
Abstract—
Polygallic acid (PGAL) has been used in vitro to protect synoviocytes from monosodium urate (MSU) crystals due to its anti-inflammatory properties. However, MSU crystals can also activate other cells of the synovial fluid (SF). We studied the impact of PGAL on the phagocytosis of MSU crystals, inflammation, and oxidative stress using an in vitro model with SF leukocytes and THP-1 monocyte cells. SF leukocytes were stimulated with PGAL and MSU crystals, proinflammatory cytokines and phagocytosis were assessed. In THP-1 cells, the effect of PGAL on the phagocytosis of MSU crystals and the levels of IL-1β, IL-6, TNF-α, and reactive oxygen species (ROS) was evaluated. PGAL was added to THP-1 cultures 24 h before MSU crystal addition as a pre-treatment, and IL-1β was measured. One-way ANOVA with Tukey's post hoc test was performed, and a P value < 0.05 was considered statistically significant. PGAL (100 µg/mL) decreased phagocytosis in SF leukocytes by 14% compared to cells exposed to crystals without PGAL. In THP-1 cells, 100 and 200 µg/mL PGAL reduced phagocytosis by 17% and 15%, respectively. In SF cells, there was a tendency to decrease IL-1β and IL-6. In THP-1 cells, decreases in IL-1β and TNF-α, as well as a slight decrease in ROS, were identified. PGAL pre-treatment resulted in a reduction of IL-1β. PGAL inhibits MSU phagocytosis by exerting an anti-inflammatory effect on cells exposed to crystals. The use of PGAL before an acute attack of gout suggests an important protective factor to control the inflammation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Gout is a chronic and disabling disease with a prevalence ranging from < 1 to 6.8% and an incidence of 0.58 to 2.89 per 1000 person-years. These rates vary according to the population studied and the methods used for its evaluation, and these values have increased due to lifestyle-related factors. [1, 2]. Gout causes severe joint pain and is produced by the deposition of monosodium urate crystals (MSU), which are the crystalline form of uric acid (UA), in the joints and soft tissues [3]. UA is soluble in plasma at concentrations < 7 mg/dL, but it crystallizes when it exceeds its saturation point, generating a state of hyperuricaemia, which plays a key role in the development of gout [4].
The pathophysiology of gout involves the recognition of these crystals by cells of the innate immune system and their phagocytosis through Toll-like cytosolic receptors (TLR)-2 and TLR-4 on monocytes/macrophages. This phagocytosis activates signalling pathways leading to inflammation. Acute joint inflammation or flares are triggered by the phagocytosis of MSU crystals by macrophages, which can activate the NOD-like receptor protein 3 (NLRP3) inflammasome [5]. Macrophage activation promotes the translocation of nuclear factor-kappa B (NF-κB), inducing the expression of pro-IL1β and activating ASC, NLRP3, and procaspase-1 proteins [6], which transform the precursor (pro-IL1β) into its active form IL-1β [7]. Additionally, IL-6, TNF-α, and chemokines such as CCL2 are released, recruiting other cells, including neutrophils [8, 9]. Oxidative stress is induced by reactive oxygen species (ROS) and reactive nitrogen species, primarily produced by neutrophils through NADPH oxidase, which can be assembled in the plasma membrane, and intracellular vesicle membranes [10]. Excessive ROS production plays a significant role in the development of acute gout, particularly promoting IL-1β production. The phagolysosomes formed undergo destabilization due to ROS, cytokines release, and myeloperoxidase, resulting in inflammation and oxidative stress leading to pyroptosis [11, 12]. The production of ROS is also influenced by TLR signalling through myeloid differentiation protein (MyD88), and NF-κB, as well as a reduction in intracellular ATP and depolarization of the mitochondrial membrane [13, 14].
Recent experimental and translational research has focussed on understanding the pathophysiology of gout and developing preventive strategies, aiming to contribute to the knowledge about its molecular mechanisms and discover new therapies. Bioactive compounds with antioxidant properties have been investigated for the treatment gout [15]. Polygallic acid (PGAL) has gained attention as an anti-inflammatory molecule with potential therapeutic applications in autoimmune diseases with inflammatory process [16], and recently against gout [17]. PGAL is synthesized by the polyoxidation of gallic acid using laccase in aqueous buffer media [18, 19], provides a highly water-soluble anionic and stable multiradical polymer (ca. 6000 Da) [20]. PGAL has shown cytoprotective effects against MSU crystals and anti-inflammatory properties in synoviocytes. These effects are likely due to electrostatic interaction of PGAL with the cell membrane, and hydrogen bonding to MSU crystals, preventing phagocytosis [17]. As the synovial fluid (SF) is the primary where crystals exert their inflammatory potential, studies are conducted to identify substances that may modulate crystal-induced inflammation [21]. MSU crystals stimulate monocytes to induce IL-6 or IL-8, factors involved in neutrophil migration [22]. Both cells types involved in the immune response to MSU crystals in the SF. Therefore, it is necessary to corroborate their biological effects and highlight PGAL as an adjuvant to gout therapy. This study aims to describes the effect of PGAL on the response of monocytes to MSU crystals, including SF, and to test its antiphagocytic, antioxidant, and anti-inflammatory properties as a potential therapeutic agent against gout.
METHODS
SF was obtained by arthrocentesis of inflamed knees of outpatients (n = 5) who were not taking medication, and they were diagnosed with indeterminate arthritis. These patients were attended to at the outpatient clinic of the Rheumatology Service of the Instituto Nacional de Rehabilitación Luis Guillermo Ibarra Ibarra (INR-LGII) in Mexico City (Mexico). SF was analysed as part of our routine procedures and examinations, including light microscopy for determining the total white blood cell count using a Neubauer chamber (Brand, Germany) [23]. We searched for crystals using compensated polarized light microscopy (Olympus BH2-RFCA, Japan, T2-102880). SF displayed 19,020 ± 10,352.5 leukocytes/mm3. The SF samples were obtained from patients with an average age of 48.6 ± 12.3 years old, of whom 80% were female. Protocol was reviewed and approved by the research committee of INRLGII, number 27/20. For the in vitro model, the human leukemic monocytic cell line THP-1 was obtained from the American Type Culture Collection (Rockville, MD, USA). PGAL was enzymatically synthesized as described elsewhere [18].
SF Cell Culture and Stimulation with PGAL and MSU Crystals
Each SF sample was centrifuged for 5 min at 3800 rpm, and the supernatant was separated. The leukocytes were stratified into four groups, each consisting of 1 × 106 cells, the control group were cells grown in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 2% fetal bovine serum (FBS) and 1% antibiotic-antimycotic (AA) (Gibco, Life Technologies Corporation). Another group of cells was stimulated with sterile MSU crystals (0.100 mg/mL; #15D14-MM InvivoGen) for 24 h. The third group were leukocytes exposed to 100 and 200 μg/mL of PGAL and 0.1 mg/mL of MSU crystals. The fourth group, the vehicle control, had cells exposed to 100 and 200 μg/mL of PGAL in RPMI at 2% of SFB and 1% AA for 24 h. All cells were incubated at 37 °C and with 5% CO2. Supernatant was collected to quantify IL-1β, TNF-α, IL-6, and the MSU phagocytosis index was quantified in cells [23]. This procedure was performed for each of SF sample.
THP-1 Cell Culture and Stimulation with PGAL and MSU Crystals
Monocytes were cultured in RPMI 1640 medium supplemented with 10% FBS and 1% AA (Gibco, Life Technologies Corporation) at 37 °C and with 5% CO2, and the medium was changed every 3 days until confluence. Cells were primed for 3 h with phorbol myristate acetate (PMA-NF-κB activator #05B09-MT; InvivoGen) at 100 ng/mL, followed by overnight incubation in RPMI medium supplemented with 10% FBS and 1% AA. THP-1 cells were stratified into four groups, with 1 × 106 cells each group. Control group consisted of THP-1 cells primed with PMA in RPMI-1640 supplemented with 2% FBS and 1% AA. The second group was stimulated with 0.1 mg/mL MSU crystals for 24 h. The third group were THP-1 cells exposed to 100 and 200 μg/mL PGAL and 0.1 mg/mL MSU crystals. The fourth group of cells was exposed to 100 and 200 μg/mL PGAL in RPMI at 2% FBS and 1% AA for 24 h. The cells were used for calcein-ethidium viability assays, oxidative stress analysis, and MSU phagocytosis assays. Proinflammatory cytokines were assessed in the culture supernatants. To evaluate the protective effect of PGAL, cells primed with PMA were treated with 100 and 200 μg/mL PGAL for 24 h, and then exposed to MSU crystals for 24 h. IL-1β was quantified in the supernatants.
Assessment of Inflammatory Status
IL-1β, IL-6, and TNF-α were quantified using the commercial Peprotech Human Standard ABTS ELISA Development Kit # 900-K95, 900-K16, and 900-K, respectively. 100 µL of the supernatant from the in vitro model with SF leukocytes and from the THP-1 experiments were placed in 96-well microplates (Elisa plate, Nest 515201) previously coated overnight with the corresponding antibodies, following the manufacturer’s instructions. Results were compared against standard curves of IL-1β, IL-6, and TNF-α and are expressed in pg/mL.
Determination of MSU Phagocytosis
The effect of PGAL on MSU phagocytosis was analyzed through their internalization by SF leukocytes and THP-1 monocytes exposed to PGAL, and crystals for 24 h. To identify the internalization of MSU crystals, a cell suspension (50 µL) was placed on a slide previously cleaned with 70% ethanol. Cells were visualized using polarized and compensated light microscopy (Olympus BH2-RFCA, Japan, T2-102880) with a 40× objective. A total of 100 cells were counted. The percentage of cells with at least one MSU in their interior was calculated relative to the total number of cells examined. This percentage is referred to as the phagocytosis index [24].
Analysis of Cell Viability and Death
Cellular viability for THP-1 monocytes was evaluated after 24 h with the designated stimuli using the Live/Dead kit (Viability/Cytotoxicity, Thermo Fisher), following the manufacturer's recommendations. Monocytes were collected in the wells of the culture plates and centrifuged for 5 min at 3800 rpm. The supernatant was removed, and cell pellet washed with PBS (1×) without calcium, and w/o magnesium (Biowest). Cells were resuspended in Hanks’ balanced solution containing 1 µM calcein and 2 µM ethidium homodimer (EthD-1) and incubated for 45 min at 37 °C in 5% CO2 and 95% humidity atmosphere. After incubation, cells were washed with PBS, resuspended, and their emitted fluorescence measured using a BD FACSAria III flow cytometer. The results were expressed as the percentage (%) of calcein- or ethidium homodimer-positive cells. Epifluorescence micrographs were obtained using an Evos epifluorescence microscope (Life Technologies Corp Bothell, WA, USA L1113-178C-173).
Assessment of Oxidative Stress Status
Oxidative stress was evaluated by measuring intracellular ROS using the CellROX Deep Red Reagent (Molecular Probes, Life Technologies C10422). THP-1 cells, after being primed with PMA, were treated with PGAL at the indicated concentrations and stimulated with MSU crystals for 24 h. CellROX Deep Red Reagent (5 µM) was added to the cells and incubated for 30 min at 37 °C, protected from light. Then, the medium was removed, and the cells were washed using PBS. Fluorescence was measured at 644/665 nm using a BD FACSCalibur flow cytometer, according to the manufacturer’s instructions. Data analysis was performed based on fluorescence intensities expressed as arbitrary fluorescence units (AFU) relative to the control. Cell micrographs were obtained using an Evos fluorescence microscope (Life Technologies Corp Bothell, WA, USA L1113-178C-173).
Statistical Analysis
All assays using THP-1 cells were performed in triplicate, and all assays with leukocytes from SF were performed with cells from at least 5 different patients. Results were grouped for one-way ANOVA with Tukey's post hoc test. A P value < 0.05 was used to identify significant differences using a Prism V.9.5.1 (528) software (GraphPad Prism).
RESULTS
Effect of PGAL on MSU Phagocytosis by SF Cells
Experiments were conducted to assess the effect of MSU crystals on synovial cell phagocytosis, which displayed a phagocytosis index of 26 ± 8.67% after 24 h of exposure. However, the addition of 100 µg/mL PGAL resulted in a significant decrease in the phagocytosis (12.75 ± 7.8%). No significant effect on phagocytosis inhibition was observed at the dose of PGAL (200 µg/mL), with a phagocytosis index of 25.5 ± 13.7%, compared to the urate group (Fig. 1).

Effect of PGAL on MSU phagocytosis in SF. PGAL inhibits MSU phagocytosis in SF cells. In cells without PGAL, a greater number of cells were observed with at least one internalized crystal. Images were taken with a 20X objective (n = 5). Bars show quantification of % MSU phagocytosis. Phagocytosis was determined with polarized and compensated light microscopy. Values are expressed as the mean ± standard deviation. *P < 0.05. One-way ANOVA with Tukey's post hoc test.
Effect of PGAL on SF Cytokines
Variability in cell morphology was observed among samples from human SFs across different treatments and the control group. The cells exhibited rounded and heterogeneous shapes, with the majority being in suspension and only a few showing adhesions. Upon the addition of MSU crystals for 24 h, the presence of cell clusters and cells with internalized crystals was observed. PGAL at either dose in cells with MSU crystals, it induced cell clusters, cells with phagocytosed crystals, showing adherence to the culture dish. In cells exposed only to PGAL, we observed an increase in adherence to the treated surface of the culture plate at 100 µg/mL. However, there were fewer adherent cells at 200 µg/mL of PGAL (Fig. 2).

Effect of PGAL on the inflammatory state of SF. SF leukocytes stimulated with MSU crystals and PGAL for 24 h. The bars show the quantification of IL-1β, IL-6, and TNF-α in SF cells (n = 5) exposed to crystals and PGAL. Cytokines were identified by ELISA. Values are expressed as the mean ± standard deviation. *P < 0.05. One-way ANOVA with Tukey's post hoc test.
SF leukocytes showed 215.75 ± 82 pg/mL of IL-1β. MSU crystals increased the production of IL-1β to 746.75 ± 286.58 pg/mL. This cytokine was inhibited when PGAL was added to the SF leukocyte culture. At a concentration of 100 μg/mL of PGAL, IL-1β quantification was 602.25 ± 357.65 pg/mL, and at 200 μg/mL of PGAL, it was 432.75 ± 389.96 pg/mL. However, neither of these doses significantly reduced IL-1β generation. PGAL doses used as vehicles produced 627.5 ± 498.25 and 618 ± 478 pg/mL, respectively (Fig. 2a).
Regarding the IL-6 response, the SF cells presented a baseline level of 2329.0 ± 3293.7 pg/mL. However, when exposed to MSU crystals, the IL-6 levels showed an increasing trend (41,88.0 ± 2771.8 pg/mL). When 100 µg/mL PGAL was added, IL-6 displayed a downward trend to 2034.0 ± 2876.5 pg/mL, while at 200 µg/mL, IL-6 was not detected, similar to that observed for the vehicle control group. Cells treated with 100 µg/mL PGAL alone exhibited 474.0 ± 670.3 pg/mL of IL-6, which is lower than the control group. Meanwhile, at 200 µg/mL PGAL alone, IL-6 was not detected, with non-statistically significant results (Fig. 2b).
The levels of TNF-α showed a tendency to increase in SF cells stimulated with MSU crystals compared to those without stimulation (2316.7 ± 1143.1 vs. 450.0 ± 200.3 pg/mL). The presence of 100 µg/mL PGAL within the cells displayed higher TNF-α concentration than that for the MSU crystal group (3150.0 ± 1425.0 vs. 2316.7 ± 1143.1 pg/mL), although this was not statistically significant. Similarly, with 200 µg/mL PGAL, TNF-α was up to 2666.0 ± 1319 pg/mL, against 2316.7 ± 1143.1 pg/mL in cells stimulated with crystals. Only PGAL at 100 µg/mL and 200 µg/mL induced 1133.3 ± 801.4 and 2016.7 ± 813.2 pg/mL, respectively (Fig. 2c).
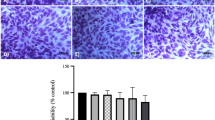
Effect of PGAL on Crystal-Induced Cell Death in THP-1 Cells
Due to the variability observed in studies using human SF samples, we proposed an in vitro model using THP-1 monocytes. Cell death increased by 24.15% in THP-1 cells exposed to MSU crystals over the control group (p < 0.05). In THP-1 cells exposed to 100 µg/mL PGAL and MSU crystals, displayed 31 ± 1.97% cell death, which was similar to that encountered when monocytes were exposed only to MSU crystals (30 ± 4.66%). Cultures stimulated with 200 µg/mL PGAL showed 29.5 ± 5.58% cell death. When PGAL was used as a vehicle, it induced 10.4 ± 0.42% cell death. Therefore, the highest concentration of PGAL (200 µg/mL) induced only 6.6 ± 1.13% cell death. Both doses of PGAL used as vehicles did not induce a significantly increase in cell death with respect to the control (Fig. 3).

Effect of PGAL on cell death induced by MSU crystals. THP-1 cells were stimulated with MSU crystals and PGAL for 24 h. MSU crystals induced an increase in cell death, and PGAL did not prevent crystal-induced cell death. The doses of PGAL used as vehicle were not cytotoxic (n = 4). Images obtained with a 20X objective using fluorescence microscopy (100-nm scale) are representative of one of four independent experiments. Cell death was quantified using ethidium and calcein homodimers by flow cytometry. The values are expressed as the mean ± standard deviation. **P < 0.001. One-way ANOVA with Tukey's post hoc test.
PGAL on the Inflammatory Status of THP-1 Cells Exposed to MSU Crystals
Exposure of monocytes to MSU crystals for 24 h generated an increase of 476 pg/mL in IL-1β levels, considering a baseline of 259 ± 302 pg/mL in THP-1 cells not exposed to MSU crystals. The presence of PGAL at both doses reduced IL-1β when MSU crystals were added to the cultures. At a concentration of 100 µg/mL PGAL, the IL-1β decreased to 706 pg/mL; however, at 200 µg/mL, IL-1β in the supernatants dropped to undetectable levels (P < 0.05). PGAL doses used as vehicle controls, 100 and 200 µg/mL, induced 139.75 ± 270 and 162 ± 273 pg/mL IL-1β, respectively, compared to the control cells (Fig. 4a).

Anti-inflammatory effect of PGAL on gouty inflammation. The use of PGAL in the in vitro model of inflammation induced by MSU crystals significantly decreased the production of IL-1β and TNF-α (n = 3). The columns show the decrements in the amounts of IL-1β, IL-6, and TNF-α induced by MSU in cells treated with PGAL. The quantification of interleukins was performed by ELISA. Values are expressed as the mean ± standard deviation. *P < 0.05. One-way ANOVA with Tukey's post hoc test.
The analysis of the IL-6 showed that the control cells had a baseline concentration of 249.12 ± 201 pg/mL. When MSU crystals were added to the cultures, IL-6 levels increased to 386.02 ± 296.86 pg/mL. However, when 100 µg/mL PGAL was added to monocytes, IL-6 decreased to 94.0 ± 82.0 pg/mL, which was similar to that with 200 µg/mL PGAL (94.8 ± 164.3), although this difference was not statistically significant. PGAL at both doses used as vehicle controls did not induce the production of IL-6 in the cultures, as the levels of IL-6 were undetectable compared to the basal production of this cytokine in the control group (Fig. 4b).
TNF-α exhibited an increasing trend when MSU crystals were added to the cultures (0.45 ± 0.12 pg/mL) compared to control group (0.08 ± 0.15 pg/mL). However, 100 µg/mL PGAL decreased this cytokine significantly, to 0.08 ± 0.06 pg/mL, although this decrease was less pronounced at 200 µg/mL PGAL (0.406 ± 0.35 µg/mL). Doses of 100 µg/mL and 200 µg/mL PGAL in the control group decreased the concentration of TNF-α to 0.01 ± 0.03 and 0.07 ± 0.12 pg/mL, respectively (Fig. 4C).
PGAL as an Inhibitor of MSU Phagocytosis by THP-1 Cells
THP-1 cells that were activated and exposed to MSU for 24 h showed a phagocytosis index for MSU of 69 ± 5.65%. Cells that received PGAL, either 100 or 200 µg/mL, had significantly fewer cells that internalized crystals. One hundred micrograms per milliliter of PGAL resulted in a phagocytosis index of 52 ± 5.73%, while at 200 µg/mL, the urate phagocytosis index was reduced to 54 ± 8.26% (Fig. 5).

Effect of PGAL on the inhibition of MSU phagocytosis by THP-1 monocytes. PGAL inhibits MSU phagocytosis in THP-1 cells. In cells without PGAL, more cells had at least one internalized crystal (n = 4). The points show the quantification of the percentage of phagocytosis in THP-1 cells stimulated with MSU and PGAL for 24 h. Phagocytosis was determined with polarized and compensated light microscopy. Values are expressed as the mean ± standard deviation. *P < 0.05. One-way ANOVA with Tukey's post hoc test.
Bioactivity of PGAL as an Antioxidant in ROS Induced by MSU Crystals
The stimulation of THP-1 cells with MSU crystals resulted in an increased ROS production of 4.36 ± 5.04 arbitrary fluorescence units (AFUs) compared to the control cells. However, when THP-1 cells were exposed to 100 µg/mL of PGAL and MSU crystals, the ROS production decreased to 2.82 ± 2.05 AFU, compared to the MSU crystals group. Similarly, PGAL at 200 µg/mL also decreased to 3.85 ± 4.65 AFU. When THP-1 cells were treated with PGAL at a concentration of 100 µg/mL, the ROS production was observed at 2.05 ± 2.37 AFU, and the highest ROS level was observed with 200 µg/mL PGAL (3.22 ± 4.47 AFU). However, these differences were not statistically significant compared to the control group (Fig. 6).

PGAL as an antioxidant in ROS induced by MSU in THP-1 monocytes. Representative photographs from one of four independent experiments. Scale bar (µm). Bars represent the mean ± standard deviation of independent experiments (n = 4). The fluorescence emitted by the ROS cells was measured by flow cytometry. One-way ANOVA with Tukey's post hoc test.
Preventive Role of PGAL in the Inhibition of Inflammation by MSU Crystals
For this experiment, the cells were pre-treated with PGAL before the stimulation with MSU crystals. For this pre-treatment approach, PGAL was placed with cells for 24 h, followed by washing and subsequent stimulation with MSU crystals for another 24 h. Supernatants were monitored for IL-1β, which had a significant decrease after both doses of PGAL pre-treatment. The cells that did not receive the previous polymer treatment, induced IL-1β secretion at 5483.4 ± 900.9 pg/mL in the supernatant when exposed to the crystals. On the other hand, those cells pre-treated with 100 µg/mL of PGAL displayed 3249.22 ± 1112.34 pg/mL concentration of IL-1β, and for the 200 µg/mL of PGAL group, the cells produced significantly less IL-1β (2906.88 ± 1190.73 pg/mL) (Fig. 7).

PGAL prevents IL-1β induction in monocytes with MSU. Bars represent the mean ± standard deviation of independent experiments (n = 3). Cytokine quantification was performed by ELISA. Values are expressed as the mean ± standard deviation. *P < 0.05. One-way ANOVA with Tukey's post hoc test.
DISCUSSION
Our findings demonstrate the anti-inflammatory and antiphagocytic properties of PGAL. These effects were observed in both the SF model and the in vitro THP-1 cell model, which mimic the conditions of an acute gout attack.
SF cell count has long been recognized as useful in the diagnosis and treatment of many types of arthritis [25]. In this regard, the measurement of SF leukocytes is the most important tool to discriminate inflammatory arthritis from non-inflammatory [25], and the cell count in our SF samples confirms the inflammatory arthritis. This investigation adds MSU crystals to assess the level of induction of inflammation and simulate the SF from gout patients. There are few reports on the use of in vitro human SF models to study of antioxidant or anti-inflammatory potential therapeutic molecules [26,27,28]. Our proposed in vitro model may be useful to determine the preventive effects of bioactive compounds on oxidative stress and inflammation since their reduction have a favorable impact to arthritis treatment [29].
In our SF experiments, PGAL demonstrated limited potential as an anti-inflammatory agent. However, it is important to note that there was no perform cell sorting to isolate specific cell types within the inflammatory infiltrate. The cellular components in the SF primarily consist of neutrophils and monocytes, which have been reported to achieve up to 22% phagocytosis in the joints of asymptomatic gout patients [30]. In a recent study, 21.1% phagocytosis was reported in gout SF samples [31]. In our experimental model using SF leukocytes, we identified a phagocytosis index of 26%. However, when treated with PGAL at a concentration of 100 µg/mL, a noticeable decrease in crystal phagocytosis was observed. The effect of PGAL at 200 µg/mL on crystal phagocytosis in SF leukocytes did not show an impact. In contrast, in the THP-1 cell model, PGAL at 200 µg/mL significantly reduced phagocytosis. The difference between both models lies in the type of cells used, comparing cells derived from SF against a transformed monocytic cell line. In the first case, the phagocytosis index was 26 ± 8.67, while in the second case, it was 69 ± 5.65, respectively. This discrepancy in phagocytosis rates between two models may be attributed to the different cell types involved and their phagocytic behaviors in response to MSU crystals.
This experimental evidence might explain the downward trend in inflammatory mediators, such as IL-1β and IL-6, when PGAL is used. Notwithstanding the expression of cytokines is related to the degree of maturation/differentiation of monocytes/macrophages, and their specific response to MSU crystals [32, 33]. The correlation between the levels of expression in differentiation markers, phagocytic capacities, and production of TNF-α is suggested as a possible mechanism. However, the latter cytokine is not necessarily linked to phagocytic activity. In this regard, cells at an intermediate stage of maturation express higher concentrations of TNF-α, whereas macrophages at a higher maturation stage are unable to produce TNF-α despite the observed phagocytosis [34].
We selected the traditional in vitro model of crystal-induced inflammation with THP-1 monocytes due to the variability in the detection of cytokines [15]. The role of PGAL in preventing the cell death of monocytes exposed to MSU under the experimental times in our model requires further revaluation since it could not reduce the death induced by MSU crystals. The doses of PGAL used as vehicles did not induce cell death, but neither exhibit a cytoprotective effect. This behavior for PGAL differs when human synoviocytes are used, showing cytoprotective characteristics [17]. This outcome provides further insight into the role that different cells play during gouty inflammation. Synovial macrophages initiate and drive inflammation, whereas monocyte-derived macrophages differentiate into M1 macrophages in response to MSU crystals [35, 36]. Therefore, it is worth to investigate the effect of each one for joint tissue inflammation. In our study with THP-1 cells, we found that PGAL downregulated IL-1β, IL-6, and TNF-α, with certain differences among doses. At 100 and 200 µg/mL of PGAL, there was a significant decrease in IL-1β and TNF-α, in addition to inhibiting MSU phagocytosis.
It is of note that crystal phagocytosis preceded IL-1β release in different cellular models of MSU crystal-induced inflammation. Baggio et al. demonstrated a positive correlation between the phagocytosis index and the levels of proinflammatory cytokines in SF, as well as a negative correlation with local inflammatory indices, such as IL-1β, and the percentage of mononuclear cells with phagocytosed crystals [28]. Even so, inhibition of IL-1β production is considered a therapeutic target in gout. Some authors have observed that pre-treatment of THP-1 cells with polyphenols can prevent their response to stimulation with MSU crystals [24, 37], which is consistent with our study where we observed a decrease in IL-1β levels with PGAL pre-treatment. These findings evidence the ability of this antioxidant to inhibit the process of crystal phagocytosis, thereby preventing inflammation.
NLRP3 has been identified as a potential predictor of inflammation in gout, making it a possible target for treatments interventions. Another is the release of ROS and pro-inflammatory mediators such as IL-1β and TNF-α leading to cell damage and lipid peroxidation. Therefore, reducing the inflammation caused by the oxidative stress could improve the symptoms. These thresholds should be considered for the development of therapeutic strategies [38]. Our results suggest that PGAL can modulate the oxidative stress during gouty inflammation, although we observed only trends in the decrease in ROS levels. Noteworthy, despite the representative benefits of this model for studying the causal relationships of oxidative stress in living systems, it has some limitations inherent to the culture conditions, which can lead to overproduction of ROS [39, 40]. A previous study demonstrated the antioxidant effect of PGAL-grafted poly-ε-caprolactone films on UV-photoinduced epithelial cells, showing a good protection against ROS [41]. Similar findings were reported for human synoviocytes and dermal fibroblasts [17, 42], in which PGAL also exhibited a good antioxidant capacity against ROS.
The polyanionic structure of PGAL may exert an electrostatic interaction possibility with cell membrane, and bonding to MSU crystals, thus preventing the crystals recognition by innate immune cells inhibiting phagocytosis [17]. Indeed, it has been proposed that protein coating could modulate the interaction between cell membranes and MSU crystals, thereby impacting their inflammatory properties [43, 44]. Additionally, further investigation is required to support that PGAL might undergo a protective barrier on cell membranes through coupling with TLRs.
We could detect an association between PGAL and the inhibition of phagocytosis, as well as of inflammation, in the in vitro model of THP-1 cells. Further work is undergoing to describe the modulation of other ROS, including the superoxide anion, hydrogen peroxide, and even nitric oxide. In addition, it is worthwhile to analyze the gene and protein expression of TLRs and NLRP3. Furthermore, ongoing molecular dynamics and docking studies at the interface of TLRs with PGAL, aiming to propose a binding model between these two molecules.
CONCLUSIONS
This work shows that the exposure of PGAL before the activation of cells of the immune system by MSU decreases phagocytosis and pro-inflammatory cytokines. This suggests an anti-inflammatory effect that could also regulate oxidative stress in acute gout attacks. The mechanisms of regulation of PGAL in gout will be identified to determine the molecular pathways by which PGAL regulates inflammatory phagocytic activity in cells. In the future, the inhibition of MSU phagocytosis by PGAL in innate immune cells could be used as a target for the control of crystal-induced inflammation.
Data Availability
The data that support the findings of this study are available on request from the corresponding author.
References
Dehlin, M., L. Jacobsson, and E. Roddy. 2020. Global epidemiology of gout: prevalence, incidence, treatment patterns and risk factors. Nature Reviews Rheumatology 16: 380–390.
Wen, P., P. Luo, B. Zhang, and Y. Zhang. 2022. Mapping knowledge structure and global research trends in gout: a bibliometric analysis from 2001 to 2021. Frontiers in Public Health 29 (10): 924676.
Ahmad, M.I., S. Masood, D.M. Furlanetto, and S. Nicolaou. 2021. Urate crystals; beyond joints. Frontiers in Medicine 8: 649505.
Oda, M., I. Hirasawa, and F. Kohori. 2020. Analysis of morphological changes in monosodium urate monohydrate crystals for gout treatment. Chemical Engineering and Technology 43: 1087–1092.
Popov, D., L. Jain, M. Alhilali, N. Dalbeth, and R.C. Poulsen. 2023. Monosodium urate crystals alter the circadian clock in macrophages leading to loss of NLRP3 inflammasome repression: Implications for timing of the gout flare. The FASEB Journal 37: e22940.
Qadri, M., G.D. Jay, L.X. Zhang, W. Wong, A.M. Reginato, C. Sun, T.A. Schmidt, and K.A. Elsaid. 2018. Recombinant human proteoglycan-4 reduces phagocytosis of urate crystals and downstream nuclear factor kappa B and inflammasome activation and production of cytokines and chemokines in human and murine macrophages. Arthritis Research & Therapy 20: 192.
Bousoik, E., M. Qadri, and K.A. Elsaid. 2020. CD44 receptor mediates urate crystal phagocytosis by macrophages and regulates inflammation in a murine peritoneal model of acute gout. Science and Reports 10: 5748.
Fernandes, M.J., and P.H. Naccache. 2018. The role of inhibitory receptors in monosodium urate crystal-induced inflammation. Frontiers in Immunology 9: 1883.
Vedder, D., M. Gerritsen, B. Duvvuri, R.F. van Vollenhoven, M.T. Nurmohamed, and C. Lood. 2020. Neutrophil activation identifies patients with active polyarticular gout. Arthritis Research & Therapy 22: 148.
Davidsson, L., A. Dahlstrand Rudin, F.P. Sanchez Klose, A. Buck, L. Björkman, K. Christenson, and J. Bylund. 2020. In vivo transmigrated human neutrophils are highly primed for intracellular radical production induced by monosodium urate crystals. International Journal of Molecular Sciences 21: 3750.
Desai, J., S. Steiger, and H.J. Anders. 2017. Molecular pathophysiology of gout. Trends in Molecular Medicine 23: 756–768.
Balik, A.R., A. Omma, S.C. Sandikci, C. Yucel, M. Kizilgun, Z.B. Balik, E.F. Oguz, S. Neselioglu, and O. Erel. 2022. Evaluation of oxidative stress in gout disease; thiol-disulfide homeostasis and ischemia-modified albumin levels. International Journal of Medical Biochemistry 5: 109–115.
Cabău, G., T.O. Crișan, V. Klück, R.A. Popp, and L.A.B. Joosten. 2020. Urate-induced immune programming: consequences for gouty arthritis and hyperuricemia. Immunological Reviews 294: 92–105.
Pan, Y.G., M.T. Huang, P. Sekar, D.Y. Huang, W.W. Lin, and S.L. Hsieh. 2021. Decoy receptor 3 inhibits monosodium urate-induced NLRP3 inflammasome activation via reduction of reactive oxygen species production and lysosomal rupture. Frontiers in Immunology 3 (12): 638676.
Scanu, A., R. Luisetto, R. Ramonda, P. Spinella, P. Sfriso, P. Galozzi, and F. Oliviero. 2022. Anti-inflammatory and hypouricemic effect of bioactive compounds: molecular evidence and potential application in the management of gout. Current Issues in Molecular Biology 44: 5173–5190.
Zamudio-Cuevas, Y., M.A. Andonegui-Elguera, A. Aparicio-Juárez, E. Aguillón-Solís, K. Martínez-Flores, E. Ruvalcaba-Paredes, et al. 2021. The enzymatic poly(gallic acid) reduces pro-inflammatory cytokines in vitro, a potential application in inflammatory diseases. Inflammation 44: 174–185.
Zamudio-Cuevas, Y., V. Martínez-López, I.A. Luján-Juárez, N. Montaño-Armendariz, K. Martínez-Flores, J. Fernández-Torres, M. Gimeno, and R. Sánchez-Sánchez. 2022. Anti-inflammatory and antioxidant effect of poly-gallic acid (PGAL) in an in vitro model of synovitis induced by monosodium urate crystals. Inflammation 45: 2066–2077.
López, J., J.M. Hernández-Alcántara, P. Roquero, C. Montiel, K. Shirai, M. Gimeno, and E. Bárzana. 2013. Trametes versicolor laccase oxidation of gallic acid toward apolyconjugated semiconducting material. Journal of Molecular Catalysis B: Enzymatic 97: 100–105.
Vona, D., G. Buscemi, R. Ragni, M. Cantore, S. Cicco, G. Farinola, and M. Trotta. 2020. Synthesis of (poly)gallic acid in a bacterial growth medium. MRS Advances 5: 957–963.
Romero-Montero, A., M. Gimeno, N. Farfán, and P. Labra-Vázquez. 2019. Enzymatic poly (gallic acid): a stable multiradical polyanion. Journal of Molecular Structure 1197: 326–335.
Scanu, A., F. Oliviero, L. Gruaz, P. Galozzi, R. Luisetto, R. Ramonda, D. Burger, and L. Punzi. 2016. Synovial fluid proteins are required for the induction of interleukin-1β production by monosodium urate crystals. Scandinavian Journal of Rheumatology 45: 384–393.
Jeong, J.H., J.H. Jung, J.S. Lee, J.S. Oh, Y.G. Kim, C.K. Lee, B. Yoo, and S. Hong. 2019. Prominent inflammatory features of monocytes/macrophages in acute calcium pyrophosphate crystal arthritis: a comparison with acute gouty arthritis. Immune Network 19: e21.
Zamudio-Cuevas, Y., G.A. Martínez-Nava, K. Martínez-Flores, L. Ventura-Ríos, J. Vazquez-Mellado, P. Rodríguez-Henríquez, C. Pineda, R. Franco-Cendejas, C.A. Lozada-Pérez, and J. Fernández-Torres. 2021. Synovial fluid analysis for the enhanced clinical diagnosis of crystal arthropathies in a tertiary care institution. Clinical Rheumatology 40: 3239–3246.
Oliviero, F., Y. Zamudio-Cuevas, E. Belluzzi, L. Andretto, A. Scanu, M. Favero, R. Ramonda, G. Ravagnan, A. López-Reyes, P. Spinella, and L. Punzi. 2019. Polydatin and resveratrol inhibit the inflammatory process induced by urate and pyrophosphate crystals in THP-1 cells. Foods 8: 560.
Huang, Q., Y. Huang, X. Guo, J. Chen, Z. Zhong, Y. Liu, W. Deng, and T. Li. 2021. The diagnostic value of synovial fluid lymphocytes in gout patients. Disease Markers 2021: 4385611.
Kundu, S., A. Bala, P. Ghosh, D. Mukhopadhyay, A. Mitra, A. Sarkar, A.K. Bauri, A. Ghosh, S. Chattopadhyay, and M. Chatterjee. 2011. Attenuation of oxidative stress by allylpyrocatechol in synovial cellular infiltrate of patients with Rheumatoid Arthritis. Free Radical Research 45: 518–526.
Hsueh, M.F., M.P. Bolognesi, S.S. Wellman, and V.B. Kraus. 2020. Anti-inflammatory effects of naproxen sodium on human osteoarthritis synovial fluid immune cells. Osteoarthritis and Cartilage 28: 639–645.
Zhang, X., L. Hu, S. Xu, C. Ye, and A. Chen. 2021. Erianin: a direct NLRP3 inhibitor with remarkable anti-inflammatory activity. Frontiers in Immunology 12: 739953.
Tsuzuki, N., Y. Kanbayashi, and K. Kusano. 2019. Markers for oxidative stress in the synovial fluid of thoroughbred horses with carpal bone fracture. Journal of Equine Science 30: 13–16.
Pascual, E., and V. Jovaní. 1995. A quantitative study of the phagocytosis of urate crystals in the synovial fluid of asymptomatic joints of patients with gout. British Journal of Rheumatology 34: 724–726.
Baggio, C., P. Sfriso, A. Cignarella, P. Galozzi, A. Scanu, F. Mastrotto, M. Favero, R. Ramonda, and F. Oliviero. 2021. Phagocytosis and inflammation in crystal-induced arthritis: a synovial fluid and in vitro study. Clinical and Experimental Rheumatology 39: 494–500.
Yagnik, D.R., P. Hillyer, D. Marshall, C.D. Smythe, T. Krausz, D.O. Haskard, and R.C. Landis. 2000. Noninflammatory phagocytosis of monosodium urate monohydrate crystals by mouse macrophages. Implications for the control of joint inflammation in gout. Arthritis and Rheumatism 43: 1779–1789.
Dalbeth, N., and D.O. Haskard. 2005. Mechanisms of inflammation in gout. Rheumatology 44: 1090–1096.
Landis, R.C., D.R. Yagnik, O. Florey, P. Philippidis, V. Emons, J.C. Mason, and D.O. Haskard. 2002. Safe disposal of inflammatory monosodium urate monohydrate crystals by differentiated macrophages. Arthritis and Rheumatism 46: 3026–3033.
Jeong, J.H., S.J. Choi, S.M. Ahn, J.S. Oh, Y.G. Kim, C.K. Lee, B. Yoo, and S. Hong. 2021. Neutrophil extracellular trap clearance by synovial macrophages in gout. Arthritis Research & Therapy 23: 88.
Liu, L., L. Zhu, M. Liu, L. Zhao, Y. Yu, Y. Xue, and L. Shan. 2022. Recent insights into the role of macrophages in acute gout. Frontiers in Immunology 13: 955806.
Vírgen Gen, J.J., R.I. Guzmán-Gerónimo, K. Martínez-Flores, G.A. Martínez-Nava, J. Fernández-Torres, and Y. Zamudio-Cuevas. 2020. Cherry extracts attenuate inflammation and oxidative stress triggered by monosodium urate crystals in THP-1 cells. Journal of Food Biochemistry 44: e13403.
Riaz, M., L.T. Al Kury, N. Atzaz, A. Alattar, R. Alshaman, F.A. Shah, and S. Li. 2022. Carvacrol alleviates hyperuricemia-induced oxidative stress and inflammation by modulating the NLRP3/NF-κB pathwayt. Drug Design, Development and Therapy 16: 1159–1170.
Furger, C. 2021. Live cell assays for the assessment of antioxidant activities of plant extracts. Antioxidants 10: 944.
Murphy, M.P., H. Bayir, V. Belousov, C.J. Chang, K.J.A. Davies, M.J. Davies, T.P. Dick, T. Finkel, et al. 2022. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nature Metabolism 4: 651–662.
Romero-Montero, A., L.J. Del Valle, J. Puiggalí, C. Montiel, R. García-Arrazola, and M. Gimeno. 2020. Poly(gallic acid)-coated polycaprolactone inhibits oxidative stress in epithelial cells. Materials Science & Engineering, C: Materials for Biological Applications 115: 111154.
Sánchez-Sánchez, R., A. Romero-Montero, C. Montiel, Y. Melgarejo-Ramírez, C. Sánchez-Ortega, H. Lugo-Martínez, B. Cabello-Arista, R. García-Arrazola, C. Velasquillo, and M. Gimeno. 2017. Cytoprotective effect of the enzyme-mediated polygallic acid on fibroblast cells under exposure of UV-irradiation. Materials Science & Engineering, C: Materials for Biological Applications 76: 417–424.
Renaudin, F., S. Sarda, L. Campillo-Gimenez, C. Séverac, T. Léger, C. Charvillat, C. Rey, F. Lioté, J.-M. Camadro, H.-K. Ea, and C. Combes. 2019. Adsorption of proteins on m-CPPD and urate crystals inhibits crystal-induced cell responses: study on albumin-crystal interaction. Journal of Functional Biomaterials 10: 18.
Scanu, A., R. Luisetto, F. Oliviero, L. Gruaz, P. Sfriso, D. Burger, and L. Punzi. 2015. High-density lipoproteins inhibit urate crystal-induced inflammation in mice. Annals of the Rheumatic Diseases 74: 587–594.
Acknowledgements
We would like to thank Jorge Cerna Cortés, Ph.D., from Instituto Politécnico Nacional (Mexico) for the reagents for carrying out cell culture assays and ELISAs.
Funding
This research received funding from the National Council of Science and Technology (CONACYT) for financing SEP-BASICA FSSEP02-C-2018-2 # A1-S-16109.
Author information
Authors and Affiliations
Contributions
YZC and JFT contributed to the conception and design of the study, data collection, analysis, and interpretation, and writing and critical revision of the manuscript. VML and ALM, cell cultures, oxidative stress experiments, and data acquisition. NMA and CALP, provision of SF samples, collection of medical records and critical revision of the manuscript. KMF and CHV, pro-inflammatory molecules quantification and acquisition of data, writing and critical revision of the manuscript. YZC and RSS, statistical analysis, data interpretation and critical revision of the manuscript. MG, data interpretation and critical revision of the manuscript. All authors contributed to data analysis and interpretation as well as writing and critical revision of the article. All authors have approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
This study was approved by the Ethics and Research Committee of the Instituto Nacional de Rehabilitación Luis Guillermo Ibarra Ibarra (registration number INR-27/20). All procedures performed in this study involving human participants were in accordance with the ethical standards of the INR-LGII-Institutional Research and Ethical Committee and with the Helsinki Declaration (1964). Informed consent was obtained from all individual participants included in the study.
Consent to Participate
Informed consent was obtained from all individual participants included in the study.
Consent for Publication
All authors have read and agreed to the published version of the manuscript.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zamudio-Cuevas, Y., Martínez-López, V., López-Macay, A. et al. Antiphagocytic Properties of Polygallic Acid with Implications in Gouty Inflammation. Inflammation 46, 1952–1965 (2023). https://doi.org/10.1007/s10753-023-01852-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-023-01852-z