
Abstract
Hepatic fibrosis (HF), a precursor to cirrhosis and hepatocellular carcinoma, is caused by abnormal proliferation of connective tissue and excessive accumulation of extracellular matrix in the liver. Notably, activation of hepatic stellate cells (HSCs) is a key link in the development of HF. Phillygenin (PHI, C21H24O6) is a lignan component extracted from the traditional Chinese medicine Forsythiae Fructus, which has various pharmacological activities such as anti-inflammatory, antioxidant and anti-tumour effects. However, whether PHI can directly inhibit HSC activation and ameliorate the mechanism of action of HF has not been fully elucidated. Therefore, the aim of the present study was to investigate the in vitro anti-HF effects of PHI and the underlying molecular mechanisms. Transforming growth factor-β1 (TGF-β1)-activated mouse HSCs (mHSCs) and human HSCs (LX-2 cells) were used as an in vitro model of HF and treated with different concentrations of PHI for 24 h. Subsequently, cell morphological changes were observed under the microscope, cell viability was analyzed by MTT assay, cell cycle and apoptosis were detected by flow cytometry, and the mechanism of anti-fibrotic effect of PHI was explored by immunofluorescence, ELISA, RT-qPCR and western blot. The results showed that PHI suppressed the proliferation of TGF-β1-activated mHSCs and LX-2 cells, arrested the cell cycle at the G0/G1 phase, decreased the levels of α-SMA, Collagen I, TIMP1 and MMP2 genes and proteins, and promoted apoptosis in activated mHSCs and LX-2 cells. Besides, PHI reduced the expression of inflammatory factors in activated mHSCs and LX-2 cells, suggesting a potential anti-inflammatory effect. Mechanically, PHI inhibited TGF-β1-induced HSC activation and inflammation, at least in part through modulation of the Bax/Bcl-2 and Wnt/β-catenin pathways. Overall, PHI has significant anti-HF effects and may be a promising agent for the treatment of HF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatic fibrosis (HF) is a progressive pathological process of abnormal proliferation of connective tissue and excessive accumulation of extracellular matrix (ECM) in the liver caused by a variety of pathogenic factors, including hepatitis viruses, alcohol, chemical toxins, drugs, autoimmune liver diseases and genetic disorders [1, 2]. Meanwhile, HF is a necessary stage for chronic liver disease to progress to cirrhosis or even liver cancer [3]. In particular, activation of hepatic stellate cells (HSCs) plays a crucial role in the development of HF [4]. Specifically, HSCs are resident mesenchymal cells located in the subendothelial space of Disse, retaining characteristics of resident fibroblasts (embedded in normal stromal matrix) and pericytes (attached to capillary endothelial cells) [5]. However, when the liver is stimulated by injurious factors, quiescent HSCs transdifferentiate into proliferative, migratory, and contractile myofibroblasts that exhibit pro-fibrogenic transcriptional and secretory properties and secrete ECM [6]. Furthermore, activated HSCs secrete metalloproteinase inhibitors that not only inhibit matrix metalloproteinase activity but also help activated HSCs escape apoptosis [7, 8]. Hence, inhibiting the activation and proliferation of HSCs and promoting apoptosis of activated HSCs are considered the main strategies against HF.
Currently, a large number of studies have demonstrated that the Wnt/β-catenin signaling pathway plays a central role in the fibrotic process and is associated with the onset and progression of fibrotic diseases in a variety of organs, including HF, renal fibrosis, pulmonary fibrosis and cardiac fibrosis [9,10,11,12]. Among them, aberrant expression of the Wnt/β-catenin signaling pathway is closely associated with HSC activation [13]. In particular, as early as 2006, Jiang et al. [14] found that several receptors, ligands, regulatory proteins, and Wnt target genes in the Wnt/β-catenin signaling pathway were highly upregulated in activated HSCs. Thus, targeting the Wnt/β-catenin pathway to inhibit HSC activation may be a novel and promising anti-HF approach.
Phillygenin (PHI, C21H24O6,) is a lignan component isolated from the traditional Chinese medicine Forsythiae Fructus, which has outstanding pharmacological activities, including anti-inflammatory, antioxidant and anti-tumour activities [15]. Interestingly, due to its excellent anti-inflammatory and antioxidant properties, PHI has been reported in recent years to have hepatoprotective effects in a variety of liver diseases, particularly in HF [16,17,18]. For example, Hu et al. [19, 20] confirmed that Forsythiae Fructuse water extract and the main active ingredient PHI attenuated carbon tetrachloride and lipopolysaccharide-induced HF by inhibiting TLR4/MyD88/NF-κB and TGF-β/Smads signaling pathways. Moreover, Wang et al. [21, 22] found that PHI exerted hepatoprotective effects on CCl4-induced HF in mice by modulating the intestinal microbiota, short-chain fatty acid metabolism and bile acid metabolism. Furthermore, Ma et al. [23] demonstrated that PHI could inhibit HSC activation and thus exert an anti-HF effect by suppressing macrophage exosome miR-125b-5p targeting Stard13. However, whether PHI can directly inhibit HSC activation and ameliorate the mechanism of action of HF has not been fully elucidated. Therefore, using TGF-β1-activated mouse HSCs (mHSCs) and human HSCs (LX-2 cells), the present study aimed to investigate the in vitro anti-HF effects of PHI and its underlying molecular mechanisms.
Materials and Methods
Materials
The experimental materials used in this study are listed in Table 1.
Cell Culture
The mouse HSC line (mHSCs) and the human HSC line (LX-2 cells) were obtained from Sichuan Bio Biological Technology Co., Ltd. (Chengdu, China) and Central South University (Changsha, China), respectively. The mHSCs and LX-2 cells were cultured in 1640 medium supplemented with 10% FBS (v/v) and 1% antibiotics (penicillin and streptomycin) at 37 °C in a humidified incubator with a chamber atmosphere of 5% CO2. The description of the experimental design of this study is shown in Fig. 1.

Description of the experimental design.
Cell Viability Assay
At 80% confluence, the mHSCs and LX-2 cells were harvested and inoculated into 96-well plates at a cell density of 5 × 103 per well for subsequent experiments. Both cells were stimulated with the indicated concentrations of TGF-β1 (0, 5, 10, 15, 20 ng/mL) and treated with different concentrations of PHI solution (0–400 μM) for 24 h. Then, 20 μL of MTT solution (5 mg/mL) was added to each well. After further incubation for 4 h in the dark, the supernatant was discarded, 150 μL of DMSO was added, and the absorbance was measured at 490 nm using a microplate analyzer.
Immunofluorescence Staining
The mHSCs and LX-2 cells were seeded at a density of 1 × 105 in confocal dishes and cultured in an incubator. At approximately 80% confluence, the cells were treated with TGF-β1 (10 ng/mL) in the presence or absence of PHI (25, 50, 100 μM) for 24 h. The cells were rinsed three times with PBS and then fixed with pre-heated 4% paraformaldehyde for 30 min. This was followed by permeabilization with Triton X-100 (weak) for 10 min, occlusion with 5% BSA for 30 min, and incubation with primary antibodies against α-SMA (1:200) and Collagen I (1:200) overnight (4 °C). Next, the cells were incubated with secondary antibody (1:400) for 1 h and stained with DAPI for 10 min in the dark (25 °C). Finally, 100 uL of anti-fluorescence quenching sealant was added to the cells, and images were captured using a laser confocal microscope.
ELISA Assay
The levels of IL-1β, IL-6 and TNF-α in the culture supernatants of mHSCs and LX-2 cells from different treatments, as well as the contents of Wnt1 and β-catenin in the cell extracts, were determined using the corresponding ELISA kits according to the manufacturer's recommended guidelines.
Flow Cytometry
The mHSCs and LX-2 cells treated according to the experimental protocol were resuspended in pre-cooled PBS solution, and 1 mL of pre-cooled 70% ethanol was added dropwise to fix the cells at 4 °C for 12 h. Subsequently, the cells were washed with pre-cooled PBS, and PI staining solution containing RNase A (50X) was added, followed by incubation in the dark at 37 °C for 30 min. Finally, flow cytometry was used to track the cells through the different phases of the cell cycle.
Similarly, the mHSCs and LX-2 cells treated according to the experimental protocol were collected and apoptosis was detected using the Annexin-V FITC kit. Briefly, both cells were digested with trypsin and suspended in binding buffer. Then, 5 μL of both Annexin V-FITC and PI were added to the cell suspension and incubated for 20 min at room temperature in the absence of light. Finally, apoptosis analysis was performed by flow cytometry.
Real‐time Quantitative PCR (RT‐qPCR) Analysis
Total RNA was obtained from mHSCs and LX-2 cells using an animal total RNA isolation kit according to the instructions. The purity of the extracted RNA was assessed using a Nucleic Acid Analyzer based on the OD260/280 value. For cDNA synthesis, 2 × RT OR-Easy™ Mix was used for reverse transcription (reaction conditions: 42 °C for 15 min, 85 °C for 5 min). The StepOnePlus Real-Time PCR System was carried out for the RT-qPCR procedure, which included the addition of 2 × Real PCR Easy™ Mix-SYBR (reaction conditions: 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s and 65 °C for 30 s). All the primers for the qPCR reactions were synthesized by Tsingke Biotechnology Co., Ltd. (Chengdu, China), and the specific primer sequences are listed in Table 2. Gene expression was assessed using the 2−ΔΔCt formula based on the amplification curve.
Western Blot Analysis
The mHSCs and LX-2 cells were washed three times with PBS and 250 μL lysis buffer (RIPA lysate: protein phosphatase inhibitor: PMSF = 100:1:1) was added. After 10 min, the cells were crushed in an ice bath using an ultrasonic cell crusher and the protein supernatants were collected by centrifugation (4 °C, 12,000 rpm, 15 min). In addition, the protein concentration was determined using the BCA Protein Assay Kit according to the manufacturer's instructions and adjusted to a consistent concentration using lysis buffer. Next, the protein loading buffer (loading buffer: total protein = 1:4) was added and heated for 10 min to denature the proteins (100 °C). 8 μL of samples were separated by 10% SDS-PAGE and then transferred to PVDF membrane. Subsequently, the PVDF membrane was immersed in TBST containing 5% BSA for 120 min and incubated with primary antibodies against α-SMA (1:1000), Collagen I (1:1000), TIMP1 (1:1000), MMP2 (1:1000), GAPDH (1:1000), Bax (1:1000), Bcl-2 (1:1000), Wnt1 (1:1000), β-catenin (1:1000), GSK-3β (1:1000), p-GSK-3β (1:1000), c-Myc (1:1000), and Cyclin D1 (1:1000) overnight (4 °C). After thorough washing with TBST, the PVDF membrane was incubated with secondary antibody (1:5000) for 90 min (25 °C). Signals were identified by ECL Kits and exposed under an E-Gel imager. Meanwhile, the net optical density was quantified using Image J software.
Statistical Analysis
All the data were statistically analyzed using SPSS software (version 25.0; SPSS Inc., Chicago, IL, USA) and the results are presented as mean ± SD. Images were drawn using GraphPad Prism 8.0, ChemDraw 20.0 and PowerPoint 16.0 software. All statistics in this study were tested using One-way analysis of variance (ANOVA) or non-parametric tests depending on whether the data were normally distributed. p < 0.05 was considered as the probability threshold for statistical significance. All experiments were performed at least three times.
Results
PHI Inhibited the Proliferation of mHSCs and LX-2 Cells
Firstly, the effects of different concentrations of PHI (0, 3.125, 6.25, 12.5, 25, 50, 100, 200, 400 μM) on the survival of mHSCs and LX-2 cells were evaluated. As shown in Fig. 2a and d, PHI at the dose of 200 μM or higher significantly reduced the survival of both cells. In contrast, PHI at the dose of 100 μM or less did not affect the survival of mHSCs and LX-2 cells. Hence, 3.125, 6.25, 12.5, 25, 50, and 100 μM PHI were selected for subsequent experimental studies. Next, to determine the optimal modeling concentration, mHSCs and LX-2 cells were stimulated with 5, 10, 15 and 20 ng/mL TGF-β1. As shown in Fig. 2b and e, 10 ng/mL or higher doses of TGF-β1 promoted the proliferation of both cells, indicating the occurrence of HSCs activation. Thus, 10 ng/mL TGF-β1 was selected as the optimal modeling concentration to induce HSCs activation.

PHI inhibited the proliferation of mHSCs and LX-2 cells. a The effects of different concentrations of PHI (0–400 μM) on the cell viability of mHSCs (n = 6). *** p < 0.001, compared with the control group. b The effect of TGF-β1 on the proliferation of mHSCs was detected by MTT assay (n = 6). *** p < 0.001, compared with the control group. c The effects of PHI and TGF-β1 treatment on mHSCs viability (n = 6). d The effects of different concentrations of PHI (0–400 μM) on the cell viability of LX-2 cells (n = 6). *** p < 0.001, compared with the control group. e The effect of TGF-β1 on the proliferation of LX-2 cells was detected by MTT assay (n = 6). *** p < 0.001, compared with the control group. f The effects of PHI and TGF-β1 treatment on LX-2 cells viability (n = 6). g The effects of PHI and TGF-β1 treatment on the cell growth of mHSCs and LX-2 cells observed under a microscope (× 100). ### p < 0.001, compared with the control group; ** p < 0.01 and *** p < 0.001, compared with the TGF-β1 group.
Subsequently, the effects of PHI on TGF-β1-induced proliferation of mHSCs and LX-2 cells were further investigated. As presented in Fig. 2c and f, the proliferative capacity of mHSCs and LX-2 cells gradually decreased with increasing concentration of PHI (3.125, 6.25, 12.5, 25, 50, 100 μM). PHI (25, 50, 100 μM) significantly inhibited the proliferation of mHSCs as compared to the model group. Similarly, PHI (12.5, 25, 50, 100 μM) significantly inhibited the proliferation of LX-2 cells compared to the model group. Therefore, 25, 50, and 100 μM PHI were selected as the optimal doses, which showed significant efficacy and did not interfere with normal cell growth. Furthermore, we observed the changes in the growth of mHSCs and LX-2 cells after 24 h of TGF-β1 stimulation and PHI treatment. As shown in Fig. 2g, both cells were in an an inactive state in the absence of TGF-β1 stimulation. After exposure to 10 ng/mL TGF-β1, both cells massively proliferated and started to undergo morphological changes, including clumping, gathering, and stretching. Interestingly, PHI treatment significantly inhibited the proliferation of both cells and significantly restored cell morphology.
PHI Induced G0/G1 Phase Cell Cycle Arrest in TGF-β1-activated mHSCs and LX-2 Cells
The cell life cycle consists of interphase and division phases, which spans the time between each cell division. Therefore, PI-staining flow cytometry was used to analyze the effect of PHI on the cell cycle of TGF-β1-activated mHSCs and LX-2 cells, and the results are shown in Fig. 3a and b. In TGF-β1-activated mHSCs, the percentage of G0/G1 phase was significantly decreased from 46.35 ± 0.75% to 40.12 ± 0.77% (p < 0.001) and the percentage of S phase was significantly increased from 36.45 ± 2.50% to 45.87 ± 1.35% (p < 0.01) compared with the control group. Compared to the TGF-β1 group, the percentage of PHI-treated (100 μM) mHSCs in G0/G1 phase was significantly increased (p < 0.001), whereas the percentage in S phase was significantly decreased (p < 0.05). Similarly, in TGF-β1-activated LX-2 cells, the percentage of G0/G1 phase was significantly reduced from 64.34 ± 0.81% to 57.49 ± 1.48% (p < 0.01) and the percentage of S phase was significantly increased from 24.73 ± 0.99% to 31.36 ± 1.44% (p < 0.001) compared to the control group. Compared to the TGF-β1 group, the percentage of PHI-treated (100 μM) LX-2 cells in G0/G1 phase was significantly increased (p < 0.01), whereas the percentage in S phase was significantly decreased (p < 0.01). These results suggest that PHI may mediate cell cycle arrest in TGF-β1-activated mHSCs and LX-2 cells, which ultimately attenuates the proliferation of mHSCs and LX-2 cells.

PHI induced G0/G1 phase cell cycle arrest in TGF-β1-activated mHSCs and LX-2 cells. a Flow cytometry assay of the cell cycle in mHSCs (n = 3). b Flow cytometry assay of the cell cycle in LX-2 cells (n = 3). ## p < 0.01 and ### p < 0.001, compared with the control group; * p < 0.05 and ** p < 0.01, compared with the TGF-β1 group.
PHI Inhibited TGF-β1-induced HSC Activation in mHSCs and LX-2 Cells
To explore the effect of PHI on TGF-β1-induced HSC activation, we examined the expression of α-SMA and Collagen I in mHSCs and LX-2 cells. Immunofluorescence results (Fig. 4a–f) showed that TGF-β1 stimulation led to a significant increase in the expression of α-SMA and Collagen I compared to the control group, confirming the successful establishment of the HF model in mHSCs and LX-2 cells. PHI treatment significantly reduced the expression of α-SMA and Collagen I compared to the TGF-β1 group.

Immunofluorescence staining of α-SMA and Collagen I in mHSCs and LX-2 cells. a Representative immunofluorescence images of α-SMA and Collagen I in mHSCs (× 400). b The relative fluorescence intensity of α-SMA in mHSCs (n = 3). c The relative fluorescence intensity of Collagen I in mHSCs (n = 3). d Representative immunofluorescence images of α-SMA and Collagen I in LX-2 cells (× 400). e The relative fluorescence intensity of α-SMA in LX-2 cells (n = 3). f The relative fluorescence intensity of Collagen I in LX-2 cells (n = 3). ### p < 0.001, compared with the control group; ** p < 0.01 and *** p < 0.001, compared with the TGF-β1 group.
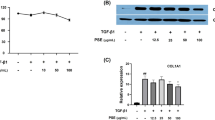
In addition, we tested the mRNA expression levels of fibrosis-associated factors in mHSCs and LX-2 cells, including α-SMA, Collagen I, TIMP1, and MMP2. RT-qPCR results (Fig. 5a and d) showed that PHI significantly down-regulated the TGF-β1-induced increase in α-SMA, Collagen I, TIMP1 and MMP2 gene expression. Consistent with the RT-qPCR results, the protein levels of α-SMA, Collagen I, TIMP1 and MMP2 were increased after TGF-β1 exposure (Fig. 5b, c, e and f). However, PHI down-regulated the increased expression of α-SMA, Collagen I, TIMP1, and MMP2 at the protein level in a dose-dependent manner. Overall, PHI inhibited TGF-β1-induced HSC activation in mHSCs and LX-2 cells.

PHI inhibited TGF-β1-induced HSC activation in mHSCs and LX-2 cells. a The expressions of α-SMA, Collagen I, TIMP1, and MMP2 mRNA in mHSCs (n = 3). b Western blotting analyses of α-SMA, Collagen I, TIMP1, and MMP2 in mHSCs. c Quantitative results of α-SMA, Collagen I, TIMP1, and MMP2 protein in mHSCs (n = 3). d The expressions of α-SMA, Collagen I, TIMP1, and MMP2 mRNA in LX-2 cells (n = 3). e Western blotting analyses of α-SMA, Collagen I, TIMP1, and MMP2 in LX-2 cells. f Quantitative results of α-SMA, Collagen I, TIMP1, and MMP2 protein in LX-2 cells (n = 3). # p < 0.05, ## p < 0.01, and ### p < 0.001, compared with the control group; * p < 0.05, ** p < 0.01 and *** p < 0.001, compared with the TGF-β1 group.
PHI Suppressed TGF-β1-induced Inflammation in mHSCs and LX-2 Cells
To investigate the effect of PHI on TGF-β1-induced inflammation, we detected the expression of inflammatory factors in the culture supernatants of mHSCs and LX-2 cells. ELISA results (Fig. 6a and c) showed that TGF-β1 stimulation significantly increased the secretion of IL-1β, IL-6 and TNF-α. However, different concentrations of PHI reversed the elevated expression of IL-1β, IL-6 and TNF-α. Similarly, we also examined the expression levels of inflammation-related genes in mHSCs and LX-2 cells using RT-qPCR, including IL-1β, IL-6 and TNF-α, which were consistent with the results of the ELISA assay (Fig. 6b and d). Collectively, these data demonstrate that PHI suppresses TGF-β1-induced inflammatory responses in mHSCs and LX-2 cells.

PHI suppressed TGF-β1-induced inflammation in mHSCs and LX-2 cells. a The contents of IL-1β, IL-6, and TNF-α in culture supernatants of mHSCs (n = 3). b The expressions of IL-1β, IL-6, and TNF-α mRNA in mHSCs (n = 3). c The contents of IL-1β, IL-6, and TNF-α in culture supernatants of LX-2 cells (n = 3). d The expressions of IL-1β, IL-6, and TNF-α mRNA in LX-2 cells (n = 3). ### p < 0.001, compared with the control group; * p < 0.05, ** p < 0.01 and *** p < 0.001, compared with the TGF-β1 group.
PHI Promoted the Cell Apoptosis of mHSCs and LX-2 Cells
To explore the influence of PHI on apoptosis in mHSCs and LX-2 cells, we performed cells apoptosis detection by flow cytometry. In particular, the Q2 and Q3 regions represented the late apoptotic and early apoptotic cells, respectively, and their combination indicated the total ratio of cell apoptosis. As shown in Fig. 7a–d, PHI promoted cell apoptosis in mHSCs and LX-2 cells in a dose-dependent manner compared with the TGF-β1 group, suggesting that PHI promotes apoptosis of TGF-β1-induced activated mHSCs and LX-2 cells.

PHI promoted the cell apoptosis of mHSCs and LX-2 cells. a Representative images of flow cytometry detection of apoptosis in mHSCs. b The total mHSCs apoptosis rate (n = 3). c Representative images of flow cytometry detection of apoptosis in LX-2 cells. d The total LX-2 cells apoptosis rate (n = 3). * p < 0.05, ** p < 0.01 and *** p < 0.001, compared with the TGF-β1 group.
PHI Promoted the Cell Apoptosis through the Bax/Bcl-2 Pathway
Subsequently, we selected the Bcl-2 protein family, an important regulator of mitochondria-mediated apoptosis, to study the effects of PHI on apoptosis in mHSCs and LX-2 cells, including the pro-apoptotic protein Bax and the anti-apoptotic protein Bcl-2. RT-qPCR results showed that the mRNA expression of Bax was down-regulated in the TGF-β1 group, while the mRNA expression of Bcl-2 was up-regulated (Fig. 8a, and e). Notably, PHI significantly increased the mRNA expression of Bax and decreased the mRNA expression of Bcl-2. Furthermore, western blot assay also confirmed that PHI reversed the decreased expression of Bax and increased expression of Bcl-2 by TGF-β1 stimulation (Fig. 8b, c, f, and g). More importantly, in mHSCs and LX-2 cells, PHI increased the Bax/Bcl-2 ratio in a concentration-dependent manner (Fig. 8d and h). The ratio is reported to reflect the degree of cell apoptosis. These results suggest that the molecular mechanism by which PHI promotes apoptosis in activated HSCs may be closely related to the regulation of the Bax/Bcl-2 pathway.

PHI promoted the cell apoptosis through the Bax/Bcl-2 pathway. a The expressions of Bax and Bcl-2 mRNA in mHSCs (n = 3). b Western blotting analyses of Bax and Bcl-2 in mHSCs. c Quantitative results of Bax and Bcl-2 protein in mHSCs (n = 3). d The Bax/Bcl-2 ratio in mHSCs (n = 3). e The expressions of Bax and Bcl-2 mRNA in LX-2 cells (n = 3). f Western blotting analyses of Bax and Bcl-2 in LX-2 cells. g Quantitative results of Bax and Bcl-2 protein in LX-2 cells (n = 3). h The Bax/Bcl-2 ratio in LX-2 cells (n = 3). # p < 0.05, ## p < 0.01, and ### p < 0.001, compared with the control group; * p < 0.05, ** p < 0.01 and *** p < 0.001, compared with the TGF-β1 group.
PHI Inhibited the Expression of Wnt1, β-catenin and Cyclin D1 in TGF-β1-induced mHSCs and LX-2 Cells
As is well known, Wnt/β-catenin signaling pathway plays an important role in the proliferation, differentiation, migration, and apoptosis of HSCs. Therefore, the effects of PHI on Wnt1, β-catenin and Cyclin D1 were investigated in TGF-β1-induced mHSCs and LX-2 cells using ELISA and RT-qPCR. ELISA results showed that TGF-β1 significantly increased the contents of Wnt1 and β-catenin in TGF-β1-induced mHSCs and LX-2 cells compared with the control group (Fig. 9a, b, d, and e). Compared to the TGF-β1 group, PHI significantly inhibited the increase of Wnt1 and β-catenin. Meanwhile, RT-qPCR results showed that PHI could significantly down-regulate the expression of Wnt1, β-catenin and Cyclin D1 mRNA, which was consistent with the ELISA results (Fig. 9c and f).

Effect of PHI on Wnt1, β-catenin, and Cyclin D1 in TGF-β1-induced mHSCs and LX-2 cells. a The contents of Wnt1 in mHSCs (n = 3). b The contents of β-catenin in mHSCs (n = 3). c The expressions of Wnt1, β-catenin, and Cyclin D1 mRNA in mHSCs (n = 3). d The contents of Wnt1 in LX-2 cells (n = 3). e The contents of β-catenin in LX-2 cells (n = 3). f The expressions of Wnt1, β-catenin, and Cyclin D1 mRNA in LX-2 cells (n = 3). # p < 0.05, ## p < 0.01, and ### p < 0.001, compared with the control group; * p < 0.05, ** p < 0.01 and *** p < 0.001, compared with the TGF-β1 group.
PHI Inhibited the Wnt/β-catenin Signaling Pathway in TGF-β1-induced mHSCs and LX-2 Cells
Subsequently, the effect of PHI on the Wnt/β-catenin signaling pathway was further investigated in TGF-β1-induced mHSCs and LX-2 cells by western bolt assay. The results showed that the expression of Wnt1, β-catenin, p-GSK-3β, c-Myc and Cyclin D1 was significantly up-regulated in the TGF-β1 group compared to the control group (Fig. 10a–d). Not surprisingly, PHI reversed the expression of proteins related to the Wnt/β-catenin signaling pathway in TGF-β1-induced mHSCs and LX-2 cells. Collectively, PHI inhibits TGF-β1-induced HSC activation at least by inhibiting the Wnt/β-catenin signaling pathway.

Effects of PHI on Wnt/β-catenin signaling pathway-related proteins. a Western blotting analyses of Wnt1, β-catenin, GSK-3β, p-GSK-3β, c-Myc, and Cyclin D1 in mHSCs. b Quantitative results of Wnt1, β-catenin, GSK-3β, p-GSK-3β, c-Myc, and Cyclin D1 protein in mHSCs (n = 3). c Western blotting analyses of Wnt1, β-catenin, GSK-3β, p-GSK-3β, c-Myc, and Cyclin D1 in LX-2 cells. d Quantitative results of Wnt1, β-catenin, GSK-3β, p-GSK-3β, c-Myc, and Cyclin D1 protein in LX-2 cells (n = 3). # p < 0.05, ## p < 0.01, and ### p < 0.001, compared with the control group; * p < 0.05, ** p < 0.01 and *** p < 0.001, compared with the TGF-β1 group.
Discussion
The establishment of appropriate and reproducible models is essential for the study of disease mechanisms and the evaluation of drug efficacy. TGF-β1 is known to be the most potent fibrotic agonist and plays an important role in mediating and promoting the development of HF [24]. Furthermore, HSCs, a major type of fibroblast, are widely used in research areas related to liver disease [5]. Therefore, we chose TGF-β1-induced mHSCs and LX-2 cells as in vitro models of HF and simultaneously intervened with different concentrations of PHI. The results showed that stimulation of mHSCs and LX-2 cells with 10 ng/mL of TGF-β1 for 24 h successfully induced their activation, which is parallel to the results of Peng et al. [25] and Liao et al. [26]. Therefore, 10 ng/mL of TGF-β1 was subsequently selected as the optimal concentration to induce HSCs activation. In addition, PHI inhibited the proliferation of activated mHSCs and LX-2 cells and significantly reversed the morphological changes in mHSCs and LX-2 cells induced by TGF-β1 stimulation, including clumping, gathering, and stretching. More importantly, PI-stained flow cytometry results showed that PHI induced G0/G1 phase cell cycle arrest in TGF-β1-activated mHSCs and LX-2 cells. Similarly, the results of Du et al. [27] showed that PHI was also able to suppress Con A-induced lymphocytes proliferation through G0/G1 cell cycle arrest. Taken together, these results suggest that PHI may mediate cell cycle arrest in TGF-β1-activated mHSCs and LX-2 cells, ultimately attenuating the proliferation of mHSCs and LX-2 cells.
Activated HSCs begin to proliferate rapidly and secrete large amounts of ECM, such as Collagen [28]. Furthermore, α-SMA is a marker of HSC activation, as it is not expressed in the resting state [8]. Therefore, α-SMA and Collagen I are often used experimentally as markers of HF in vitro. The results of immunofluorescence, RT-qPCR and western blot all showed that PHI significantly reversed the TGF-β1-induced increase in α-SMA and Collagen I gene and protein levels. Furthermore, activated HSCs secrete large amounts of metalloproteinase inhibitors (such as TIMP1) to inhibit ECM degradation by matrix metalloproteinases (such as MMP2) [29]. Thus, the level of TIMP1 and MMP2 expression represents the degree of progression of HF. Similarly, both RT-qPCR and western blot results showed that PHI significantly down-regulated the expression levels of TIMP1 and MMP2 genes and proteins. Collectively, these results further suggest that PHI inhibits TGF-β1-induced HSC activation in mHSCs and LX-2 cells.
Activated HSCs secrete not only ECM and metalloproteinase inhibitors, but also a variety of pro-inflammatory cytokines [30]. Subsequently, the increased levels of pro-inflammatory cytokines further promote the activation and proliferation of HSCs, allowing the synthesis of large amounts of ECM, which in turn accelerates the progression of HF [31, 32]. Thus, the inflammatory response plays a very important role in the activation, proliferation and apoptosis of HSCs. Due to the superior anti-inflammatory activity of PHI, we investigated the effect of PHI on TGF-β1-induced inflammation in mHSCs and LX-2 cells. ELISA and RT-qPCR results showed that TGF-β1 stimulation significantly increased the secretion of inflammatory factors, including IL-1β, IL-6, and TNF-α, in the supernatants of mHSCs and LX-2 cell cultures, which was consistent with the results of Zhou et al. [33]. Unsurprisingly, different concentrations of PHI reversed the elevated expression of these inflammatory factors in mHSCs and LX-2 cells induced by TGF-β1 stimulation. Similarly, previous studies have shown that PHI significantly inhibited lipopolysaccharide-induced increases in IL-1β, IL-6 and TNF-α secretion in LX-2 cells and RAW264.7 cells [20, 34]. In a word, these results suggest that PHI attenuates the inflammatory response of activated HSCs and has a potent anti-inflammatory effect.
Under normal conditions, most myofibroblasts actively undergo apoptosis after repair is complete [35]. In contrast, in chronic liver disease, activated HSCs can escape apoptosis by paracrine and autocrine cytokines, which not only prevents the regression of HF, but also promotes its progression to cirrhosis [1, 36]. Hence, the induction of HSC apoptosis has emerged as an important molecular mechanism for the reversibility of HF. Flow cytometry assay showed that PHI significantly promoted apoptosis in activated mHSCs and LX-2 cells. As is well known, the Bax/Bcl-2 signaling pathway is an important signaling molecule in the regulation of apoptosis-dependent pathways [37]. Among them, Bax and Bcl-2 are important intracellular regulators of apoptosis, and their functions are antagonistic to each other [38]. In brief, Bax is pro-apoptotic and Bcl-2 is anti-apoptotic, and together they are involved in the apoptotic process [39]. Subsequently, the results of RT-qPCR and western blot further indicated that PHI could regulate the Bax/Bcl-2 signaling pathway and thus exert pro-apoptotic effects. More importantly, PHI increased the Bax/Bcl-2 ratio in TGF-β1-induced mHSCs and LX-2 cells in a concentration-dependent manner. According to reports, this ratio reflects the degree of cell apoptosis [40]. Taken together, these results suggest that PHI promotes apoptosis in activated HSCs and that the mechanism of apoptosis is at least partially related to the Bax/Bcl-2 pathway.
In recent years, there has been increasing evidence that the Wnt/β-catenin signaling pathway is associated with fibrosis in a variety of organ systems, particularly in the onset and development of HF [10, 12]. Specifically, the Wnt/β-catenin signaling pathway may intervene in the process of HF by modulating inflammatory responses, apoptosis and oxidative stress [13, 41]. Notably, β-catenin is a key transcription factor in the Wnt/β-catenin signaling pathway and an important regulator of cell proliferation and differentiation that mediates signal transduction and cell adhesion [42]. In the resting state, β-catenin is subject to sustained phosphorylation by the degradation complex (consisting of GSK-3β, APC, CK1 and Axin) to the extent that it is present at low levels in the cytoplasm [43]. However, when the Wnt ligand binds to the receptor and co-receptor, dishevelled is recruited and activated, axonin is inactivated, and GSK-3β is phosphorylated (GSK-3β is inactivated), which in turn leads to the escape of degradation of β-catenin and its entry into the nucleus [14, 44]. Subsequently, the activated Wnt/β-catenin signaling pathway overexpresses a series of downstream target genes, such as MMP2, which plays an important role in ECM metabolism and remodeling, c-Myc, which is indispensable in regulating apoptosis in tumour cells, and Cylcin D1, which has a role in regulating the cell cycle, thereby promoting the activation and proliferation of HSCs and further exacerbating HF [45, 46].
Moreover, cytokines such as TGF-β can activate the Wnt/β-catenin pathway, which not only promotes HSC activation and proliferation, but also inhibits apoptosis [47]. Therefore, blocking the Wnt/β-catenin pathway to inhibit HSC proliferation and activation may be a novel therapeutic strategy to attenuate HF. The results of ELISA, RT-qPCR and western blot showed that TGF-β1 significantly increased the contents of Wnt1 and β-catenin in mHSCs and LX-2 cells, which was consistent with the results of Xiong et al. [48], Zhang et al. [49] and Liu et al. [50]. However, PHI treatment significantly suppressed the increase in Wnt1 and β-catenin. In addition, TGF-β1 significantly increased the gene and protein expression of p-GSK-3β, c-Myc and Cyclin D1 in the Wnt/β-catenin signaling pathway. As expected, PHI significantly inhibited the expression of these genes/proteins, suggesting that PHI inhibits TGF-β1-induced HSC activation and inflammation, at least in part, by blocking the Wnt/β-catenin signaling pathway, thereby exerting an anti-HF effect. In general, these results provide a novel anti-fibrotic mechanism for PHI, which may be related to its ability to inhibit the Wnt/β-catenin signaling pathway.
Based on the above, we proposed a potential mechanism for PHI to counteract TGF-β1-induced HSC activation and inflammation (Fig. 11). PHI inhibits TGF-β1-induced HSC activation and inflammation by modulating the Bax/Bcl-2 and Wnt/β-catenin pathways.

The potential mechanisms of PHI inhibition of TGF-β1-induced HSC activation and inflammation.
Conclusions
This study demonstrated that PHI exerted anti-HF effects by inhibiting TGF-β1-induced proliferation of mHSCs and LX-2 cells, arresting the cell cycle at the G0/G1 phase, reducing the expression of several HF markers and pro-inflammatory cytokines, and promoting apoptosis of activated HSCs. Mechanically, PHI inhibited TGF-β1-induced HSC activation and inflammation, at least in part, through the Bax/Bcl-2 and Wnt/β-catenin pathways.
Data Availability
The analyzed data sets generated during the present study will be provided by the corresponding author on reasonable request.
Abbreviations
- HF:
-
Hepatic fibrosis
- ECM:
-
Extracellular matrix
- HSCs:
-
Hepatic stellate cells
- TGF-β1:
-
Transforming growth factor-β1
- PHI:
-
Phillygenin
- mHSCs:
-
Mouse hepatic stellate cells
- FBS:
-
Fetal bovine serum
- MTT:
-
3-(4,5-Dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide
- DMSO:
-
Dimethyl sulfoxide
- PI:
-
Propidium iodide
- IL-1β:
-
Interleukin 1β
- IL-6:
-
Interleukin 6
- TNF-α:
-
Tumor necrosis factor α
- BSA:
-
Bovine serum albumin
- GSK-3β:
-
Glycogen synthase kinase 3β
- α-SMA:
-
α-Smooth muscle actin
- TIMP1:
-
Tissue inhibitors of metalloproteinase 1
- Bax:
-
Bcl-2 associated X
- Bcl-2:
-
B-cell lymphoma 2
- ELISA:
-
Enzyme-linked immunosorbent assay
- RT-qPCR:
-
Real-time quantitative PCR
References
Kisseleva, T., and D. Brenner. 2021. Molecular and cellular mechanisms of liver fibrosis and its regression. Nature Reviews Gastroenterology & Hepatology 18 (3): 151–166.
Berumen, J., J. Baglieri, T. Kisseleva, and K. Mekeel. 2021. Liver fibrosis: Pathophysiology and clinical implications. WIREs Mechanisms of Disease 13 (1): e1499.
Cogliati, B., C.N. Yashaswini, S. Wang, D. Sia, and S.L. Friedman. 2023. Friend or foe? The elusive role of hepatic stellate cells in liver cancer. Nature Reviews Gastroenterology & Hepatology 20 (10): 647–661.
Kumar, S., Q. Duan, R. Wu, E.N. Harris, and Q. Su. 2021. Pathophysiological communication between hepatocytes and non-parenchymal cells in liver injury from NAFLD to liver fibrosis. Advanced Drug Delivery Reviews 176: 113869.
Higashi, T., S.L. Friedman, and Y. Hoshida. 2017. Hepatic stellate cells as key target in liver fibrosis. Advanced Drug Delivery Reviews 121: 27–42.
Friedman, S.L. 2008. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiological Reviews 88 (1): 125–172.
Tsuchida, T., and S.L. Friedman. 2017. Mechanisms of hepatic stellate cell activation. Nature Reviews Gastroenterology & Hepatology 14 (7): 397–411.
Parola, M., and M. Pinzani. 2019. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Molecular Aspects of Medicine 65: 37–55.
Guo, Y., L. Xiao, L. Sun, and F. Liu. 2012. Wnt/beta-catenin signaling: A promising new target for fibrosis diseases. Physiological Research 61 (4): 337–346.
Dees, C., and J.H. Distler. 2013. Canonical Wnt signalling as a key regulator of fibrogenesis - implications for targeted therapies? Experimental Dermatology 22 (11): 710–713.
Hu, H.H., G. Cao, X.Q. Wu, N.D. Vaziri, and Y.Y. Zhao. 2020. Wnt signaling pathway in aging-related tissue fibrosis and therapies. Ageing Research Reviews 60: 101063.
Schunk, S.J., J. Floege, D. Fliser, and T. Speer. 2021. WNT-β-catenin signalling - a versatile player in kidney injury and repair. Nature Reviews Nephrology 17 (3): 172–184.
Nishikawa, K., Y. Osawa, and K. Kimura. 2018. Wnt/β-Catenin Signaling as a Potential Target for the Treatment of Liver Cirrhosis Using Antifibrotic Drugs. International Journal of Molecular Sciences 19 (10): 3103.
Jiang, F., C.J. Parsons, and B. Stefanovic. 2006. Gene expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. Journal of Hepatology 45 (3): 401–409.
Wang, C., R. Wu, S. Zhang, L. Gong, K. Fu, C. Yao, C. Peng, and Y. Li. 2023. A comprehensive review on pharmacological, toxicity, and pharmacokinetic properties of phillygenin: Current landscape and future perspectives. Biomedicine & Pharmacotherapy 166: 115410.
Song, W., J. Wu, L. Yu, and Z. Peng. 2018. Evaluation of the Pharmacokinetics and Hepatoprotective Effects of Phillygenin in Mouse. BioMed Research International 2018: 7964318.
Zhou, W., X. Yan, Y. Zhai, H. Liu, L. Guan, Y. Qiao, J. Jiang, and L. Peng. 2022. Phillygenin ameliorates nonalcoholic fatty liver disease via TFEB-mediated lysosome biogenesis and lipophagy. Phytomedicine 103: 154235.
Guo, J., W.R. Yan, J.K. Tang, X. Jin, H.H. Xue, T. Wang, L.W. Zhang, Q.Y. Sun, and Z.X. Liang. 2022. Dietary phillygenin supplementation ameliorates aflatoxin B(1)-induced oxidative stress, inflammation, and apoptosis in chicken liver. Ecotoxicology and Environmental Safety 236: 113481.
Hu, N., C. Guo, X. Dai, C. Wang, L. Gong, L. Yu, C. Peng, and Y. Li. 2020. Forsythiae Fructuse water extract attenuates liver fibrosis via TLR4/MyD88/NF-κB and TGF-β/smads signaling pathways. Journal of Ethnopharmacology 262: 113275.
Hu, N., C. Wang, X. Dai, M. Zhou, L. Gong, L. Yu, C. Peng, and Y. Li. 2020. Phillygenin inhibits LPS-induced activation and inflammation of LX2 cells by TLR4/MyD88/NF-κB signaling pathway. Journal of Ethnopharmacology 248: 112361.
Wang, C., C. Ma, K. Fu, L.H. Gong, Y.F. Zhang, H.L. Zhou, and Y.X. Li. 2021. Phillygenin Attenuates Carbon Tetrachloride-Induced Liver Fibrosis via Modulating Inflammation and Gut Microbiota. Frontiers in Pharmacology 12: 756924.
Wang, C., C. Ma, K. Fu, Y. Liu, L. Gong, C. Peng, and Y. Li. 2022. Hepatoprotective effect of phillygenin on carbon tetrachloride-induced liver fibrosis and its effects on short chain fatty acid and bile acid metabolism. Journal of Ethnopharmacology 296: 115478.
Ma, C., C. Wang, Y. Zhang, Y. Li, K. Fu, L. Gong, H. Zhou, and Y. Li. 2023. Phillygenin inhibited M1 macrophage polarization and reduced hepatic stellate cell activation by inhibiting macrophage exosomal miR-125b-5p. Biomedicine & Pharmacotherapy 159: 114264.
Kim, K.K., D. Sheppard, H.A. Chapman. 2018. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb Perspect Biol 10(4).
Peng, F., Y. Tian, J. Ma, Z. Xu, S. Wang, M. Tang, J. Lei, G. Gong, and Y. Jiang. 2020. CAT1 silencing inhibits TGF-β1-induced mouse hepatic stellate cell activation in vitro and hepatic fibrosis in vivo. Cytokine 136: 155288.
Liao, J., Z. Zhang, Q. Yuan, L. Luo, and X. Hu. 2022. The mouse Anxa6/miR-9-5p/Anxa2 axis modulates TGF-β1-induced mouse hepatic stellate cell (mHSC) activation and CCl(4)-caused liver fibrosis. Toxicology Letters 362: 38–49.
Du, B., L. Zhang, Y. Sun, G. Zhang, J. Yao, M. Jiang, L. Pan, and C. Sun. 2019. Phillygenin exhibits anti-inflammatory activity through modulating multiple cellular behaviors of mouse lymphocytes. Immunopharmacology and Immunotoxicology 41 (1): 76–85.
Jiao, J., S.L. Friedman, and C. Aloman. 2009. Hepatic fibrosis. Current Opinion in Gastroenterology 25 (3): 223–229.
Arthur, M.J., J.P. Iredale, and D.A. Mann. 1999. Tissue inhibitors of metalloproteinases: Role in liver fibrosis and alcoholic liver disease. Alcoholism, Clinical and Experimental Research 23 (5): 940–943.
Pinzani, M., and F. Marra. 2001. Cytokine receptors and signaling in hepatic stellate cells. Seminars in Liver Disease 21 (3): 397–416.
Koyama, Y., and D.A. Brenner. 2017. Liver inflammation and fibrosis. The Journal of Clinical Investigation 127 (1): 55–64.
Hammerich, L., and F. Tacke. 2023. Hepatic inflammatory responses in liver fibrosis. Nature Reviews Gastroenterology & Hepatology 20 (10): 633–646.
Zhou, M., X. Zhao, L. Liao, Y. Deng, M. Liu, J. Wang, X. Xue, and Y. Li. 2022. Forsythiaside A Regulates Activation of Hepatic Stellate Cells by Inhibiting NOX4-Dependent ROS. Oxidative Medicine and Cellular Longevity 2022: 9938392.
Wang, C., Y. Liu, L. Gong, X. Xue, K. Fu, C. Ma, and Y. Li. 2023. Phillygenin Ameliorates Carbon Tetrachloride-Induced Liver Fibrosis: Suppression of Inflammation and Wnt/β-Catenin Signaling Pathway. Inflammation 46 (4): 1543–1560.
Schuster, R., F. Younesi, M. Ezzo, and B. Hinz. 2023. The Role of Myofibroblasts in Physiological and Pathological Tissue Repair. Cold Spring Harbor Perspectives in Biology 15 (1): a041231.
Hinz, B., and D. Lagares. 2020. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nature Reviews Rheumatology 16 (1): 11–31.
Green, D.R. 2022. The Mitochondrial Pathway of Apoptosis Part II: The BCL-2 Protein Family. Cold Spring Harbor Perspectives in Biology 14 (6): a041046.
Gibson, C.J., and M.S. Davids. 2015. BCL-2 Antagonism to Target the Intrinsic Mitochondrial Pathway of Apoptosis. Clinical Cancer Research 21 (22): 5021–5029.
Edlich, F. 2018. BCL-2 proteins and apoptosis: Recent insights and unknowns. Biochemical and Biophysical Research Communications 500 (1): 26–34.
Jarskog, L.F., E.S. Selinger, J.A. Lieberman, and J.H. Gilmore. 2004. Apoptotic proteins in the temporal cortex in schizophrenia: High Bax/Bcl-2 ratio without caspase-3 activation. American Journal of Psychiatry 161 (1): 109–115.
Perugorria, M.J., P. Olaizola, I. Labiano, A. Esparza-Baquer, M. Marzioni, J.J.G. Marin, L. Bujanda, and J.M. Banales. 2019. Wnt-β-catenin signalling in liver development, health and disease. Nature Reviews. Gastroenterology & Hepatology 16 (2): 121–136.
Lee, J.M., J. Yang, P. Newell, S. Singh, A. Parwani, S.L. Friedman, K.N. Nejak-Bowen, and S.P. Monga. 2014. β-Catenin signaling in hepatocellular cancer: Implications in inflammation, fibrosis, and proliferation. Cancer Letters 343 (1): 90–97.
Miao, C.G., Y.Y. Yang, X. He, C. Huang, Y. Huang, L. Zhang, X.W. Lv, Y. Jin, and J. Li. 2013. Wnt signaling in liver fibrosis: Progress, challenges and potential directions. Biochimie 95 (12): 2326–2335.
Liu, J., Q. Xiao, J. Xiao, C. Niu, Y. Li, X. Zhang, Z. Zhou, G. Shu, and G. Yin. 2022. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduction and Targeted Therapy 7 (1): 3.
Nusse, R., and H. Clevers. 2017. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 169 (6): 985–999.
Koehler, A., J. Schlupf, M. Schneider, B. Kraft, C. Winter, and J. Kashef. 2013. Loss of Xenopus cadherin-11 leads to increased Wnt/β-catenin signaling and up-regulation of target genes c-myc and cyclin D1 in neural crest. Developmental Biology 383 (1): 132–145.
Ahmed, H., M.I. Umar, S. Imran, F. Javaid, S.K. Syed, R. Riaz, and W. Hassan. 2022. TGF-β1 signaling can worsen NAFLD with liver fibrosis backdrop. Experimental and Molecular Pathology 124: 104733.
Xiong, Y., H. Lu, and H. Xu. 2020. Galangin Reverses Hepatic Fibrosis by Inducing HSCs Apoptosis via the PI3K/Akt, Bax/Bcl-2, and Wnt/β-Catenin Pathway in LX-2 Cells. Biological &/and Pharmaceutical Bulletin 43 (11): 1634–1642.
Zhang, C., X.Q. Liu, H.N. Sun, X.M. Meng, Y.W. Bao, H.P. Zhang, F.M. Pan, and C. Zhang. 2018. Octreotide attenuates hepatic fibrosis and hepatic stellate cells proliferation and activation by inhibiting Wnt/β-catenin signaling pathway, c-Myc and cyclin D1. International Immunopharmacology 63: 183–190.
Liu, Z., S. Zhou, Y. Zhang, and M. Zhao. 2022. Rat bone marrow mesenchymal stem cells (BMSCs) inhibit liver fibrosis by activating GSK3β and inhibiting the Wnt3a/β-catenin pathway. Infectious Agents and Cancer 17 (1): 17.
Funding
This study was supported by National Natural Science Foundation of China (No: 81891012, 81630101, and U19A2010), Sichuan TCM Science and Technology Industry Innovation Team (No: 2022C001), Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (No: ZYYCXTD-D-202209), Sichuan Province Science and Technology Support Program (No: 2021JDRC0041, 2022ZYD0088).
Author information
Authors and Affiliations
Contributions
Cheng Wang: conceptualization, methodology, writing-original draft, writing-reviewing and editing. Shenglin Zhang: writing-original draft. Cheng Wang and Yanzhi Li: project administration. Lihong Gong and Chenhao Yao: software, validation. Ke Fu: data curation. Yunxia Li: writing-reviewing and editing, funding acquisition. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
The whole experimental scheme was approved by the Animal Experiment Ethics Committee of Chengdu University of traditional Chinese medicine and carried out under its supervision (Permit number: SYXK 2020-124).
Consent for Publication
All authors have read the manuscript and authorized its publication.
Competing Interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, C., Zhang, S., Li, Y. et al. Phillygenin Inhibits TGF-β1-induced Hepatic Stellate Cell Activation and Inflammation: Regulation of the Bax/Bcl-2 and Wnt/β-catenin Pathways. Inflammation 47, 1403–1422 (2024). https://doi.org/10.1007/s10753-024-01984-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-024-01984-w