
Abstract
The mammalian Transient Receptor Potential Canonical (TRPC) subfamily comprises seven transmembrane proteins (TRPC1–7) forming cation channels in the plasma membrane of mammalian cells. TRPC channels mediate Ca2+ and Na+ influx into the cells. Amongst TRPCs, TRPC6 deficiency or increased activity due to gain-of-function mutations has been associated with a multitude of diseases, such as kidney disease, pulmonary disease, and neurological disease. Indeed, the TRPC6 protein is expressed in various organs and is involved in diverse signalling pathways. The last decade saw a surge in the investigative studies concerning the physiological roles of TRPC6 and describing the development of new pharmacological tools modulating TRPC6 activity. The current review summarizes the progress achieved in those investigations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
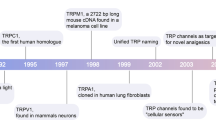
Transient Receptor Potential Canonical (TRPC) channels belong to the TRP superfamily of cation channels and constitute a group of calcium-permeable cation channels [1]. Based on the structural and functional similarities, TRPC channels have been subdivided into four major subgroups, namely: TRPC1, TRPC2, TRPC4/5, and TRPC3/6/7. The first mammalian TRPC1 channel was cloned in 1995 [2, 3]. Since then, numerous investigations have shown the involvement of these channels in various biological pathways.
TRPC channels are expressed in many cell types and are activated by agonists of heterotrimeric G-protein-coupled receptors or by intracellular calcium store depletion. They integrate multiple signals involving the activation of phospholipase C (PLC), which catalyses the breakdown of phosphatidylinositol 4,5-bisphosphate (PIP2) to produce inositol 1,4,5-trisphosphate (InsP3) and diacylglycerol (DAG). These channels allow the influx of Ca2+ and Na+ ions into the cells via a pore formed by four subunits with six transmembrane domains each [4,5,6,7,8,9,10]. TRPC channels are regulated by multiple cellular signalling pathways, and they contribute to the pathogenesis of multiple diseases.
The current review discusses the roles of TRPC6 in the progression or development of various diseases and the signalling pathways involving in regulating TRPC6 activity. The human TRPC6 gene is located on chromosome 11 [11]; therefore, mutations in the gene may equally affect both males and females. TRPC6 proteins are widely expressed in the heart, lungs, kidneys, muscles, and the brain [12,13,14,15,16,17,18,19,20]. TRPC6 is a DAG-gated channel [21] which is involved in mediating a significant amount of Ca2+ influx into the cell. Past and recent studies in human and mouse models have shown that TRPC6 dysfunction may contribute to the pathogenesis of various diseases and is associated with a facilitated disease progression. Figure 1 summarizes the roles of TRPC6 in pathological conditions ranging from increased vascular endothelial permeability, cardiac pathology, nephrological ailments, to cancer.

Involvement of TRPC6 signalling in various diseases and pathological conditions. The structure of TRPC6 protein was redrawn from PDB:7dxf.
KIDNEY DISEASE
Familial Focal Segmental Glomerulosclerosis
The earliest reports about the involvement of TRPC6 in the pathogenesis of a human disease were published in 2005 [18, 22] and concerned familial focal segmental glomerulosclerosis, a kidney glomerular disease. It was demonstrated that the TRPC6 channel is localized to podocyte foot processes that are important for regulating glomerular permeability in the kidney. The authors identified several gain-of-function mutations in TRPC6, such as P112Q, S270T, N143S, R895C, and E897K, that resulted in increased TRPC6 functional activity and increased glomerular permeability leading to the development of proteinuria. Consistently, Kim and Dryer later found that TRPC6 inactivation had a protective effect against glomerulonecrosis but not age-related renal fibrosis in aged mice [23].
Diabetic Kidney Disease
An increased expression of TRPC6 was also reported in podocytes of patients with diabetic kidney disease (DKD). TRPC6 activation has been directly linked to angiotensin II [24], which along with ROS generation is responsible for podocyte hypertrophy and associated outcomes. It was demonstrated that TRPC6 may function in conjugation with Protease-Activated Receptors (PARs) and GPCRs, contributing towards the development of glomerular injury and kidney disease in the patients presenting with diabetes. Notably, Wang et al. found that the simultaneous induction of hypertension and streptozotocin-induced moderate diabetes in the same animal leads to markedly increased albuminuria and kidney injury compared to the conditions when hypertension and diabetes were induced separately, with genetic ablation of TRPC6 significantly reducing albuminuria and kidney injury [25]. Consistently, Spires et al. also found that TRPC6 plays an important role in the development of DKD [26]. Uniquely, the authors genetically deleted TRPC6 in Dahl salt-sensitive rats (Dahl SSTrpc6−/− rats) and then induced type 1 diabetes in both wild-type Dahl SS rats and SSTrpc6−/− rats using streptozotocin. Albuminuria was not different in streptozotocin-treated Dahl SS and SSTrpc6−/− rats; however, the loss of TRPC6 was associated with attenuated damage to foot processes of podocytes during the development of DKD in the SSTrpc6−/− rats [26].
Proteinuria and Podocytopenia
Activation of TRPC6 has been directly connected with mechanical stretch and actin reorganization in the podocytes. This in turn changes the glomerular permeability barrier, thereby adversely affecting the kidney function. TRPC6 plays a critical role in the development and advancement of proteinuria by regulating its actin cytoskeleton rearrangement in podocytes.
The pathological changes in glomerular morphology and permeability are due to the increase in TRPC6-mediated Ca2+ influx in podocytes, and podocyte injury has been linked to diabetic kidney disease. Overactivation of TRPC6 has also been found to cause glomerular damage as this plays a role in abnormal podocyte deficiency termed “podocytopenia.” In patients with diabetes, angiotensin II levels have been found to be elevated, and it was reported that angiotensin II causes activation of TRPC6 in podocytes [27].
Glomerular Proteinuria
It was reported that podocyte-specific overexpression of TRPC6 led to a Ca2+-mediated increase in RhoA activity, whilst genetic deletion of TRPC6 resulted in increased Rac1 activity and podocyte motility. The podocytes are in turn dependent on the highly organized actin skeleton for their normal structure and function. This outlines an important role for intracellular Ca2+ in regulating the actin cytoskeleton of podocytes. The changes in podocyte number or morphology have been demonstrated to be the main cause leading to glomerular proteinuria. As the TRPC6-mediated calcium influx increases, the size selectivity of the glomerular filtration barrier by podocytes finely regulated by Ca2+ signals gets altered due to actin cytoskeleton rearrangement [28]. These events are the initial point of the commence of proteinuria formation. Angiotensin II is the major effector molecule which plays a pivotal role in the progression of glomerulosclerosis. It was revealed that angiotensin II increased TRPC6 expression and activated the channel leading to excessive calcium influx into the podocytes, causing the dysfunction and breakdown of the glomerular filtration barrier (GFB) with an increase in albuminuria [29]. This was further verified in other studies where angiotensin II induced NADPH oxidase 4 (NOX4) activation, thereby releasing high amount of H2O2, which in turn activates calcium influx via TRPC6 channels in podocytes. TRPC6 is a redox-sensitive channel, and its activation by Reactive Oxygen Species (ROS) overproduction may trigger injury in multiple cells including podocytes [30]. The contributions of TRPC6 channels in glomerular disease progression have been reviewed by Staruschenko et al. [31].
Some in vitro studies disclosed that high glucose levels increased TRPC6 mRNA and protein expression. Recent reports also revealed that an increased proteinuria along with reduced renal function leading to enhanced glomerular fibrosis has been directly associated with the elevated expression of TRPC6 in the model of ischaemia–reperfusion acute kidney injury [32]. Contribution of TRPC6-mediated calcium influx in podocyte injury during preeclampsia has also been proposed [33].
Kidney Fibrosis
A study by Wu et al. [34] showed that inhibiting TRPC6 channels can help in recovery from kidney fibrosis. However, a more recent study by Kim and Dryer [35] revealed that TRPC6 inactivation can alleviate glomerulosclerosis but not renal interstitial fibrosis. A study conducted on unilateral ureteral obstruction (UUO) mouse model demonstrated that TRPC6 knockout mice had much less fibrosis compared to the wild type. In the same model, it has been shown that TRPC6 inhibition by BI-749327 ameliorates renal fibrosis [36].
In conclusion, mutations in TRPC6, as well as upregulation of its expression and activity, are directly linked to renal abnormalities such as albuminuria and podocyte cell loss that ultimately contributes to kidney damage (Fig. 2). Thus, TRPC6 is a potential therapeutic target for the treatment of kidney diseases given its widespread involvement in multiple disease-instigating pathways in the kidney of diabetic patients and experimental models.

NEUROLOGICAL DISORDERS
TRPC6 is abundantly expressed in various regions of the Central Nervous System (CNS) [16, 37]. As a regulator of Ca2+ influx, TRPC6 is involved in the survival of neurons, synaptic plasticity, nerve growth cone guidance, spine morphology changes, and the regulation of neurite length [38,39,40,41,42]. Dysregulation of TRPC6 activity may trigger a series of downstream signalling events leading to many neurobiological disorders (Fig. 3). TRPC6 along with TRPC3 play an important role in brain-derived neurotrophic factor (BDNF)-induced axon guidance and neuron survival [38]. Consistently, one study reported an increased expression of TRPC6 in hippocampus during the postnatal development [40].

Alzheimer’s Disease
Neurodegenerative diseases, such as Alzheimer’s disease, are major causes of morbidity in aging population. Though the pathophysiological mechanisms may differ, Ca2+ dyshomeostasis is common in neurodegenerative diseases [40, 43]. A significant reduction in overall expression of TRPC6 has been found in patients presenting with Alzheimer’s disease and cognitive disorders [44]. Investigators discovered that TRPC6 may specifically inhibit the cleavage of amyloid precursor protein (APP) by γ-secretase and can subsequently reduce β-amyloid formation [45]. It has also been found that TRPC6 activation leads to the reduction of accumulation of beta-amyloid (Aβ) plaques in the aged brain due to increased cerebrovascular P-glycoprotein [46]. Additionally, a recent study demonstrated that repeated moderate hypoglycaemia increases the rate of Alzheimer’s disease progression by decreasing TRPC6 expression and causing impairment of the TRPC6/GLUT3 pathway [47].
Autism Spectrum Disorders
TRPC6 also plays a role in developmental psychiatric conditions such as autism. Griesi-Oliveira et al. reported [48] a decreased activity of TRPC6 in various models, including stem cell-derived neuronal cells and mouse models. They demonstrated that the reduction of TRPC6 function contributes to changes in neuronal development, morphology, and function. The authors also found that it is possible to prevent the changes in neuronal phenotypes by TRPC6 complementation in conjunction with the channel activation by insulin-like growth factor-1 or hyperforin (purported TRPC6 agonists). The authors also sequenced TRPC6 in 1041 autism spectrum disorders (ASD) individuals and 2872 controls and uncovered a significant level of mutations in the ASD population, with some of those mutations being loss-of-function alterations. A more recent study by Palacios-Muñoz et al. [49] reported the consequences of TRPC6 mutations on nervous system physiology using a Drosophila melanogaster model. Their findings demonstrated that null mutations in TRPγ (the homologous gene of TRPC6 in the fly) led to a variety of behavioural alterations that simulated characteristics seen in ASD patients. These included deficient social interactions, impaired sleep homeostasis, defects in memory and learning, and hyperactivity. Similar to the study by Griesi-Oliveira et al. [48], this group found that treatment with hyperforin diminished the behavioural deficiencies displayed by the fruit flies.
Cerebral Ischaemic Injury
TRPC6 overexpression has been found to protect neurons from ischaemia [50, 51]. The role of TRPC6 function was investigated using TRPC6 agonist (HYP9) and antagonist (SKF96365) in oxygen–glucose deprivation cell models and the middle cerebral artery occlusion (MCAO) mouse model of stroke [37, 52]. HYP9, which inhibited the downregulation of TRPC6 in a dose-dependent manner, led to reduced ischaemic responses, including decreased astrocytic apoptosis and cytotoxicity [52]. Conversely, SKF96365 further increased the ischaemia-associated damage. Inhibition of TRPC6 degradation by calpain inhibitors prevented ischaemic neuronal death and provided neuroprotection through the Ras/MEK/ERK/CREB pathway [37]. It has been identified in preclinical studies that TRPC6 activation may reduce neuronal death [37].
CANCER
The TRPC6 channel has been highly expressed by proliferating cells, including cancer cells. Several studies have highlighted the role of TRPC6 in cancer, especially in breast cancer cells [53], which express high levels of TRPC6 [54]. Besides breast cancer, TRPC6 has also been shown to be involved in gastro-oesophageal (GE) [55], glial [56], liver [57], gastric [58, 59], renal [60], lung [61], head and neck squamous cell [62], and cervical [63] cancers.
Breast Cancer
The role of calcium signalling in breast cancer is unquestionable. Several reports link TRPC activity with breast cancer progression [53]. Besides TRPC6 [19], TRPC1 and TRPC5 have also been implicated in the pathogenesis of breast cancer [64]. However, TRPC6 expression is the highest amongst all TRPCs in the samples of human breast ductal adenocarcinomas. Additionally, high expression of TRPC6 has been found in some breast cancer cell lines, such as MCF-7 and MDA-MB-231, that may underlie at least in part the cell lines’ increased proliferation rate [65].
TRPC6 mediates multiple interactions with various proteins; some of which are directly linked to increased breast cancer progression. For example, TRPC6 associates with possible breast cancer oncogenes like large-conductance Ca2+-activated K+ (BKCa) channels, creating conditions for tumour growth [66]. TRPC6 also interacts with Fyn and Src tyrosine kinases, which may in turn activate the channel via phosphorylation events [67]. These kinases have also been reported to be overexpressed in breast cancer cell lines [68, 69]. Furthermore, the identification of TRPC6 interaction with the human myxovirus resistance protein 1 (MxA) highly expressed in triple-negative breast cancer tumours strongly suggests its role in breast cancer progression [70].
Oesophageal Cancer
In a clinical study, high levels of TRPC6 expression were associated with poor prognosis in oesophageal squamous cell carcinoma (ESCC) patients [71]. Indeed, Shi et al. reported that TRPC6 is overexpressed in ESCC cells, and that TRPC6 is a key factor to control G2 phase transition in the tumourigenesis of oesophageal cancers [55]. Thus, inhibition of TRPC6-mediated Ca2+ influx would be beneficial to prevent ESCC progression.
Glial Cancer
TRPC6 in association with a Ca2+-activated channel, KCa1.1, has been considered as a potential therapeutic target for malignant glioma. Studies revealed the modulating role of TRPC6 in the increased expression and current density of KCa1.1 by interacting with the latter in vitro and in vivo [72]. The anti-proliferative effects of ionizing radiation had been found to be increased after inhibiting TRPC6 in cellular models. Also, inhibition of TRPC6 led to a reduced tumour volume in a subcutaneous mouse model of xenografted human tumours and increased mean survival in mice in an intracranial model [56].
Liver Cancer
Hepatocellular carcinoma (HCC) cells exhibit increased SOCE possibly due to TRPC6 activity. Wen et al. [73] reported that in a xenograft model of HCC, inhibition of TRPC6 enhanced the efficacy of anti-cancer drug, doxorubicin. The authors provided evidence that the increased intracellular calcium entry via TRPC6 may be in part responsible for multidrug resistance in HCC cells.
Renal Cancer
TRPC6 expression is markedly increased in renal cell carcinoma (RCC) specimens, and TRPC6 activity plays an important role in human renal adenocarcinoma (ACHN) cell proliferation [60]. Kim et al. reported [74] that lysine-deficient protein kinase 1 (WNK1) can activate TRPC6 via the PI4KIIIα — Gαq-coupled receptor/PLC-β in clear-cell renal cell carcinoma (ccRCC). TRPC6-mediated Ca2+ influx in turn resulted in calcineurin-dependent activation of the nuclear factor of activated T cells cytoplasmic 1 (NFATc1) signalling in ccRCC cells, leading to their increased proliferation and migration. Importantly, inhibition of the WNK1-TRPC6-mediated Ca2+ influx decreased NFATc1 activation and slowed down renal tumour progression. Remarkably, NFATc1 activity has been found to be elevated in tumour tissues compared to the normal tissues. These data support the hypothesis that increased TRPC6 activity may promote renal tumourigenesis.
Lung Cancer
Several reports provided evidence that TRPC6 activity may contribute to lung cancer pathogenesis. It was demonstrated that TRPC6-mediated Ca2+ influx in non-small-cell lung cancer (NSCLC) cells leads to increased proliferation of the cells by promoting cell cycle progression [75]. Yang et al. demonstrated that TRPC6 activity was greater in the detached cells compared to the still attached NSCLC cells and that inhibition of TRPC6 attenuated NSCLC cell proliferation and invasion. Conversely, Wang et al. reported that nicotine exposure increased TRPC6 mRNA expression and activation in non‑small-cell lung cancer (NSCLC) A549 cells via a HIF‑1α-dependent pathway leading to increased rate of A549 cell proliferation [76].
Head and Neck Squamous Cell Carcinoma
Bernaldo de Quirós et al. [62] established the role of TRPC6 in head and neck squamous cell carcinomas (HNSCC). They found a significantly elevated level of TRPC6 gene transcription in HNSCC-derived cells. The siRNA-induced knockdown of TRPC6 dramatically decreased HNSCC cell invasion.
Prostate Cancer
The Prevarskaya laboratory was the first to demonstrate that TRPC6-mediated Ca2+ influx is important for proliferation of human prostate cancer epithelial (hPCE) cells [77] and that silencing TRPC6 by anti-sense hybrid depletion decreased proliferation of hPCE cells. The authors found that alpha1-adrinergic signalling coupled to the activation of TRPC6 and NFAT was critical for hPCE cell proliferation. Later, Yue et al. found that TRPC6 was highly expressed in human androgen-dependent and androgen-independent malignant prostate cancers, and its expression correlated with prostate cancer Gleason score and extraprostatic extension [20]. Consistently, Wang et al. demonstrated that TRPC6 upregulation is common in migrating prostate cancer cells, further supporting the role of TRPC6 in metastatic prostate cancer [78].
The pathological involvement of TRPC6 channels was also identified in the hepatocyte growth factor (HGF)-induced prostate cancer cells [79]. The study demonstrated that TRPC6 was highly expressed in DU145 and PC3 prostate cancer cell lines and its inhibition arrested DU145 and PC3 cells in the G2/M phase, thereby suppressing HGF-induced cell proliferation. Interestingly, Bernichtein et al. reported that dietary vitamin D supplementation can reduce calcium-triggered, TRPC6-mediated acceleration in the progression of prostate intraepithelial neoplasia, cell proliferation, micro-invasion, and tissue inflammation in early-stage prostate cancer [80]. Thus, vitamin D supplementation may potentially slow down prostate cancer progression by attenuating calcium-triggered tumourigenesis.
Ovarian and Cervical Cancers
Zeng et al. reported that splice variants of TRPC6 channels with exon 3 and 4 deletions were highly expressed in ovarian cancer cells along with TRPC1, TRPC3, and TRPC4 channels [81]. Inhibitors of TRPC channels, siRNA targeting TRPC6, and blocking antibodies targeting TRPC channels decreased ovarian cancer cell proliferation, whereas overexpression of TRPC6 increased ovarian cancer cell colony growth. Therefore, the authors concluded that TRPC6 may be involved in the tumourigenesis of ovarian cancer [81]. Similarly, a high expression level of TRPC6 has been linked to increased proliferation and migration of HeLa and SiHa cervical cancer cells, further supporting the hypothesis that TRPC6 may play a pathogenic role in cervical cancers [82].
CARDIOVASCULAR PATHOLOGY
Hypertrophy and Heart Failure
Myocardial stretch increases cardiomyocytes’ intracellular Ca2+ levels, thereby promoting cardiac hypertrophy. It was reported that the activation of TRPC3 and TRPC6 contributes to myocardial stretch-associated slow force response (SFR) and increased Ca2+ transient and twitch force during stretch [83]. It was also demonstrated that DAG-induced Ca2+ signalling pathway is essential for angiotensin II-induced NFAT activation and cardiac hypertrophy during TRPC3 and TRPC6 activation. It was reported that TRPC6 can counteract hyperglycaemia-induced heart failure by disrupting the TRPC3-Nox2 complex formation [84]. Conversely, Lin et al. [36] reported that BI 749,327, a TRPC6 inhibitor, improved left heart function, reduces volume/mass ratio, and decreased expression of profibrotic genes in sustained pressure overload mice. TRPC6 was upregulated in response to activated calcineurin and pressure overload, in failing human and mouse hearts [85]. TRPC6 responsiveness to cardiac stress in a calcineurin-dependent manner is likely attributed to two conserved NFAT consensus sites in the promoter region of the TRPC6 gene. Transgenic mice overexpressing TRPC6 in cardiac tissue had increased sensitivity to stress and heart failure [86]. It was reported that NFAT-dependent expression of beta-myosin heavy chain was increased in models of hypertrophy. TRPC6-calcineurin-NFAT signalling was implicated in pathologic cardiac remodelling resulting in fibrosis and hypertrophy in spontaneously hypertensive rats with 5/6 nephrectomy [87]. Furthermore, transgenic mice overexpressing TRPC6 in the heart developed hypertrophy and exhibited heart failure death due to cardiomyopathy [88]. Zhou et al. [89] demonstrated an increased TRPC6 expression in a 1-month post-Myocardial Infraction (MI) rat model. The administration of plant-chemical danshensu protects against ischaemia–reperfusion injury (IRI) by reducing TRPC6 expression via the c-Jun N-terminal kinase (JNK) signalling pathway. Thus, TRPC6 is a possible target for cardioprotection and its inhibition might have anti-hypertrophic effects via various cardiac signalling pathways including the ANP/BNP-GC-A [90].
Atrial Fibrillation
Atrial fibrillation (AF) is the most common heart arrhythmia which is directly associated with mechanical stretch. Nikolova-Krstevski et al. [91] have provided evidence that TRPC6 act as an atrial mechano-sensor. The authors proposed that the stretch-sensitive TRPC6 channel may prevent atrial fibrillation under physiological conditions in the human heart by regulating paracrine crosstalk between the atrial endocardium and atrial contractile cardiomyocytes. However, chronic stretch may lead to the atrial endocardial TRPC6 internalization, precipitating atrial arrhythmia.
Hypertrophic Cardiomyopathy
Xie et al. demonstrated that heart-specific overexpression of TRPC6 was associated with cardiac remodelling and cardiac hypertrophy [88]. The same study showed that deletion of TRPC6 in Klotho-deficient mice led to attenuation of stress-induced cardiac remodelling and cardiac hypertrophy. Overexpression of Klotho also prevented hypertrophic cardiomyopathy in these mice, improving their survival rate. The authors concluded that the Klotho’s ability of reducing TRPC6 activity in cardiac muscle cells via inhibition of phosphoinositide-3-kinase (PI3K)-dependent exocytosis of TRPC6 channels may underlie the Klotho’s beneficial effect in hypertrophic cardiomyopathy.
Pulmonary Hypertension
TRPC6 is a key regulator of hypoxia-mediated pulmonary vasoconstriction and pulmonary hypertension. A unique genetic variation in the promoter region of the TRPC6 gene was linked to the facilitated progression of pulmonary vascular abnormalities in idiopathic pulmonary arterial hypertension (PAH) [92].
Coronary Artery Disease
Upregulated TRPC6 expression was found in the smooth muscle layer of coronary arteries isolated from metabolic syndrome pigs [93], thereby showing a correlation between TRPC6 expression and increased coronary artery contractility.
Thus, the role of TRPC6 in cardiovascular pathology (Fig. 4) is beyond doubt, and hence, multiple groups all over the world are developing novel small molecules targeting TRPC6 to modulate TRPC6 activity.

PULMONARY DISORDERS
The TRPC6 channel is expressed in human airway smooth muscle cells (HASMCs) and has an important role in the development of an array of lung diseases (Fig. 5). TRPC6 contributes to the pathogenesis of pulmonary diseases such as cystic fibrosis, chronic obstructive pulmonary disease (COPD), asthma, lung oedema, and lung fibrosis [94,95,96,97,98]. The activation of Toll-like Receptor 4 (TLR4) and generation of DAG lead to TRPC6-dependent Ca2+ influx into lung endothelial cells. This is the basic mechanism proven to be involved in endotoxin-induced lung inflammation [99].

Chronic Obstructive Pulmonary Disease
Long-term smoking causes chronic obstructive pulmonary disease (COPD), at least in part, by increasing airway smooth muscle cell proliferation. It was reported that nicotine can enhance airway smooth muscle cell proliferation via the α7 nAChR-PI3K/Akt-TRPC6 signalling pathway [100]. Furthermore, the pathology may also arise due to excessive alveolar macrophage activation that depends on calcium influx. Finney-Hayward et al. reported that human lung tissue macrophages expressed increased levels of TRPC6 mRNA and protein as compared with monocytes and monocyte-derived macrophages and that TRPC6 mRNA expression was significantly elevated in alveolar macrophages from patients with COPD compared to control subjects [95].
Lung Oedema
Lung ischaemia–reperfusion-induced oedema occurs due to increased endothelial permeability that in turns results from increased activation of endothelial TRPC6. Consistently, no lung ischaemia–reperfusion-induced oedema was noted in TRPC6-deficient mice [98]. Samapati et al. reported that platelet-activating factor dependent activation of acid sphingomyelinase followed by recruitment of TRPC6 channels to caveolae increased lung endothelial permeability, with TRPC6 inhibitors preventing and direct activation of TRPC6 mimicking platelet-activating factor-induced lung oedema [101]. Furthermore, Jiang et al. reported that prostaglandin E2-PGE3 receptor 3-dependent pulmonary permeability results from TRPC6 activation in a Gi-PLC-SrcFK-dependent manner [102].
Fibrosis
Published evidence indicates that TRPC6 is responsible for increased vascular permeability in the lungs which might in turn help circulating fibrocytes to migrate to the injured areas. Indeed, the TRPC6-deficient lungs exhibited a less severe pulmonary fibrosis compared to the wild-type lungs, and normal lung function was closely associated with low level of TRPC6 expression in the lungs [97].
COVID-19
COVID-19 pathology has also been associated with TRPC6 channels. In a small clinical study including four COVID-19 patients presenting with pneumonia, TRPC6 expression was found to be markedly elevated in fibrotic lung tissue, inflammatory lesions, and cellular infiltrates [103]. Although the authors did not elucidate the underlying mechanisms for TRPC6 upregulation, these findings suggest a pathological role of TRPC6 in developing COVID-19-induced complications.
Airway Inflammation
Studies utilizing TRPC6−/− mice and TRPC6-selective inhibitor revealed that TRPC6 contributes to ozone (O3) inhalation-induced airway inflammation. It has been shown to regulate oxidative inflammatory responses induced by O3 or H2O2 through activating the ERK signalling pathway [104]. Elevated activity of platelet activating factor was also associated with increased lung vascular permeability, oedema, and inflammation. The effect was mediated via activation of acid sphingomyelinase that resulted in recruitment of TRPC6 to caveolae and the channel activation. This in turn led to increased calcium influx into endothelial cells and consequently decreased endothelial barrier function [101].
Hypoxic Pulmonary Vasoconstriction and Pulmonary Hypertension
Increased pulmonary smooth muscle cell contractions and proliferation are long known to contribute to hypoxia-induced pulmonary vasoconstriction. In 2006, Wang et al. provided evidence that increased calcium influx observed in hypoxic pulmonary smooth muscle cells was due to an increased activity of TRPC6 channels [105]. Chronic hypoxia not only stimulated calcium influx through TRPC6 in pulmonary smooth muscle cells, but also increased the expression rate of TRPC6 via a hypoxia-inducible factor 1 transcription factor-dependent mechanism. Similar findings were simultaneously reported by Weissmann et al. who used TRPC6−/− mice and demonstrated that hypoxic pulmonary vasoconstriction was not observed in this TRPC6 knockout mouse model. In contrast to wild-type mice expressing TRPC6, TRPC6−/− mice developed regional hypoventilation-induced severe arterial hypoxemia due to the lack of hypoxic pulmonary vasoconstriction. Notably, no hypoxia-induced cation influx was observed in smooth muscle cells from precapillary pulmonary arteries of TRPC6−/− mice despite increased hypoxia-dependent DAG accumulation in the cells. Conversely, wild-type pulmonary smooth muscle cells exhibited significant hypoxia-activated cation currents requiring DAG accumulation [106].
Weissmann et al. did not find any correlation between TRPC6 activity/expression and chronic hypoxia-induced pulmonary hypertension [106] and later provided evidence that TRPC1, rather than TRPC6, might be responsible for inducing hypoxic pulmonary hypertension [107]. However, several other studies demonstrated that hypoxia-induced TRPC6 activation and/or upregulation may also contribute to the pathogenesis of hypoxic pulmonary hypertension [108,109,110]. Such disagreement may stem from the fact that cultured pulmonary artery smooth muscle cells may exhibit different properties depending on culturing conditions. Recently, Zhao et al. has demonstrated that both TRPC6 and Piezo1, which is another mechanosensitive cation channel present in lung endothelial cells, are responsible for membrane stretch-mediated influx of Ca2+ ions in human pulmonary arterial endothelial cells (PAECs) accounting for mechanotransduction-led vascular remodelling in patients with pulmonary arterial hypertension [111].
TRPC6 MODULATORS
The rapidly expanding role of TRPC6 in the pathogenesis of several human diseases has been fuelling the progress of TRPC6 modulator development. There have been multiple reports on various TRPC6 modulators, which include both agonists and antagonists. The usage of CryoEM approach for solving TRPC6 structures in the presence or absence of modulators allowed researchers to markedly facilitate the process of drug discovery. For example, Bai et al. have recently provided the structural insights on the TRPC6 protein interaction with its antagonist (AM-1473) and agonist (AM-0883) [112]. It appears that the antagonist makes both hydrophilic and hydrophobic interactions at multiple sites of S1–S4 and nearby helices and loops. The authors demonstrated that the positively charged piperidine moiety may form hydrogen bonds with Glu509 and Asp530 of the helices S2 and S3, respectively. The benzonitrile group may be involved in a cation-π interaction with Arg758 on the reentrant loop as well as aromatic-stacking interactions with His446 on S1 and Tyr753 on the TRP helix, whilst the indene double ring makes van der Waal interactions with Tyr612 on S4. Notably, the antagonist is 36-fold more selective for TRPC6 over its homolog TRPC3 (IC50 ≈ 8.0 nM). Important residues on TRPC3 and TRPC6, interacting with the antagonist, are identical, except for Arg758 of TRPC6 that is replaced by a lysine in TRPC3. The indispensability of this Arg758 for TRPC6 antagonist binding was tested by the authors [112].
Antagonists
There have been several studies focusing on the ability of various small molecules to inhibit TRPC6 function. Such small-molecule inhibitors are critical to counteract the deleterious effect of gain-of-function mutations in the TRPC6 protein that may lead to diseases. BI 749,327 is one of the mostly used TRPC6 inhibitors. It was shown that BI 749,327 can prevent fibrosis as well as various dysfunctions in cardiac and renal diseases [113]. Notably, some derivatives of BI 749,327 are already being investigated in phase I and II clinical trials (https://clinicaltrials.gov/: NCT04665700, NCT03854552, NCT04176536). Whilst NCT04665700 and NCT03854552 are phase I clinical studies investigating BI 764,198 tolerance at different doses in healthy individuals, the NCT04176536 clinical study investigates the pharmacokinetics of a single dose of BI 764,198 in patients with moderate and severe renal impairment in comparison to a group of matched healthy individuals. Another study (NCT04604184) investigates whether BI 764,198 can improve lung health in hospitalized patients suffering from acute severe COVID-19. Another inhibitor of TRPC6, SAR7334, was used to suppress TRPC6-dependent acute hypoxic pulmonary vasoconstriction (HPV) in perfused mouse lungs [114].
A phytochemical ( +)-larixol originating from European larch (Larix decidua) resin has been modified to derive its acetylated derivatives as selective TRPC6 inhibitors. Further, one of its methylcarbamate congeners, SH045, was shown to ameliorate lung ischaemia–reperfusion oedema in lung preparations [115]. It is a selective TRPC6 inhibitor which also attenuates renal fibrosis in an animal model of metabolic syndrome [116]. Similarly, 1-benzilpiperadine derivative (1-BP), a selective inhibitor for TRPC3 and TRPC6 channels, was effective to reduce peripheral artery disease (PAD) [117]. BTDM [(2-(benzo[d][1,3]dioxol-5-ylamino)thiazol-4-yl)((3 S,5 R)-3,5-dimethylpiperidin-1-yl)methanone] is a high-affinity antagonist for the human TRPC6 channel. Its interaction with the latter was detailed by Tang et al., 2018, who were able to determine TRPC6 structure in its presence. The authors demonstrated that BTDM binds between the S5-S6 pore domain and the voltage sensor-like domain thereby inhibiting pore opening [6]. Later, Bai et al. published two highly informative structures of TRPC6 bound with an antagonist (AM-1473) and an agonist (AM-0883), respectively. The conformational changes due to antagonist/agonist binding in the two structures allowed the authors to understand the conformational changes TRPC6 undergoes during these transition states [112]. SAR7334 designed by Maier et al. specifically inhibited TRPC6 at lower concentrations; however, at higher doses it was also inhibiting other TRPC isoforms. Its inhitory effects on acute hypoxic pulmonary vasoconstriction (HPV) and systemic BP were highly encouraging [114].
Agonists
Because depending on the disease, both inhibition and activation of TRPC6 may be clinically useful, many research groups focused on identifying not only specific antagonists of TRPC6 but also specific agonists. For example, agonists may be useful either for treating some neurological conditions or may help in scrutinizing the role of TRPC6 activation during various in vivo experiments. Bai et al. indeed identified a relatively specific TRPC6 activator, AM-0883, that was as effective as OAG, but has a considerably higher potency. In another study, Yang et al. identified two additional agonists: M085 and GSK1702934A, which can directly activate TRPC6 via a mechanism involving residues of the pore helix (PH) and transmembrane (TM) helix S6 [118]. Recently, the pyrazolopyrimidine skeleton was identified as a TRPC6 agonist by Qu et al. using a cell-based high-throughput screening approach [119]. Additionally, the authors identified a series of potent and selective TRPC agonists. The discovery of a naturally occurring secondary plant metabolite, hyperforin, as a putative TRPC6 agonist added another aspect of targeting this channel using natural compounds [120]. Hyperforin is the main component of St. John’s wort extract. It exhibits some anti-depressant properties. In addition, it was shown that hyperforin can modulate intracellular Ca2+ levels possibly via activating TRPC6 channels. However, the ability of hyperforin to activate TRPC6 was later disputed [121].
CONCLUSION
The role of TRPC6 channels in calcium ion permeability makes it central to various signalling pathways and thereby in disease progression. Findings from animal and in vitro cell culture experiments have shown TRPC6 involvement in various diseases including cardiac, neurological, and nephrological diseases. The history of TRPC6 in disease pathology dates to more than 2 decades back when the first reports were published on the contribution of TRPC6 upregulation in cardiomyocyte pathophysiology in cardiac hypertrophy, cardiac fibrosis, and heart failure. Now we know that TRPC6 is involved not only in cardiac fibrosis, but also in pulmonary and kidney fibrosis. Deletion of the TRPC6 gene impacted expression of other pro-fibrotic genes in the knockout model [122]. It has been clearly demonstrated that TRPC6 can be a therapeutic target for treatment of cardiac fibrosis. Research carried out by Lin et al. (2019) found that BI 749327, a bioavailable antagonist of TRPC6, can ameliorate renal and cardiac fibrosis [36]. Role of TRPC6 has also being studied in cystic fibrosis and pulmonary fibrosis [94, 97]. Kurahara et al. (2015) has reported its role in stenotic fibrosis, a complication frequently observed in Crohn’s disease [36].
Due to the critical involvement of TRPC6 in various diseases, many novel and potent antagonists have been designed, which proved to be efficacious for the cause. As this aspect is another interesting subject but beyond the scope of the current review, the readers are advised to refer to the literature for drug discovery of TRPC6 modulators.
Many recent reviews have detailed the role of TRPC6 in cardiac, pulmonary, renal, and neurological diseases. However there has been no “one-stop shop,” where consolidated information on the role of TRPC6 in various diseases is available. Here, we provide such broad review of the literature with a bird view on many human diseases which TRPC6 may be involved in.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
References
Chen, X., G. Sooch, I.S. Demaree, F.A. White, and A.G. Obukhov. 2020. Transient Receptor Potential Canonical (TRPC) channels: then and now, Cells. 9.
Wes, P.D., J. Chevesich, A. Jeromin, C. Rosenberg, G. Stetten, and C. Montell. 1995. TRPC1, a human homolog of a Drosophila store-operated channel. Proceedings of the National Academy of Sciences USA 92: 9652–9656.
Zhu, X., P.B. Chu, M. Peyton, and L. Birnbaumer. 1995. Molecular cloning of a widely expressed human homologue for the Drosophila trp gene. FEBS Letters 373: 193–198.
Fan, C., W. Choi, W. Sun, J. Du, and W. Lu. 2018. Structure of the human lipid-gated cation channel TRPC3. Elife. 7.
Sierra-Valdez, F., C.M. Azumaya, L.O. Romero, T. Nakagawa, and J.F. Cordero-Morales. 2018. Structure-function analyses of the ion channel TRPC3 reveal that its cytoplasmic domain allosterically modulates channel gating. Journal of Biological Chemistry 293: 16102–16114.
Tang, Q., W. Guo, L. Zheng, J.X. Wu, M. Liu, X. Zhou, X. Zhang, and L. Chen. 2018. Structure of the receptor-activated human TRPC6 and TRPC3 ion channels. Cell Research 28: 746–755.
Azumaya, C.M., F. Sierra-Valdez, J.F. Cordero-Morales, and T. Nakagawa. 2018. Cryo-EM structure of the cytoplasmic domain of murine transient receptor potential cation channel subfamily C member 6 (TRPC6). Journal of Biological Chemistry 293: 10381–10391.
Vinayagam, D., T. Mager, A. Apelbaum, A. Bothe, F. Merino, O. Hofnagel, C. Gatsogiannis, and S. Raunser. 2018. Electron cryo-microscopy structure of the canonical TRPC4 ion channel. Elife 7.
Duan, J., J. Li, B. Zeng, G.L. Chen, X. Peng, Y. Zhang, J. Wang, D.E. Clapham, Z. Li, and J. Zhang. 2018. Structure of the mouse TRPC4 ion channel. Nature Communications 9: 3102.
Duan, J., J. Li, G.L. Chen, Y. Ge, J. Liu, K. Xie, X. Peng, W. Zhou, J. Zhong, Y. Zhang, J. Xu, C. Xue, B. Liang, L. Zhu, W. Liu, C. Zhang, X.L. Tian, J. Wang, D.E. Clapham, B. Zeng, Z. Li, and J. Zhang. 2019. Cryo-EM structure of TRPC5 at 2.8-A resolution reveals unique and conserved structural elements essential for channel function. Science Advances 5: eaaw7935.
D’Esposito, M., M. Strazzullo, M. Cuccurese, C. Spalluto, M. Rocchi, M. D’Urso, and A. Ciccodicola. 1998. Identification and assignment of the human Transient Receptor Potential Channel 6 gene TRPC6 to chromosome 11q21–>q22. Cytogenetics and Cell Genetics 83: 46–47.
Hassock, S.R., M.X. Zhu, C. Trost, V. Flockerzi, and K.S. Authi. 2002. Expression and role of TRPC proteins in human platelets: Evidence that TRPC6 forms the store-independent calcium entry channel. Blood 100: 2801–2811.
Buess, M., O. Engler, H.H. Hirsch, and C. Moroni. 1999. Search for oncogenic regulators in an autocrine tumor model using differential display PCR: Identification of novel candidate genes including the calcium channel mtrp6. Oncogene 18: 1487–1494.
Dalrymple, A., D.M. Slater, D. Beech, L. Poston, and R.M. Tribe. 2002. Molecular identification and localization of Trp homologues, putative calcium channels, in pregnant human uterus. Molecular Human Reproduction 8: 946–951.
Wang, J., L.G. Laurier, S.M. Sims, and H.G. Preiksaitis. 2003. Enhanced capacitative calcium entry and TRPC channel gene expression in human LES smooth muscle. American Journal of Physiology. Gastrointestinal and Liver Physiology 284: G1074–G1083.
Riccio, A., A.D. Medhurst, C. Mattei, R.E. Kelsell, A.R. Calver, A.D. Randall, C.D. Benham, and M.N. Pangalos. 2002. mRNA distribution analysis of human TRPC family in CNS and peripheral tissues. Brain Research. Molecular Brain Research 109: 95–104.
Yu, Y., I. Fantozzi, C.V. Remillard, J.W. Landsberg, N. Kunichika, O. Platoshyn, D.D. Tigno, P.A. Thistlethwaite, L.J. Rubin, and J.X. Yuan. 2004. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proceedings of the National Academy of Sciences USA 101: 13861–13866.
Reiser, J., K.R. Polu, C.C. Moller, P. Kenlan, M.M. Altintas, C. Wei, C. Faul, S. Herbert, I. Villegas, C. Avila-Casado, M. McGee, H. Sugimoto, D. Brown, R. Kalluri, P. Mundel, P.L. Smith, D.E. Clapham, and M.R. Pollak. 2005. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nature Genetics 37: 739–744.
Guilbert, A., I. Dhennin-Duthille, Y.E. Hiani, N. Haren, H. Khorsi, H. Sevestre, A. Ahidouch, and H. Ouadid-Ahidouch. 2008. Expression of TRPC6 channels in human epithelial breast cancer cells. BMC Cancer 8: 125.
Yue, D., Y. Wang, J.Y. Xiao, P. Wang, and C.S. Ren. 2009. Expression of TRPC6 in benign and malignant human prostate tissues. Asian Journal of Andrology 11: 541–547.
Hofmann, T., A.G. Obukhov, M. Schaefer, C. Harteneck, T. Gudermann, and G. Schultz. 1999. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397: 259–263.
Winn, M.P., P.J. Conlon, K.L. Lynn, M.K. Farrington, T. Creazzo, A.F. Hawkins, N. Daskalakis, S.Y. Kwan, S. Ebersviller, J.L. Burchette, M.A. Pericak-Vance, D.N. Howell, J.M. Vance, and P.B. Rosenberg. 2005. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308: 1801–1804.
Kim, E.Y., and S.E. Dryer. 2021. Effects of TRPC6 inactivation on glomerulosclerosis and renal fibrosis in aging rats. Cells 10.
Staruschenko, A., D. Spires, and O. Palygin. 2019. Role of TRPC6 in progression of diabetic kidney disease. Current Hypertension Reports 21: 48.
Wang, Z., Y. Fu, and do Carmo, J. M., da Silva, A. A., Li, X., Mouton, A., Omoto, A. C. M., Sears, J. & Hall, J. E. 2022. Transient receptor potential cation channel 6 contributes to kidney injury induced by diabetes and hypertension. American Journal of Physiology. Renal Physiology 322: F76–F88.
Spires, D., D.V. Ilatovskaya, V. Levchenko, P.E. North, A.M. Geurts, O. Palygin, and A. Staruschenko. 2018. Protective role of TRPC6 knockout in the progression of diabetic kidney disease. American Journal of Physiology. Renal Physiology 315: F1091–F1097.
Ilatovskaya, D.V., V. Levchenko, A. Lowing, L.S. Shuyskiy, O. Palygin, and A. Staruschenko. 2015. Podocyte injury in diabetic nephropathy: Implications of angiotensin II-dependent activation of TRPC channels. Science and Reports 5: 17637.
Hunt, J.L., M.R. Pollak, and B.M. Denker. 2005. Cultured podocytes establish a size-selective barrier regulated by specific signaling pathways and demonstrate synchronized barrier assembly in a calcium switch model of junction formation. Journal of the American Society of Nephrology 16: 1593–1602.
Ilatovskaya, D.V., O. Palygin, V. Chubinskiy-Nadezhdin, Y.A. Negulyaev, R. Ma, L. Birnbaumer, and A. Staruschenko. 2014. Angiotensin II has acute effects on TRPC6 channels in podocytes of freshly isolated glomeruli. Kidney International 86: 506–514.
Ilatovskaya, D.V., G. Blass, O. Palygin, V. Levchenko, T.S. Pavlov, M.N. Grzybowski, K. Winsor, L.S. Shuyskiy, A.M. Geurts, A.W. Cowley Jr., L. Birnbaumer, and A. Staruschenko. 2018. A NOX4/TRPC6 pathway in podocyte calcium regulation and renal damage in diabetic kidney disease. Journal of the American Society of Nephrology 29: 1917–1927.
Staruschenko, A., R. Ma, O. Palygin, and S.E. Dryer. 2023. Ion channels and channelopathies in glomeruli. Physiological Reviews 103: 787–854.
Chen, Y., L. Lin, X. Tao, Y. Song, J. Cui, and J. Wan. 2019. The role of podocyte damage in the etiology of ischemia-reperfusion acute kidney injury and post-injury fibrosis. BMC Nephrology 20: 106.
Yu, Y., L. Zhang, G. Xu, Z. Wu, Q. Li, Y. Gu, and J. Niu. 2018. Angiotensin II type i receptor agonistic autoantibody induces podocyte injury via activation of the TRPC6-calcium/calcineurin pathway in pre-eclampsia. Kidney & Blood Pressure Research 43: 1666–1676.
Wu, Y.L., J. Xie, S.W. An, N. Oliver, N.X. Barrezueta, M.H. Lin, L. Birnbaumer, and C.L. Huang. 2017. Inhibition of TRPC6 channels ameliorates renal fibrosis and contributes to renal protection by soluble klotho. Kidney International 91: 830–841.
Kim, E.Y., and S.E. Dryer. 2021. RAGE and alphaVbeta3-integrin are essential for suPAR signaling in podocytes. Biochimica et Biophysica Acta, Molecular Basis of Disease 1867: 166186.
Lin, B.L., D. Matera, J.F. Doerner, N. Zheng, D. Del Camino, S. Mishra, H. Bian, S. Zeveleva, X. Zhen, N.T. Blair, J.A. Chong, D.P. Hessler, D. Bedja, G. Zhu, G.K. Muller, M.J. Ranek, L. Pantages, M. McFarland, M.R. Netherton, A. Berry, D. Wong, G. Rast, H.S. Qian, S.M. Weldon, J.J. Kuo, A. Sauer, C. Sarko, M.M. Moran, D.A. Kass, and S.S. Pullen. 2019. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proceedings of the National Academy of Sciences USA 116: 10156–10161.
Du, W., J. Huang, H. Yao, K. Zhou, B. Duan, and Y. Wang. 2010. Inhibition of TRPC6 degradation suppresses ischemic brain damage in rats. The Journal of Clinical Investigation 120: 3480–3492.
Li, Y., Y.C. Jia, K. Cui, N. Li, Z.Y. Zheng, Y.Z. Wang, and X.B. Yuan. 2005. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature 434: 894–898.
Jia, Y., J. Zhou, Y. Tai, and Y. Wang. 2007. TRPC channels promote cerebellar granule neuron survival. Nature Neuroscience 10: 559–567.
Zhou, J., W. Du, K. Zhou, Y. Tai, H. Yao, Y. Jia, Y. Ding, and Y. Wang. 2008. Critical role of TRPC6 channels in the formation of excitatory synapses. Nature Neuroscience 11: 741–743.
Quick, K., J. Zhao, N. Eijkelkamp, J.E. Linley, F. Rugiero, J.J. Cox, R. Raouf, M. Gringhuis, J.E. Sexton, J. Abramowitz, R. Taylor, A. Forge, J. Ashmore, N. Kirkwood, C.J. Kros, G.P. Richardson, M. Freichel, V. Flockerzi, L. Birnbaumer, and J.N. Wood. 2012. TRPC3 and TRPC6 are essential for normal mechanotransduction in subsets of sensory neurons and cochlear hair cells. Open Biology 2: 120068.
Kumar, S., S. Chakraborty, C. Barbosa, T. Brustovetsky, B. Brustovetsky, and A.G. Obukhov. 2011. Mechanisms controlling neurite outgrowth in a pheochromocytoma cell line: The role of TRPC channels. Journal of Cellular Physiology 227: 1408–1409.
Lu, R., J. Wang, R. Tao, J. Wang, T. Zhu, W. Guo, Y. Sun, H. Li, Y. Gao, W. Zhang, C.J. Fowler, Q. Li, S. Chen, Z. Wu, C.L. Masters, C. Zhong, N. Jing, Y. Wang, and Y. Wang. 2018. Reduced TRPC6 mRNA levels in the blood cells of patients with Alzheimer’s disease and mild cognitive impairment. Molecular Psychiatry 23: 767–776.
Chen, J.M., Q.W. Li, J.S. Liu, G.X. Jiang, J.R. Liu, S.D. Chen, and Q. Cheng. 2019. TRPC6 mRNA levels in peripheral leucocytes of patients with Alzheimer’s disease and mild cognitive impairment: A case-control study. Progress in Neuro-Psychopharmacology and Biological Psychiatry 92: 279–284.
Wang, J., R. Lu, J. Yang, H. Li, Z. He, N. Jing, X. Wang, and Y. Wang. 2015. TRPC6 specifically interacts with APP to inhibit its cleavage by gamma-secretase and reduce Abeta production. Nature Communications 6: 8876.
Hartz, A.M., D.S. Miller, and B. Bauer. 2010. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Molecular Pharmacology 77: 715–723.
He, C., Q. Li, Y. Cui, P. Gao, W. Shu, Q. Zhou, L. Wang, L. Li, Z. Lu, Y. Zhao, H. Ma, X. Chen, H. Jia, H. Zheng, G. Yang, D. Liu, M. Tepel, and Z. Zhu. 2022. Recurrent moderate hypoglycemia accelerates the progression of Alzheimer’s disease through impairment of the TRPC6/GLUT3 pathway. JCI Insight 7.
Griesi-Oliveira, K., A. Acab, A.R. Gupta, D.Y. Sunaga, T. Chailangkarn, X. Nicol, Y. Nunez, M.F. Walker, J.D. Murdoch, S.J. Sanders, T.V. Fernandez, W. Ji, R.P. Lifton, E. Vadasz, A. Dietrich, D. Pradhan, H. Song, G.L. Ming, X. Gu, G. Haddad, M.C. Marchetto, N. Spitzer, M.R. Passos-Bueno, M.W. State, and A.R. Muotri. 2015. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Molecular Psychiatry 20: 1350–1365.
Palacios-Munoz, A., D. de Paula Moreira, V. Silva, I.E. Garcia, F. Aboitiz, M. Zarrei, G. Campos, O. Rennie, J.L. Howe, E. Anagnostou, P. Ambrozewic, S.W. Scherer, M.R. Passos-Bueno, and J. Ewer. 2022. Mutations in trpgamma, the homologue of TRPC6 autism candidate gene, causes autism-like behavioral deficits in Drosophila. Molecular Psychiatry 27: 3328–3342.
Shen, H., J. Pan, L. Pan, and N. Zhang. 2013. TRPC6 inhibited NMDA current in cultured hippocampal neurons. Neuromolecular Medicine 15: 389–395.
Li, W., F. Yang, J. Gao, Y. Tang, J. Wang, and Y. Pan. 2019. Over-expression of TRPC6 via CRISPR based synergistic activation mediator in BMSCs ameliorates brain injury in a rat model of cerebral ischemia/reperfusion. Neuroscience 415: 147–160.
Lin, Y., J.C. Zhang, J. Fu, F. Chen, J. Wang, Z.L. Wu, and S.Y. Yuan. 2013. Hyperforin attenuates brain damage induced by transient middle cerebral artery occlusion (MCAO) in rats via inhibition of TRPC6 channels degradation. Journal of Cerebral Blood Flow and Metabolism 33: 253–262.
Jardin, I., R. Diez-Bello, J.J. Lopez, P.C. Redondo, G.M. Salido, T. Smani, and J.A. Rosado. 2018. TRPC6 channels are required for proliferation, migration and invasion of breast cancer cell lines by modulation of Orai1 and Orai3 surface exposure. Cancers (Basel). 10.
Dhennin-Duthille, I., M. Gautier, M. Faouzi, A. Guilbert, M. Brevet, D. Vaudry, A. Ahidouch, H. Sevestre, and H. Ouadid-Ahidouch. 2011. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: Correlation with pathological parameters. Cellular Physiology and Biochemistry 28: 813–822.
Shi, Y., X. Ding, Z.H. He, K.C. Zhou, Q. Wang, and Y.Z. Wang. 2009. Critical role of TRPC6 channels in G2 phase transition and the development of human oesophageal cancer. Gut 58: 1443–1450.
Ding, X., Z. He, K. Zhou, J. Cheng, H. Yao, D. Lu, R. Cai, Y. Jin, B. Dong, Y. Xu, and Y. Wang. 2010. Essential role of TRPC6 channels in G2/M phase transition and development of human glioma. Journal of the National Cancer Institute 102: 1052–1068.
El Boustany, C., G. Bidaux, A. Enfissi, P. Delcourt, N. Prevarskaya, and T. Capiod. 2008. Capacitative calcium entry and Transient Receptor Potential Canonical 6 expression control human hepatoma cell proliferation. Hepatology 47: 2068–2077.
Ge, P., L. Wei, M. Zhang, B. Hu, K. Wang, Y. Li, S. Liu, J. Wang, and Y. Li. 2018. TRPC1/3/6 inhibition attenuates the TGF-beta1-induced epithelial-mesenchymal transition in gastric cancer via the Ras/Raf1/ERK signaling pathway. Cell Biology International 42: 975–984.
Cai, R., X. Ding, K. Zhou, Y. Shi, R. Ge, G. Ren, Y. Jin, and Y. Wang. 2009. Blockade of TRPC6 channels induced G2/M phase arrest and suppressed growth in human gastric cancer cells. International Journal of Cancer 125: 2281–2287.
Song, J., Y. Wang, X. Li, Y. Shen, M. Yin, Y. Guo, L. Diao, Y. Liu, and D. Yue. 2013. Critical role of TRPC6 channels in the development of human renal cell carcinoma. Molecular Biology Reports 40: 5115–5122.
Jiang, H.N., B. Zeng, Y. Zhang, N. Daskoulidou, H. Fan, J.M. Qu, and S.Z. Xu. 2013. Involvement of TRPC channels in lung cancer cell differentiation and the correlation analysis in human non-small cell lung cancer. PLoS ONE 8: e67637.
Bernaldo de Quiros, S., A. Merlo, P. Secades, I. Zambrano, I.S. de Santa Maria, N. Ugidos, E. Jantus-Lewintre, R. Sirera, C. Suarez, and M.D. Chiara. 2013. Identification of TRPC6 as a possible candidate target gene within an amplicon at 11q21–q22.2 for migratory capacity in head and neck squamous cell carcinomas. BMC Cancer 13: 116.
Wan, Q., A. Zheng, X. Liu, Y. Chen, and L. Han. 2012. Expression of Transient Receptor Potential Channel 6 in cervical cancer. Oncotargets and Therapy 5: 171–176.
Ma, X., Y. Cai, D. He, C. Zou, P. Zhang, C.Y. Lo, Z. Xu, F.L. Chan, S. Yu, Y. Chen, R. Zhu, J. Lei, J. Jin, and X. Yao. 2012. Transient Receptor Potential Channel TRPC5 is essential for P-glycoprotein induction in drug-resistant cancer cells. Proceedings of the National Academy of Sciences USA 109: 16282–16287.
Aydar, E., S. Yeo, M. Djamgoz, and C. Palmer. 2009. Abnormal expression, localization and interaction of canonical transient receptor potential ion channels in human breast cancer cell lines and tissues: A potential target for breast cancer diagnosis and therapy. Cancer Cell International 9: 23.
Girault, A., A. Ahidouch, and H. Ouadid-Ahidouch. 2020. Roles for Ca(2+) and K(+) channels in cancer cells exposed to the hypoxic tumour microenvironment. Biochimica et Biophysica Acta, Molecular Cell Research 1867: 118644.
Hisatsune, C., Y. Kuroda, K. Nakamura, T. Inoue, T. Nakamura, T. Michikawa, A. Mizutani, and K. Mikoshiba. 2004. Regulation of TRPC6 channel activity by tyrosine phosphorylation. Journal of Biological Chemistry 279: 18887–18894.
Xie, Y.G., Y. Yu, L.K. Hou, X. Wang, B. Zhang, and X.C. Cao. 2016. FYN promotes breast cancer progression through epithelial-mesenchymal transition. Oncology Reports 36: 1000–1006.
Bagu, E.T., S. Miah, C. Dai, T. Spriggs, Y. Ogunbolude, E. Beaton, M. Sanders, R.K. Goel, K. Bonham, and K.E. Lukong. 2017. Repression of Fyn-related kinase in breast cancer cells is associated with promoter site-specific CpG methylation. Oncotarget 8: 11442–11459.
Lussier, M.P., S. Cayouette, P.K. Lepage, C.L. Bernier, N. Francoeur, M. St-Hilaire, M. Pinard, and G. Boulay. 2005. MxA, a member of the dynamin superfamily, interacts with the ankyrin-like repeat domain of TRPC. Journal of Biological Chemistry 280: 19393–19400.
Zhang, S.S., J. Wen, F. Yang, X.L. Cai, H. Yang, K.J. Luo, Q.W. Liu, R.G. Hu, X. Xie, Q.Y. Huang, J.Y. Chen, J.H. Fu, and Y. Hu. 2013. High expression of transient potential receptor C6 correlated with poor prognosis in patients with esophageal squamous cell carcinoma. Medical Oncology 30: 607.
Yin, H., H. Cheng, P. Li, and Z. Yang. 2022. TRPC6 interacted with K(Ca)1.1 channels to regulate the proliferation and apoptosis of glioma cells. Archives of Biochemistry and Biophysics 725: 109268.
Wen, L., C. Liang, E. Chen, W. Chen, F. Liang, X. Zhi, T. Wei, F. Xue, G. Li, Q. Yang, W. Gong, X. Feng, X. Bai, and T. Liang. 2016. Regulation of multi-drug resistance in hepatocellular carcinoma cells is TRPC6/calcium dependent. Science and Reports 6: 23269.
Kim, J.H., K.H. Hwang, M. Eom, M. Kim, E.Y. Park, Y. Jeong, K.S. Park, and S.K. Cha. 2019. WNK1 promotes renal tumor progression by activating TRPC6-NFAT pathway. The FASEB Journal 33: 8588–8599.
Yang, L.L., B.C. Liu, X.Y. Lu, Y. Yan, Y.J. Zhai, Q. Bao, P.W. Doetsch, X. Deng, T.L. Thai, A.A. Alli, D.C. Eaton, B.Z. Shen, and H.P. Ma. 2017. Inhibition of TRPC6 reduces non-small cell lung cancer cell proliferation and invasion. Oncotarget 8: 5123–5134.
Wang, Y., J. He, H. Jiang, Q. Zhang, H. Yang, X. Xu, C. Zhang, C. Xu, J. Wang, and W. Lu. 2018. Nicotine enhances store-operated calcium entry by upregulating HIF-1alpha and SOCC components in non-small cell lung cancer cells. Oncology Reports 40: 2097–2104.
Thebault, S., M. Flourakis, K. Vanoverberghe, F. Vandermoere, M. Roudbaraki, V. Lehen’kyi, C. Slomianny, B. Beck, P. Mariot, J.L. Bonnal, B. Mauroy, Y. Shuba, T. Capiod, R. Skryma, and N. Prevarskaya. 2006. Differential role of transient receptor potential channels in Ca2+ entry and proliferation of prostate cancer epithelial cells. Cancer Research 66: 2038–2047.
Wang, D., X. Li, J. Liu, J. Li, L.J. Li, and M.X. Qiu. 2014. Effects of TRPC6 on invasibility of low-differentiated prostate cancer cells. Asian Pacific Journal of Tropical Medicine 7: 44–47.
Wang, Y., D. Yue, K. Li, Y.L. Liu, C.S. Ren, and P. Wang. 2010. The role of TRPC6 in HGF-induced cell proliferation of human prostate cancer DU145 and PC3 cells. Asian Journal of Andrology 12: 841–852.
Bernichtein, S., N. Pigat, N. Barry Delongchamps, F. Boutillon, V. Verkarre, P. Camparo, E. Reyes-Gomez, A. Mejean, S.M. Oudard, E.M. Lepicard, M. Viltard, J.C. Souberbielle, G. Friedlander, T. Capiod, and V. Goffin. 2017. Vitamin D3 prevents calcium-induced progression of early-stage prostate tumors by counteracting TRPC6 and calcium sensing receptor upregulation. Cancer Research 77: 355–365.
Zeng, B., C. Yuan, X. Yang, S.L. Atkin, and S.Z. Xu. 2013. TRPC channels and their splice variants are essential for promoting human ovarian cancer cell proliferation and tumorigenesis. Current Cancer Drug Targets 13: 103–116.
Bai, L.P., Y.L. Chen, and A. Zheng. 2022. Pharmacological targeting Transient Receptor Potential Canonical channel 6 modulates biological behaviors for cervical cancer HeLa and SiHA cell. Cancer Cell International 22: 145.
Yamaguchi, Y., G. Iribe, M. Nishida, and K. Naruse. 2017. Role of TRPC3 and TRPC6 channels in the myocardial response to stretch: Linking physiology and pathophysiology. Progress in Biophysics and Molecular Biology 130: 264–272.
Oda, S., T. Numaga-Tomita, N. Kitajima, T. Toyama, E. Harada, T. Shimauchi, A. Nishimura, T. Ishikawa, Y. Kumagai, L. Birnbaumer, and M. Nishida. 2017. TRPC6 counteracts TRPC3-Nox2 protein complex leading to attenuation of hyperglycemia-induced heart failure in mice. Science and Reports 7: 7511.
Kuwahara, K., Y. Wang, J. McAnally, J.A. Richardson, R. Bassel-Duby, J.A. Hill, and E.N. Olson. 2006. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. The Journal of Clinical Investigation 116: 3114–3126.
Nishida, M., and H. Kurose. 2008. Roles of TRP channels in the development of cardiac hypertrophy. Naunyn-Schmiedeberg’s Archives of Pharmacology 378: 395–406.
Bogdanova, E., O. Beresneva, O. Galkina, I. Zubina, G. Ivanova, M. Parastaeva, N. Semenova, and V. Dobronravov. 2021. Myocardial hypertrophy and fibrosis are associated with cardiomyocyte beta-catenin and TRPC6/calcineurin/NFAT signaling in spontaneously hypertensive rats with 5/6 nephrectomy. International Journal of Molecular Sciences 22.
Xie, J., S.K. Cha, S.W. An, O.M. Kuro, L. Birnbaumer, and C.L. Huang. 2012. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nature Communications 3: 1238.
Zhou, R., P. Hang, W. Zhu, Z. Su, H. Liang, and Z. Du. 2011. Whole genome network analysis of ion channels and connexins in myocardial infarction. Cellular Physiology and Biochemistry 27: 299–304.
Kinoshita, H., K. Kuwahara, M. Nishida, Z. Jian, X. Rong, S. Kiyonaka, Y. Kuwabara, H. Kurose, R. Inoue, Y. Mori, Y. Li, Y. Nakagawa, S. Usami, M. Fujiwara, Y. Yamada, T. Minami, K. Ueshima, and K. Nakao. 2010. Inhibition of TRPC6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A signaling in the heart. Circulation Research 106: 1849–1860.
Nikolova-Krstevski, V., S. Wagner, Z.Y. Yu, C.D. Cox, J. Cvetkovska, A.P. Hill, I.G. Huttner, V. Benson, A.A. Werdich, C. MacRae, M.P. Feneley, O. Friedrich, B. Martinac, and D. Fatkin. 2017. Endocardial TRPC-6 channels act as atrial mechanosensors and load-dependent modulators of endocardial/myocardial cross-talk. JACC: Basic to Translational Science 2: 575–590.
Yu, Y., S.H. Keller, C.V. Remillard, O. Safrina, A. Nicholson, S.L. Zhang, W. Jiang, N. Vangala, J.W. Landsberg, J.Y. Wang, P.A. Thistlethwaite, R.N. Channick, I.M. Robbins, J.E. Loyd, H.A. Ghofrani, F. Grimminger, R.T. Schermuly, M.D. Cahalan, L.J. Rubin, and J.X. Yuan. 2009. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 119: 2313–2322.
Li, W., X. Chen, A.M. Riley, S.C. Hiett, C.J. Temm, E. Beli, X. Long, S. Chakraborty, M. Alloosh, F.A. White, M.B. Grant, M. Sturek, and A.G. Obukhov. 2017. Long-term spironolactone treatment reduces coronary TRPC expression, vasoconstriction, and atherosclerosis in metabolic syndrome pigs. Basic ResCardiol. 112: 54.
Antigny, F., C. Norez, L. Dannhoffer, J. Bertrand, D. Raveau, P. Corbi, C. Jayle, F. Becq, and C. Vandebrouck. 2011. Transient Receptor Potential Canonical channel 6 links Ca2+ mishandling to cystic fibrosis transmembrane conductance regulator channel dysfunction in cystic fibrosis. American Journal of Respiratory Cell and Molecular Biology 44: 83–90.
Finney-Hayward, T.K., M.O. Popa, P. Bahra, S. Li, C.T. Poll, M. Gosling, A.G. Nicholson, R.E. Russell, O.M. Kon, G. Jarai, J. Westwick, P.J. Barnes, and L.E. Donnelly. 2010. Expression of transient receptor potential C6 channels in human lung macrophages. American Journal of Respiratory Cell and Molecular Biology 43: 296–304.
Wang, Y.X., and Y.M. Zheng. 2011. Molecular expression and functional role of canonical transient receptor potential channels in airway smooth muscle cells. Advances in Experimental Medicine and Biology 704: 731–747.
Hofmann, K., S. Fiedler, S. Vierkotten, J. Weber, S. Klee, J. Jia, W. Zwickenpflug, V. Flockerzi, U. Storch, A.O. Yildirim, T. Gudermann, M. Konigshoff, and A. Dietrich. 2017. Classical Transient Receptor Potential 6 (TRPC6) channels support myofibroblast differentiation and development of experimental pulmonary fibrosis. Biochimica et Biophysica Acta, Molecular Basis of Disease 1863: 560–568.
Weissmann, N., A. Sydykov, H. Kalwa, U. Storch, B. Fuchs, and Mederos y Schnitzler, M., Brandes, R. P., Grimminger, F., Meissner, M., Freichel, M., Offermanns, S., Veit, F., Pak, O., Krause, K. H., Schermuly, R. T., Brewer, A. C., Schmidt, H. H., Seeger, W., Shah, A. M., Gudermann, T., Ghofrani, H. A. & Dietrich, A. 2012. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nature Communications 3: 649.
Tauseef, M., N. Knezevic, K.R. Chava, M. Smith, S. Sukriti, N. Gianaris, A.G. Obukhov, S.M. Vogel, D.E. Schraufnagel, A. Dietrich, L. Birnbaumer, A.B. Malik, and D. Mehta. 2012. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. Journal of Experimental Medicine 209: 1953–1968.
Hong, W., G. Peng, B. Hao, B. Liao, Z. Zhao, Y. Zhou, F. Peng, X. Ye, L. Huang, M. Zheng, J. Pu, C. Liang, E. Yi, H. Peng, B. Li, and P. Ran. 2017. Nicotine-induced airway smooth muscle cell proliferation involves TRPC6-dependent calcium influx via alpha7 nAChR. Cellular Physiology and Biochemistry 43: 986–1002.
Samapati, R., Y. Yang, J. Yin, C. Stoerger, C. Arenz, A. Dietrich, T. Gudermann, D. Adam, S. Wu, M. Freichel, V. Flockerzi, S. Uhlig, and W.M. Kuebler. 2012. Lung endothelial Ca2+ and permeability response to platelet-activating factor is mediated by acid sphingomyelinase and Transient Receptor Potential Classical 6. American Journal of Respiratory and Critical Care Medicine 185: 160–170.
Jiang, T., R. Samapati, S. Klassen, D. Lei, L. Erfinanda, V. Jankowski, S. Simmons, J. Yin, C. Arenz, A. Dietrich, T. Gudermann, D. Adam, M. Schaefer, J. Jankowski, V. Flockerzi, R. Nusing, S. Uhlig, and W.M. Kuebler. 2022. Stimulation of the EP(3) receptor causes lung oedema by activation of TRPC6 in pulmonary endothelial cells. European Respiratory Journal 60.
Leidinger, G., F. Flockerzi, J. Hohneck, R.M. Bohle, A. Fieguth, and T. Tschernig. 2022. TRPC6 is altered in COVID-19 pneumonia. Chemico-Biological Interactions 362: 109982.
Chen, Q., Y. Zhou, L. Zhou, Z. Fu, C. Yang, L. Zhao, S. Li, Y. Chen, Y. Wu, Z. Ling, Y. Wang, J. Huang, and J. Li. 2020. TRPC6-dependent Ca(2+) signaling mediates airway inflammation in response to oxidative stress via ERK pathway. Cell Death & Disease 11: 170.
Wang, J., L. Weigand, W. Lu, J.T. Sylvester, G.L. Semenza, and L.A. Shimoda. 2006. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circulation Research 98: 1528–1537.
Weissmann, N., A. Dietrich, B. Fuchs, H. Kalwa, M. Ay, R. Dumitrascu, A. Olschewski, U. Storch, M. Schnitzler, H.A. Ghofrani, R.T. Schermuly, O. Pinkenburg, W. Seeger, F. Grimminger, and T. Gudermann. 2006. Classical Transient Receptor Potential Channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proceedings of the National Academy of Sciences USA 103: 19093–19098.
Malczyk, M., C. Veith, B. Fuchs, K. Hofmann, U. Storch, R.T. Schermuly, M. Witzenrath, K. Ahlbrecht, C. Fecher-Trost, V. Flockerzi, H.A. Ghofrani, F. Grimminger, W. Seeger, T. Gudermann, A. Dietrich, and N. Weissmann. 2013. Classical Transient Receptor Potential Channel 1 in hypoxia-induced pulmonary hypertension. American Journal of Respiratory and Critical Care Medicine 188: 1451–1459.
He, X., S. Song, R.J. Ayon, A. Balisterieri, S.M. Black, A. Makino, W.G. Wier, W.J. Zang, and J.X. Yuan. 2018. Hypoxia selectively upregulates cation channels and increases cytosolic [Ca(2+)] in pulmonary, but not coronary, arterial smooth muscle cells. American Journal of Physiology. Cell Physiology 314: C504–C517.
Xia, Y., X.R. Yang, Z. Fu, O. Paudel, J. Abramowitz, L. Birnbaumer, and J.S. Sham. 2014. Classical Transient Receptor Potential 1 and 6 contribute to hypoxic pulmonary hypertension through differential regulation of pulmonary vascular functions. Hypertension 63: 173–180.
Lin, M.J., G.P. Leung, W.M. Zhang, X.R. Yang, K.P. Yip, C.M. Tse, and J.S. Sham. 2004. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: A novel mechanism of hypoxic pulmonary hypertension. Circulation Research 95: 496–505.
Zhao, T., S. Parmisano, Z. Soroureddin, M. Zhao, L. Yung, P.A. Thistlethwaite, A. Makino, and J.X. Yuan. 2022. Mechanosensitive cation currents through TRPC6 and Piezo1 channels in human pulmonary arterial endothelial cells. American Journal of Physiology. Cell Physiology 323: C959–C973.
Bai, Y., X. Yu, H. Chen, D. Horne, R. White, X. Wu, P. Lee, Y. Gu, S. Ghimire-Rijal, D.C. Lin, and X. Huang. 2020. Structural basis for pharmacological modulation of the TRPC6 channel. Elife 9.
Lin, B.L., J.Y. Shin, W.P. Jeffreys, N. Wang, C.A. Lukban, M.C. Moorer, E. Velarde, O.A. Hanselman, S. Kwon, S. Kannan, R.C. Riddle, C.W. Ward, S.S. Pullen, A. Filareto, and D.A. Kass. 2022. Pharmacological TRPC6 inhibition improves survival and muscle function in mice with Duchenne muscular dystrophy. JCI Insight 7.
Maier, T., M. Follmann, G. Hessler, H.W. Kleemann, S. Hachtel, B. Fuchs, N. Weissmann, W. Linz, T. Schmidt, M. Lohn, K. Schroeter, L. Wang, H. Rutten, and C. Strubing. 2015. Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol-sensitive TRPC cation channels. British Journal of Pharmacology 172: 3650–3660.
Hafner, S., F. Burg, M. Kannler, N. Urban, P. Mayer, A. Dietrich, D. Trauner, J. Broichhagen, and M. Schaefer. 2018. A (+)-larixol congener with high affinity and subtype selectivity toward TRPC6. ChemMedChem 13: 1028–1035.
Zheng, Z., Y. Xu, U. Krugel, M. Schaefer, T. Grune, B. Nurnberg, M.B. Kohler, M. Gollasch, D. Tsvetkov, and L. Marko. 2022. In vivo inhibition of TRPC6 by SH045 attenuates renal fibrosis in a New Zealand Obese (NZO) mouse model of metabolic syndrome. International Journal of Molecular Sciences 23.
Shimauchi, T., T. Numaga-Tomita, Y. Kato, H. Morimoto, K. Sakata, R. Matsukane, A. Nishimura, K. Nishiyama, A. Shibuta, Y. Horiuchi, H. Kurose, S.G. Kim, Y. Urano, T. Ohshima, and M. Nishida. 2022. A TRPC3/6 channel inhibitor promotes arteriogenesis after hind-limb ischemia. Cells 11.
Yang, P.L., X.H. Li, J. Wang, X.F. Ma, B.Y. Zhou, Y.F. Jiao, W.H. Wang, P. Cao, M.X. Zhu, P.W. Li, Z.H. Xiao, C.Z. Li, C.R. Guo, Y.T. Lei, and Y. Yu. 2021. GSK1702934A and M085 directly activate TRPC6 via a mechanism of stimulating the extracellular cavity formed by the pore helix and transmembrane helix S6. Journal of Biological Chemistry 297: 101125.
Qu, C., M. Ding, Y. Zhu, Y. Lu, J. Du, M. Miller, J. Tian, J. Zhu, J. Xu, M. Wen, A. Er-Bu, J. Wang, Y. Xiao, M. Wu, O.B. McManus, M. Li, J. Wu, H.R. Luo, Z. Cao, B. Shen, H. Wang, M.X. Zhu, and X. Hong. 2017. Pyrazolopyrimidines as potent stimulators for Transient Receptor Potential Canonical 3/6/7 channels. Journal of Medicinal Chemistry 60: 4680–4692.
Leuner, K., J.H. Heiser, S. Derksen, M.I. Mladenov, C.J. Fehske, R. Schubert, M. Gollasch, G. Schneider, C. Harteneck, S.S. Chatterjee, and W.E. Muller. 2010. Simple 2,4 diacylphloroglucinols as TRPC6 activators — identification of a novel pharmacophore. Molecular Pharmacology 77: 368–377.
Sell, T.S., T. Belkacemi, V. Flockerzi, and A. Beck. 2014. Protonophore properties of hyperforin are essential for its pharmacological activity. Science and Reports 4: 7500.
Kong, W., T.N. Haschler, B. Nurnberg, S. Kramer, M. Gollasch, and L. Marko. 2019. Renal fibrosis, immune cell infiltration and changes of TRPC channel expression after unilateral ureteral obstruction in TRPC6−/− mice. Cellular Physiology and Biochemistry 52: 1484–1502.
ACKNOWLEDGEMENTS
The authors thank the Department of Life Science, DAVV for the facilities.
Funding
Financial assistance from UGC-DSKPDF (BL/20–21/0482 (S-90)) was provided to Dr. Uzma Saqib.
Author information
Authors and Affiliations
Contributions
U. S., K. H., and A. G. O. designed the review. S. S., O. M., S. B., H. S., and S. M. were responsible for drafting the manuscript. U. S. and A.G.O. were responsible for the figures. M. S. B., A. G. O., and K. H. edited the manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Saqib, U., Munjuluri, S., Sarkar, S. et al. Transient Receptor Potential Canonical 6 (TRPC6) Channel in the Pathogenesis of Diseases: A Jack of Many Trades. Inflammation 46, 1144–1160 (2023). https://doi.org/10.1007/s10753-023-01808-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-023-01808-3