.0067003.
Absher, D.M., X. Li, L.L. Waite, A. Gibson, K. Roberts, J. Edberg, et al. 2013. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS Genetics 9 (8): e1003678. https://doi.org/10.1371/journal.pgen.1003678.
Heyn, H., S. Moran, I. Hernando-Herraez, S. Sayols, A. Gomez, J. Sandoval, et al. 2013. DNA methylation contributes to natural human variation. Genome Research 23 (9): 1363–1372. https://doi.org/10.1101/gr.154187.112.
Fan, H., G. Zhao, D. Ren, F. Liu, G. Dong, and Y. Hou. 2017. Gender differences of B cell signature related to estrogen-induced IFI44L/BAFF in systemic lupus erythematosus. Immunology Letters 181: 71–78. https://doi.org/10.1016/j.imlet.2016.12.002.
Yen, E.Y., M. Shaheen, J.M.P. Woo, N. Mercer, N. Li, D.K. McCurdy, et al. 2017. 46-year trends in systemic lupus erythematosus mortality in the United States, 1968 to 2013: A nationwide population-based study. Annals of Internal Medicine 167 (11): 777–785. https://doi.org/10.7326/m17-0102.
Lewis, M.J., and A.S. Jawad. 2017. The effect of ethnicity and genetic ancestry on the epidemiology, clinical features and outcome of systemic lupus erythematosus. Rheumatology (Oxford). 56 (suppl_1): i67-i77. https://doi.org/10.1093/rheumatology/kew399.
Almaani, S., A. Meara, and B.H. Rovin. 2017. Update on lupus nephritis. Clinical Journal of the American Society of Nephrology 12 (5): 825–835. https://doi.org/10.2215/cjn.05780616.
Nakamura, H., Y. Fujieda, S. Yasuda, M. Nakai, and T. Atsumi. 2018. Remission of Nephrotic Syndrome after therapy for chronic hepatitis C virus infection in a patient with systemic lupus erythematosus. Annals of Internal Medicine 169 (5): 352–353. https://doi.org/10.7326/l17-0759.
Sim, J.J.L., and C.C. Lim. 2022. Influenza vaccination in systemic lupus erythematosus: Efficacy, effectiveness, safety, utilization, and barriers. American Journal of Medicine 135 (3): 286-296.e289. https://doi.org/10.1016/j.amjmed.2021.08.038.
Chasset, F., C. Ribi, M. Trendelenburg, U. Huynh-Do, P. Roux-Lombard, D.S. Courvoisier, et al. 2020. Identification of highly active systemic lupus erythematosus by combined type I interferon and neutrophil gene scores vs classical serologic markers. Rheumatology (Oxford) 59 (11): 3468–3478. https://doi.org/10.1093/rheumatology/keaa167.
He, Z., S. Zhou, M. Yang, Z. Zhao, Y. Mei, Y. Xin, et al. 2022. Comprehensive analysis of epigenetic modifications and immune-cell infiltration in tissues from patients with systemic lupus erythematosus. Epigenomics 14 (2): 81–100. https://doi.org/10.2217/epi-2021-0318.
Pope, J.E., C.P. Denton, S.R. Johnson, A. Fernandez-Codina, M. Hudson, and T. Nevskaya. 2023. State-of-the-art evidence in the treatment of systemic sclerosis. Nature Reviews Rheumatology 19 (4): 212–226. https://doi.org/10.1038/s41584-023-00909-5.
Perelas, A., R.M. Silver, A.V. Arrossi, and K.B. Highland. 2020. Systemic sclerosis-associated interstitial lung disease. Lancet Respiratory Medicine 8 (3): 304–320. https://doi.org/10.1016/s2213-2600(19)30480-1.
Denton, C.P., and D. Khanna. 2017. Systemic sclerosis. Lancet 390 (10103): 1685–1699. https://doi.org/10.1016/s0140-6736(17)30933-9.
Marie, I., J.F. Gehanno, M. Bubenheim, A.B. Duval-Modeste, P. Joly, S. Dominique, et al. 2014. Prospective study to evaluate the association between systemic sclerosis and occupational exposure and review of the literature. Autoimmunity Reviews 13 (2): 151–156. https://doi.org/10.1016/j.autrev.2013.10.002.
Ramos, P.S., K.D. Zimmerman, S. Haddad, C.D. Langefeld, T.A. Medsger Jr., and C.A. Feghali-Bostwick. 2019. Integrative analysis of DNA methylation in discordant twins unveils distinct architectures of systemic sclerosis subsets. Clinical Epigenetics 11 (1): 58. https://doi.org/10.1186/s13148-019-0652-y.
Zheng, J.N., Y. Li, Y.M. Yan, H. Shi, T.T. Zou, W.Q. Shao, et al. 2020. Identification and validation of key genes associated with systemic sclerosis-related pulmonary hypertension. Frontiers in Genetics 11: 816. https://doi.org/10.3389/fgene.2020.00816.
Steen, V.D., and T.A. Medsger. 2007. Changes in causes of death in systemic sclerosis, 1972–2002. Annals of the Rheumatic Diseases 66 (7): 940–944. https://doi.org/10.1136/ard.2006.066068.
Le Pavec, J., M. Humbert, L. Mouthon, and P.M. Hassoun. 2010. Systemic sclerosis-associated pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine 181 (12): 1285–1293. https://doi.org/10.1164/rccm.200909-1331PP.
Imgenberg-Kreuz, J., A. Rasmussen, K. Sivils, and G. Nordmark. 2021. Genetics and epigenetics in primary Sjögren’s syndrome. Rheumatology (Oxford) 60 (5): 2085–2098. https://doi.org/10.1093/rheumatology/key330.
Shiboski, C.H., S.C. Shiboski, R. Seror, L.A. Criswell, M. Labetoulle, T.M. Lietman, et al. 2017. 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Primary Sjögren’s Syndrome: A consensus and data-driven methodology involving three international patient cohorts. Arthritis & Rheumatology 69 (1): 35–45. https://doi.org/10.1002/art.39859.
Joachims, M.L., B. Khatri, C. Li, K.L. Tessneer, J.A. Ice, A.M. Stolarczyk, et al. 2022. Dysregulated long non-coding RNA in Sjögren's disease impacts both interferon and adaptive immune responses. RMD Open. 8 (2). https://doi.org/10.1136/rmdopen-2022-002672.
Nocturne, G., and X. Mariette. 2013. Advances in understanding the pathogenesis of primary Sjögren’s syndrome. Nature Reviews Rheumatology 9 (9): 544–556. https://doi.org/10.1038/nrrheum.2013.110.
Psianou, K., I. Panagoulias, A.D. Papanastasiou, A.L. de Lastic, M. Rodi, P.I. Spantidea, et al. 2018. Clinical and immunological parameters of Sjögren’s syndrome. Autoimmunity Reviews 17 (10): 1053–1064. https://doi.org/10.1016/j.autrev.2018.05.005.
Wang-Renault, S.F., S. Boudaoud, G. Nocturne, E. Roche, N. Sigrist, C. Daviaud, et al. 2018. Deregulation of microRNA expression in purified T and B lymphocytes from patients with primary Sjögren’s syndrome. Annals of the Rheumatic Diseases 77 (1): 133–140. https://doi.org/10.1136/annrheumdis-2017-211417.
Brauner, S., L. Folkersen, M. Kvarnström, S. Meisgen, S. Petersen, M. Franzén-Malmros, et al. 2017. H1N1 vaccination in Sjögren’s syndrome triggers polyclonal B cell activation and promotes autoantibody production. Annals of the Rheumatic Diseases 76 (10): 1755–1763. https://doi.org/10.1136/annrheumdis-2016-210509.
Swiecki, M., and M. Colonna. 2010. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunological Reviews 234 (1): 142–162. https://doi.org/10.1111/j.0105-2896.2009.00881.x.
Maria, N.I., E.C. Steenwijk, I.J. AS, C.G. van Helden-Meeuwsen, P. Vogelsang, W. Beumer, et al. 2017. Contrasting expression pattern of RNA-sensing receptors TLR7, RIG-I and MDA5 in interferon-positive and interferon-negative patients with primary Sjögren's syndrome. Annals of the Rheumatic Diseases. 76 (4): 721-730https://doi.org/10.1136/annrheumdis-2016-209589
Moschonas, A., M. Ioannou, and A.G. Eliopoulos. 2012. CD40 stimulates a “feed-forward” NF-κB-driven molecular pathway that regulates IFN-β expression in carcinoma cells. Journal of Immunology 188 (11): 5521–5527. https://doi.org/10.4049/jimmunol.1200133.
Rodríguez Murúa, S., M.F. Farez, and F.J. Quintana. 2022. The immune response in multiple sclerosis. Annual Review of Pathology 17: 121–139. https://doi.org/10.1146/annurev-pathol-052920-040318.
Dobson, R., and G. Giovannoni. 2019. Multiple sclerosis - a review. European Journal of Neurology 26 (1): 27–40. https://doi.org/10.1111/ene.13819.
Oh, J., A. Vidal-Jordana, and X. Montalban. 2018. Multiple sclerosis: Clinical aspects. Current Opinion in Neurology 31 (6): 752–759. https://doi.org/10.1097/wco.0000000000000622.
Jakimovski, D., C. Kolb, M. Ramanathan, R. Zivadinov, and B. Weinstock-Guttman. 2018. Interferon β for multiple sclerosis. Cold Spring Harbor Perspectives in Medicine. 8 (11). https://doi.org/10.1101/cshperspect.a032003.
Cohan, S.L., B.A. Hendin, A.T. Reder, K. Smoot, R. Avila, J.P. Mendoza, et al. 2021. Interferons and multiple sclerosis: Lessons from 25 years of clinical and real-world experience with intramuscular interferon Beta-1a (Avonex). CNS Drugs 35 (7): 743–767. https://doi.org/10.1007/s40263-021-00822-z.
Wellen, J., J. Walter, P. Jangouk, H.P. Hartung, and M. Dihné. 2009. Neural precursor cells as a novel target for interferon-beta. Neuropharmacology 56 (2): 386–398. https://doi.org/10.1016/j.neuropharm.2008.09.011.
Pachner, A.R., K. Narayan, and E. Pak. 2006. Multiplex analysis of expression of three IFNbeta-induced genes in antibody-positive MS patients. Neurology 66 (3): 444–446. https://doi.org/10.1212/01.wnl.0000196467.71646.72.
Serrano-Fernández, P., S. Möller, R. Goertsches, H. Fiedler, D. Koczan, H.J. Thiesen, et al. 2010. Time course transcriptomics of IFNB1b drug therapy in multiple sclerosis. Autoimmunity 43 (2): 172–178. https://doi.org/10.3109/08916930903219040.
Pachner, A.R., J.D. Warth, A. Pace, and S. Goelz. 2009. Effect of neutralizing antibodies on biomarker responses to interferon beta: The INSIGHT study. Neurology 73 (18): 1493–1500. https://doi.org/10.1212/WNL.0b013e3181bf98db.
Pietrzak, A., A. Kalinowska-Łyszczarz, K. Osztynowicz, A. Khamidulla, W. Kozubski, and S. Michalak. 2020. A long-term follow-up study on biochemical and clinical biomarkers of response to interferon beta-1b treatment in relapsing-remitting multiple sclerosis. Advances in Clinical and Experimental Medicine. 29 (7): 841–851. https://doi.org/10.17219/acem/121063.
Malhotra, S., M.F. Bustamante, F. Pérez-Miralles, J. Rio, M.C. Ruiz de Villa, E. Vegas, et al. 2011. Search for specific biomarkers of IFNβ bioactivity in patients with multiple sclerosis. PLoS ONE 6 (8): e23634. https://doi.org/10.1371/journal.pone.0023634.
Yuan, Z., and B. Ding. 2024. Coronavirus hijacks STX18-ATG14 axis-regulated lipophagy to evade an anti-viral effect. Autophagy. 1–2. https://doi.org/10.1080/15548627.2024.2330039.
Yu, H., M. Yu, Z. Li, E. Zhang, and H. Ma. 2022. Identification and analysis of mitochondria-related key genes of heart failure. Journal of Translational Medicine 20 (1): 410. https://doi.org/10.1186/s12967-022-03605-2.
Pan, M., T. Hu, J. Lyu, Y. Yin, J. Sun, Q. Wang, et al. 2023. CSNK1A1/CK1α suppresses autoimmunity by restraining the CGAS-STING1 signaling. Autophagy. 1–18. https://doi.org/10.1080/15548627.2023.2256135.
Funding
This work was supported by Zhejiang College Students Innovative Entrepreneurial Training Program (New Young Talent Program) (2023R413022), National Innovation and Entrepreneurship Training Program for College Students (202310343026), Natural Science Foundation of Zhejiang Province (LTGY23H100001), and Science and Technology Plan Project of Wenzhou, China (Y20220389, Y20220045).
Author information
Authors and Affiliations
Contributions
Manuscript conceptualization CH, SG; Literature search and articles acquisition SC, JY, YL, WC, QY and HQ; Funding acquisition, HQ, CH and SG; Writing original manuscript draft SC, JY, YL, WC; Figures drawing, SC, JY and SH; Final manuscript review and editing, CH, SG, SC, JY, YL, WC, SH, QY and HQ. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Consent for Publication
All authors have consented to the publication.
Open Access
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view acopy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, S., Ye, J., Lin, Y. et al. Crucial Roles of RSAD2/viperin in Immunomodulation, Mitochondrial Metabolism and Autoimmune Diseases. Inflammation (2024). https://doi.org/10.1007/s10753-024-02076-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10753-024-02076-5