
Abstract—
Hidradenitis suppurativa (HS) is a complex and debilitating inflammatory skin disease for which no effective treatment is available currently. This is partly because of the lack of adequate human or animal models for defining the pathobiology of the disease. Here, we describe the development of air–liquid (A-L) interface, liquid-submersion (L-S), and bioreactor (Bio) ex vivo skin culture models. All three ex vivo platforms were effective for culturing skin samples for up to 14 days. Tissue architecture and integrity remained intact for at least 3 days for healthy skin and 14 days for HS skin. Up to day 3, no significant differences were observed in % early apoptotic cells among all three platforms. However, late apoptotic/necrotic cell death was increased in HS skin at day 3 in A-L and Bio culture. These cultures efficiently support the growth of various cells populations, including keratinocytes and immune cells. Profiling inflammatory gene signatures in HS skin from these ex vivo cultures showed dynamic changes in expression at day 3 and day 14. All three culture platforms were necessary to represent the inflammatory gene status of HS skin at day 0, suggesting that not all gene clusters were identically altered in each culture method. Similarly, cytokine/chemokine profiling of the supernatants from vehicle- and drug-treated ex vivo HS cultures again showed a better prediction of drug efficacy against HS. Overall, development of these three culture systems collectively provides a powerful tool to uncover the pathobiology of HS progression and screen various drugs against HS.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition with painful lesions, including nodules, abscesses, draining tracts, and fibrotic scars. This debilitating disease affects 1–4% of the Western population [1, 2]. So far, no effective treatments have been identified particularly for severe disease. The pathologic process of HS is thought to develop from occlusion and rupture of defective hair follicles and the subsequent release of contents including bacteria into the dermis [3, 4]. A complex inflammatory response by surrounding neutrophils and lymphocytes is known to accelerate the formation of abscess and causes the subsequent destruction of the pilosebaceous unit [5, 6]. Although the presence of abscesses and malodorous drainage has implicated a role of microorganisms in HS pathogenesis, only certain antimicrobial therapies have been found temporarily and/or partially effective. In addition, the risk of drug resistance is high, if administered on a long-term basis [7]. Keratinocyte hyperproliferation is also a hallmark of HS [8]. Unlike other inflammatory skin diseases, the immunobiology of HS is poorly understood, which has also hindered the development of effective therapies. Studies defining the elevation of inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukins (IL)-1β and IL17A, and various others in HS patients, have provided possible therapeutic targets for emerging treatments [9,10,11,12]. This is also based on the known success of biologics against these targets for therapeutic intervention of various other inflammatory diseases [13]. The TNF-α antagonist adalimumab is currently the only biological approved by the United States Food and Drug Administration [13, 14] and by European Medicines Agency for treatment of HS [15]. The lack of optimal pre-clinical models of HS has hampered the pace of development of novel treatments to effectively target immune dysregulation and the associated pathobiology of HS disease progression [16].
In this study, we report the establishment of three different ex vivo HS skin culture models that enable efficient characterization of immune infiltrates and hyperproliferative keratinocytes in HS tissues alone and following treatment with various drugs. These models include air–liquid interface (A-L) culture, liquid submersion (L-S) culture, and culture using a three-dimensional perfusion bioreactor (Bio) platform with well-defined culture condition. L-S culture is very similar to traditional cell culture, in which the entire three-dimensional tissue is submerged in culture media. A-L culture better mimics the native tissue environment, with the apical tissue surface exposed to air and the basal surface covered by media. Both the A-L culture and the L-S culture are static culture platforms, whereas the Bio platform allows for continuous perfusion of oxygen/nutrients to mimic the human circulatory system. Thus, each of these culture methods has some benefits over others as well as some disadvantages. We demonstrate that the tissue architecture and viability of both healthy and HS skin tissues are maintained ex vivo in these models, over a period of 14 days. CD4+ and CD8+ lymphocyte populations, including the regulatory T cells and memory T cells, as well as keratinocytes and monocytes/macrophages could be characterized and enumerated by FACS analysis in these models. Importantly, we assess the feasibility of these models to test the effects of drugs on the inflammatory cytokines/chemokine signatures and signaling pathways associated with immune dysfunction in HS. Utilization of multiple ex vivo HS skin culture platforms will facilitate the development of novel therapeutics and unravel unique mechanisms underlying the disease pathogenesis.
METHODS
Human Tissue
The Institutional Review Boards of the University of Alabama at Birmingham approved the protocol (IRB-300005214) for obtaining discarded surgically removed skin tissues from Healthy and HS (Hurley stage 2 or stage 3) subjects.
Sample Processing and Ex Vivo Culture
Clinical specimen were divided into ~ 5 mm × 5 mm tissue pieces. Tissue pieces were then either (1) placed on to a 0.4-µm transwell filter (Merck Millipore) with a thin layer of extracellular matrix (ECM, 90% collagen type 1 (Advanced Biomatrix, USA) + 10% growth factor reduced Matrigel (Corning, USA)) for stability or (2) placed into the central chamber of a polydimethylsiloxane (PDMS, Krayden, USA) bioreactor containing a mixture of ECM for structural support as previously described [17]. The tissue/ECM volume within the bioreactor was penetrated with five 400-µm Teflon-coated stainless steel wires to generate through-channels for tissue perfusion. Following ECM polymerization, the wires were removed and the through-channels were filled with tissue culture media (1:1 mixture of X-Vivo15 and bronchial epithelial growth media (Lonza, USA) with antibiotics (MP Biomedicals, USA)). The bioreactor was then connected to a perfusion system that contained a media reservoir, peroxide-cured silicon tubing (Cole Parmer, USA), a collection reservoir, and peristaltic pump (ESI, USA), and tissue culture media was perfused through the tissue volume for 3 to 14 days (37 °C, 5% CO2), with media changed every 3 days. Following ECM polymerization, tissue culture media was added to the transwell filters and to the bottom of the well to generate L-S (tissue fully submerged in media) and A-L (the media volume did not cover the top tissue layer) cultures. When applicable, treatment with lenalidomide (1.5 mg/mL, Selleckchem), CPI-0610 (1.5 mg/mL, Selleckchem), or vehicle control (dimethyl sulfoxide (DMSO)) occurred daily for 3 days of culture. At the end of each experiment, a portion of each tissue was fixed for histologic processing, digested with collagenase B (Roche, Switzerland) for flow cytometry analysis, or stored in RNAlater (Sigma Aldrich, USA) for RNA extraction. The supernatant culture media was collected for assaying cytokine/chemokines levels.
Histologic Processing and Analysis
Following ex vivo culture, a portion of each cultured tissue was fixed with neutral buffered formalin, processed to paraffin, and histological sections were prepared, as previously described [17]. Five-micron sections were stained with hematoxylin and eosin (H&E) to evaluate tissue morphology.
Analysis of Cell Viability via Flow Cytometry
The PE Annexin V Apoptosis Detection Kit I (BD, Germany) was used to analyze cell viability following manufacturer’s instructions. Analyses were performed on FACSymphony A3 Cell Analyzer with FACSDiva software version 8.0.1 (BD, Germany). Data were analyzed with FlowJo 10.7.1 (Treestar, USA).
Multiparametric Flow Cytometry
The following antibodies were used for multiparametric flow cytometry for T cell analysis: anti-CD3-Alexa Fluor 700 (Clone: UCHT1); anti-CD4-FITC (Clone: RPA-T4); anti-CD69-BV563 (Clone: FN50); and anti-CD25-BV510 (Clone: M-A251) from BD Biosciences (Germany). Anti-CD3-PE-Cy7 (Clone: UCHT1); anti-CD8-APC (Clone: 53–6.7); anti-CD4-PE-Cy7 (Clone: SK3); anti-FoxP3-PerCP-Cy5.5 (Clone: PCH101); anti-CD8-FITC (Clonse: SK1); anti-CD-127-APC (Clone: eBioRDR5) from eBioscience (Thermo Fisher, Germany). Anti-Ki-67-Dylight350 (Clone: 1297A) from Novus (USA). Anti-CD45-APC-Cy7 (Clone: 2D1); anti-CCR7-Pacific Blue (Clone: G043H7); anti-CD45RA-BV510 (Clone: HI100); anti-CD45RO-PerCy-Cy5.5 (Clone: UCHL1); anti-CD62L-BV650 (Clone: DREG-56); anti-CD103-PE (Clone: Ber-ACT8); and anti-CD154-PE/Dazzle (Clone: 24–31) from Biolegend (USA). The following antibodies were used for multiparametric flow cytometry for analysis of resident immune and structural cells: anti-CD64-PerCp-eFluor710 (Clone: 10.1); anti-CD11b-APC-Cy7 (Clone: ICRF44); anti-HLA-DR-FITC (Clone: LN3); anti-EpCAM (CD326)-Alexafluor 594 (Clone:9C4); anti-CD14-PE-Cy7 (Clone:61D3); anti-CD163-PE (Clone: GHI/61); anti-CD49f-PE (Clone: GoH3); anti-E-Cadherin-PerCP-eFluor 710 (Clone: DECMA-1); and anti-CD16-FITC (Clone: eBioCB16) from eBioscience (Thermo Fisher, Germany). Anti-CD45-Pacific Blue (Clone: HI30); anti-CD15-Alexa Fluor 700 (Clone: WCD3) from Biolegend (USA). Anti-PanCytokerain-APC (Clone: C-11) and anti-Ki-67-Dylight350 (Clone: 1297A) from Novus Biologicals (USA). The Foxp3/transcription factor staining buffer set (Thermo Fisher, Germany) and the cytofix/cytoperm fixation/permeablization kit (BD, Germany) were used according to the manufacturer’s protocol to stain for intracellular molecules (intranuclear and cytoplasmic molecules, respectively). Analyses were performed on FACSymphony A3 Cell Analyzer with FACSDiva software version 8.0.1 (BD, Germany). Data were analyzed with FlowJo 10.7.1 (Treestar, USA). For analysis of cell populations, initial gating on the forward scatter (FSC) vs. side scatter (SSC) plot was completed (as shown in Sup. Figs. 1 and 2) to remove dead cells and cell debris from analysis.
Expression Profiling of Inflammatory Markers
To assess the expression of inflammation-related genes, we employed the TaqMan-based human inflammation open array panel (Cat # 4475389, Thermo Fischer Scientific, USA). This panel contained 607 targets, 586 inflammatory genes, and 21 endogenous controls. For this, total RNA from tissues from various ex vivo cultures of HS skins was isolated using TRIzol reagent (Cat#15596018, Ambion). A total of 2 µg of RNA was reverse transcribed into cDNA using SuperScript® VILO™ cDNA Synthesis Kit (Cat #11754250, Life Technologies, USA). Following the reverse transcription reaction, the mixture was denatured by incubating it at 85 °C. Pre-amplification of cDNA using custom TaqMan preamp primer pools (Cat#4441856, Thermo Fischer Scientific, USA) was performed in 25 µl total volume containing 12.5 µl of TaqMan PreAmp master mixture (Cat#4391128), 2.5-µl PreAmp primer pools, 2.5-µl reverse transcription product, and the remaining volume was adjusted by nuclease-free water. Pre-amplification thermal cycling conditions include incubation of products at 95 °C for 10 min followed by 12 × cycles at 95 °C for 15 s and 4 min at 60 °C. The pre-amplification products obtained after the cycling were incubated for 10 min at 99.9 °C. A 20-fold diluted pre-amplification product was used in the final amplification reaction as per the manufacturer’s instructions. OpenArray chip containing the pre-coated primers for 607 targets was read on a 12-K Flex RT-PCR machine (Thermo Fisher Scientific, Life Technologies Corporation, Grand Island, New York). Data analysis was performed through the online available ExpressionSuite v 1.3 software (Thermo Fisher Scientific, Life Technologies Corporation, Grand Island, NY) using global gene normalization method. Bioinformatics analysis was carried out using ingenuity pathway analysis (IPA, Qiagen) application.
Chemokine Profiling
To identify the drug response on HS skin, multiplex cytokine/chemokine analysis was performed using the conditioned medium procured from various ex vivo cultures. Cytokine/chemokine/growth factor 45-Plex Human ProcartaPlex™ Panel 1 (Cat# EPX450-12171–901, Thermo Fisher, USA) was used. Briefly, the conditioned medium was centrifuged at 10,000 rpm for 10 min and stored at −80 °C until assayed. A total of forty-five target proteins containing (i) Th1/Th2 markers: GM-CSF, IFN gamma, IL-1 beta, IL-2, IL-4, IL-5, IL-6, IL-8, IL-12p70, IL-13, IL-18, TNF alpha, and LIF; (ii) Th9/Th17/Th22/Treg markers: IL-9, IL-10, IL-17A (CTLA-8), IL-21, IL-22, IL-23, and IL-27; (iii) Inflammatory cytokines: IFNα, IL-1α, IL-1RA, IL-7, IL-15, IL-31, and TNFβ; (iv) chemokines: eotaxin (CCL11), GROα (CXCL1), IP-10 (CXCL10), MCP-1 (CCL2), MIP-1α (CCL3), MIP-1β (CCL4), RANTES (CCL5), SDF-1 alpha; and (v) growth factors: BDNF, EGF, FGF-2, HGF, NGF beta, PDGF-BB, PlGF-1, SCF, VEGF-A, and VEGF-D were assessed. 50 µl of condition medium was used for multiplexing on the Luminex 200 instrument (Luminex Corporation, USA) as described earlier [18].
Statistical Analysis
Statistical analysis was performed using GraphPad Prism software (La Jolla, CA, USA). Data are presented as mean ± standard error of the mean unless indicated otherwise in the figure legend. A p-value less than 0.05 was considered statistically significant. A two-tailed unpaired Student’s t-test was used to evaluate statistical differences between two groups. One-way ANOVA with Tukey’s multiple comparison testing was utilized to evaluate statistical difference for data with more than two groups.
RESULTS
Tissue Integrity
Skin tissue specimen were collected from healthy individuals (without evidence of any skin disease) and patients with HS. Three culture platforms were established and tested for ex vivo maintenance of tissues. These culture platforms include air–liquid interface culture (A-L), liquid submersion culture (L-S), and perfusion bioreactor culture (Bio), which we have previously utilized for human lung tissue (Fig. 1A–C) [19]. Histologic analysis of tissues demonstrates that all three platforms are effective for culturing these skin samples, with maintenance of tissue integrity for at least 3 days (Fig. 2A, left). When the culture period was further extended to 14 days, the healthy skin tissues showed some signs of loss of tissue architecture, whereas HS skin specimen maintained most of the tissue structure and tissue integrity when H&E stained cross-sections were analyzed (Fig. 2A, right).

Ex Vivo Culture Platforms for healthy and HS skin. Schematics of three ex vivo culture platforms utilized to culture human skin (top) with images of each platform (bottom). A Air–liquid interface (A-L) culture utilized 0.4-µm transwell filters in which an extracellular matrix (ECM) scaffold stabilized tissue specimen for static culture. Media was then added to the top and bottom chambers but did not fully cover the tissue so that the apical surface had exposure to air. B Liquid submersion culture (L-S) was set up in a similar method to A-L, with tissues stabilized within ECM on transwell filters; however, media was added to cover the entire tissue. C For perfusion bioreactor culture (Bio), a portion of tissue was placed into the central chamber of the polydimethylsiloxane (PDMS) bioreactor which contained an ECM scaffold for stabilization. Through-channels were generated in the tissue/ECM volume using five 400-µm Teflon-coated stainless steel wires during ECM polymerization for continuous tissue perfusion. Wires were removed following ECM polymerization, and the bioreactor was then connected to a perfusion system, in which a serum-free defined tissue culture media was perfused from a media reservoir through the tissue volume and collected in a collection reservoir.

Tissue histology to demonstrate architecture and viability following ex vivo culture. A Histologic architecture of ex vivo cultured tissue is similar to that observed in starting tissue for both healthy and HS skin. B, C When early apoptosis B and late apoptosis/necrosis C were evaluated via flow cytometry, HS skin was found to have a higher proportion of early apoptotic cells when compared to healthy skin at day 3 with L-S and Bio culture. The proportion of late apoptotic/necrotic cells did not differ between healthy and HS skin. D, E Following 14 days of culture, early apoptosis D and late apoptosis/necrosis E were increased in healthy skin. n = 3–14. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.005, ****p ≤ 0.0001.
Apoptosis Analysis
Flow cytometry data did not show significant differences in early apoptosis among the three culture platforms up to day 3. However, the percentage of early apoptotic cells was higher in HS skin culture (L-S: 32.8% ± 4.26%, Bio: 26.4% ± 2.14%) relative to healthy skin (L-S: 12.2% ± 2.9%, Bio: 9.47% ± 2.1%; p = 0.019 L-S culture, p = 0.017 Bio culture) (Fig. 2B). No significant differences were observed in the percentage of late apoptotic/necrotic cells among the three platforms of healthy skin cultures, but late apoptotic/necrotic cells were significantly higher in Bio culture (3.83% ± 0.69%) of HS skin relative to A-L culture (2.29% ± 0.4%; p = 0.026) at 3 days (Fig. 2C). Interestingly, at day 14, a moderate increase in apoptotic cells was observed in the healthy skin cultures relative to HS skin cultures. When healthy and HS skin cultures were compared, a significant increase in apoptotic cells was observed in A-L culture (36.37% ± 9.27%) and Bio culture (40.8% ± 15.7%) from healthy skin when compared to A-L culture (8.9% ± 2.26%) and Bio culture (12.18% ± 3.2%) of HS skin (A-L culture: p = 0.0074, Bio culture: p = 0.0069) (Fig. 2D). Similarly, an increase in the late apoptotic population stained with Annexin V+ 7-amino-actinomycin D+ (7-AAD) was observed at day 14 relative to day 3 of healthy skin cultures. L-S culture of healthy skin contained the highest proportion of late apoptotic cells (14.5% ± 1.83%) when compared to A-L culture (7.17% ± 2.38%; p = 0.0001) and Bio culture (4.68% ± 0.15%; p < 0.0001). Additionally, when healthy and HS skin culture were compared, a significant increase in the late apoptotic population was observed in healthy skin samples when using all culture platforms (Fig. 2E).
Characterization of Various Cell Populations
T cell populations were evaluated following 3 days of culture. Although the CD4+ population was very high in HS skin (11.27 ± 3.86) relative to healthy skin (2.95 ± 0.168; p = 0.037) at day 0, no significant difference was observed in the proportion of CD8+ T cells between healthy and HS skin (Fig. 3A and B). Upon further analysis, Bio culture was shown to best represent the CD4+ T cell populations observed in the starting tissue of healthy skin, while a significant decrease in this population was found with A-L (2.92 ± 1.08; p = 0.011) and L-S (2.65 ± 0.93; p = 0.011) culture at day 3 relative to HS skin at day 0 (11.27 ± 3.86). Interestingly, CD4+ tissue-resident memory (TRM) cells, regulatory T cells (Treg), and central memory T cells were expanded in all culture platforms in HS specimen when compared to their levels in the starting tissue (Fig. 3C), whereas TRM, effector, and central memory T cells were decreased in healthy skin. When CD8+ T cell populations were compared in healthy skin, the A-L and L-S culture platforms were found to best maintain similar proportions to those seen in the starting tissue. However, Treg increased and TRM and central memory T cells decreased when using all culture platforms (Fig. 3D). In HS skin, CD8+ T cell populations remained similar to the starting tissue across culture platforms; however, slight increases in Treg and central memory T cells, as well as slight decreases in effector memory cells, were seen with culture. When activated T cell populations were evaluated, a significant increase in activated CD4+ T cells (A-L culture: 16.83 ± 4.37, p = 0.0001; L-S culture: 12.03 ± 2.07, p = 0.035; Bio culture: 12.07 ± 3.19, p = 0.034) was observed following culture at day 3 relative to healthy skin tissues at day 0 (2.94 ± 0.62) (Fig. 3E). Similarly, activated CD8+ T cells were elevated with culture, but the number did not reach significance in either tissue type or across culture platforms (Fig. 3F). Proliferating CD4+ T cells were increased with Bio culture in HS tissue, but no other changes were observed and no changes in proliferating CD8+ T cells were found (Fig. 3G and H).

Cellular heterogeneity is maintained with ex vivo culture. A, B CD4+ T cells are increased in HS skin when compared to healthy skin at day 0, but loss of this population is seen with culture (A), whereas CD8+ T cells are maintained (B). C, D Subpopulations of CD4+ (C) and CD8+ (D) T cells vary somewhat with culture platform. E, F Activated CD4+ T cells increased with culture in healthy skin (E), but the changes observed did not reach statistical significance when activated CD8+ T cells were examined (F). G, H Proliferating CD4+ T cells (G) were increased with Bio culture in HS skin, whereas no changes were observed in CD8+ T cells (H). I The proportion of keratinocytes was similar between healthy and HS skin. J Langerhans cells showed no significant change between healthy and HS skin or with culture. K-L The proportion of macrophages (K), M2-like macrophages (L), neutrophils (M), and natural killer (NK) cells (N) were small in both healthy and HS skin. n = 3–12. *p ≤ 0.05, **p ≤ 0.01.
When the proportions of keratinocytes and proliferating keratinocytes were evaluated, a significant increase in keratinocytes was found in A-L culture (11.74 ± 2.17) relative to the starting tissue (4.6 ± 0.48) in HS skin (p = 0.035); however, no other significant changes were observed between tissue type or across culture platforms (Fig. 3I and Sup. Fig. 3A). Similarly, no changes were observed in the proportions of Langerhans cells (Fig. 3J and Sup. Fig. 3B). Although the proportion of macrophages and proliferating macrophages were small within both healthy and HS skin specimen, these cell populations were found to increase with Bio culture in healthy skin (Fig. 3K and Sup. Fig. 3C). No significant changes were observed in M2 macrophages and proliferating M2 macrophages (Fig. 3L and Sup. Fig. 3D). Similarly, the proportions of neutrophils (Fig. 3M) and natural killer (NK) cells (Fig. 3N) were found to be small within both healthy and HS skin. While no significant differences were observed in the NK cell population, a significant decrease in neutrophils was observed with all culture platforms in healthy skin tissue when compared to the starting tissue (Fig. 3M).
Status of Inflammatory Markers Genes in Ex Vivo HS Models
Next, we carried out mRNA expression profiling of 586 inflammatory genes using the TaqMan-based human inflammation open array panel to assess the dynamic regulation of the expression of inflammatory genes in these culture models. Data collected from inflammatory arrays demonstrate that HS skin tissue maintain the dynamics of the inflammatory signatures in ex vivo skin culture platforms (Fig. 4A), although these changes were not identical across the three culture platforms. The expression of many inflammatory signatures was progressively elevated at day 14, as shown by the heat map (Fig. 4A). Pathway analysis using Ingenuity Pathway Analysis (IPA) showed that the osteoarthritis pathway, IL-6 signaling, pathways associated with the role of IL-17A in psoriasis, acute phase response signaling, STAT3, NFκB, and TLR signaling remained elevated in all ex vivo culture platforms up to day 14 (Fig. 4B). On the other hand, downregulated gene signatures affected pathways such as LXR/RXR activation, PPARα/RXRα activation, antioxidant action of vitamin C, CREB signaling in neurons, PI3K/AKT signaling in the pathogenesis of influenza, PTEN signaling, phagosome formation, eicosanoids signaling, and crosstalk between dendritic and NK cells (Fig. 4C). Many pathways were unchanged in culture at both time-points, including the G1/S checkpoint regulation pathway, Gαi signaling, apoptosis signaling, coagulation system, endocannabinoid cancer inhibition pathway, and the ferroptosis-signaling pathway (Fig. 4D).

Comparison of the inflammatory gene profile of HS skin before and after ex vivo culture. A (i) Heatmap showing the change in expression of various inflammatory genes in three different ex vivo culture platforms at day 3 and day 14 of HS skin culture relative to inflammatory signature at day 0 of HS skin excision. (ii) Heat Map of HS skin at day 0. (iii) Stitched heatmap derived from the most resemble segment of heatmaps of various ex vivo cultures either at day 3 or day 14 after dividing all the heat maps into 8 segments. Based on gene expression profile, we divided the heatmap into eight gene clusters. This demonstrates that any of the three different culture methods alone was not sufficient to represent the initial HS inflammatory gene signature, but all three together provide great value as a tool to understand molecular pathogenesis of HS and its treatment; we stitched clusters that match with HS skin at day 0. As can be seen that four of the day 3 clusters and four of the day 14 clusters from three different culture method match the gene expression pattern in HS at day 0. Thus, a 14-day drug treatment to demonstrate its efficacy is possible under these conditions. B Heatmap showing upregulated pathways at all time-points of ex vivo culture of HS skin, relative to HS skin at day 0. C Heatmap showing downregulated pathways at all-time points of ex vivo culture of HS skin, relative to HS skin at day 0. D Heatmap showing pathways remained unchanged during ex vivo culture of HS skin, relative to HS skin at day 0.
Inflammatory pathways, including hepatic fibrosis signaling, cardiac hypertrophy signaling, production of NO and ROS in macrophages, dendritic cell maturation, TEC kinase signaling, HER-2 signaling in breast cancer, ILK signaling, type I diabetes mellitus signaling, iNOS signaling, HMGB1 signaling, and IL9 signaling (Fig. 5A), were upregulated at day 3 in various cultures, but downregulated and/or returned to basal levels by day 14 of culture. The signaling pathways, which remained highly activated at day 14 of various ex vivo HS skin cultures were the osteoarthritis pathway, the pathway associated with the role of IL-17A in psoriasis, and LPS/IL-1-mediated inhibition of RXR function. Interestingly, Fcγ receptor-mediated phagocytosis in macrophage and monocytes was only upregulated in various ex vivo cultures at day 14 (Fig. 5B). Overall, the data from the inflammatory array demonstrate that these ex vivo HS skin cultures are suitable to defining the molecular pathogenesis of inflammation in HS skin.

Most inflammatory pathways were upregulated at day 3 of ex vivo culture, but few could be sustained at day 14. A Heatmap showing the pathways significantly upregulated or downregulated at day 3 of ex vivo culture. B Heatmap showing the pathways significantly upregulated or downregulated at day 14 of ex vivo culture.
Verification of Suitability of Ex Vivo HS Skin Culture Models for Drug Screening
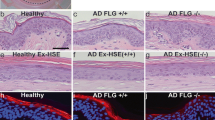
To verify the suitability of these ex vivo skin cultures to predict treatment efficacy, we employed an immunomodulatory drug, lenalidomide, and a bromodomain and extra-terminal (BET) inhibitor, CPI-0610, in all three ex vivo culture platforms for 3 days of culture. Both of these drugs are expected to alter the inflammatory profile by different mechanisms [20, 21]. Histopathologic evaluation using H&E staining did not reflect major differences in tissue architecture due to drug treatment or associated toxicity in healthy skin when using any of the three culture methods (Fig. 6A and Sup. Fig. 4). Moreover, no significant change in early apoptotic (Annexin V+ 7AAD−) or late apoptotic/necrotic (Annexin V+ 7AAD+) cell populations was observed in comparisons between drug-treated- and vehicle-treated groups using any of all these culture platforms at day-3 (Fig. 6B). However, an overall increase in late apoptosis/necrosis in HS tissues was observed. Nonetheless, we did not establish a dose–response relationship for any of these drugs at this stage (Fig. 6C).

Effects of drug treatment on tissue histologic architecture and viability following ex vivo culture. A Histologic architecture of ex vivo cultured tissues treated with lenalidomide or CPI-0610 is similar to that observed in vehicle control-treated tissue for both healthy and HS skin when cultured using the Bio platform. B When early apoptosis was evaluated via flow cytometry, no significant difference was noted with treatment or culture platform in healthy or HS skin. C Late apoptosis/necrosis was increased in HS skin when compared to healthy skin; however, no changes in response to treatment or culture platform were observed. n = 4 per group.
Employing Luminex analysis to evaluate cytokines/chemokines and growth factors in the supernatants procured from drug- and vehicle-treated cultures, we observed that both drugs effectively blocked the secretion of various cytokines/chemokines and growth factors in the supernatants (Fig. 7). Both drugs were able to reduce the levels of Th1/Th2/Th9/Treg associated cytokines such as IL2, IL4, IL5, IL9, and IL21, while inflammatory cytokines/chemokines such as IL-8, IFN-α, MIP-1α, SDF-1α, MCP-1, GROα/KC, etc., and various growth factors, namely, PDGF-1, VEGFd, and SCF, were also diminished in the supernatant procured from A-L ex vivo HS skin cultures at day 3 relative to vehicle-treated controls (Fig. 7A). Secretion of some inflammatory cytokines/chemokines such as MIP-1α, IL-1RA, IL-6, and IFN-α were also blocked in the supernatant of ex vivo HS skin from Bio culture at day 3 of culture (Fig. 7B). However, only CPI-0610 was effective in blocking many cytokines/chemokines and growth factors, as assessed in the supernatant of ex vivo HS skin tissue of L-L culture at day 3 with exception to IL-2, IL-4, MCP-1, IL-9, and VEGFd (Fig. 7C). Interestingly, these were not inhibited in the supernatant of A-L ex vivo HS skin cultures at day 3. Multiple studies have also evaluated cytokines and other inflammatory proteins in lesional, peri-lesional skin wound exudate, and serum of HS patients to find out the importance of these proteins in pathogenesis and treatment [22,23,24,25].

Effect of drug treatment on anti-inflammatory cytokine secretion in culture supernatant of ex vivo models of HS. Changes in the levels of various cytokine/chemokines and growth factors in the supernatant procured from either Vehicle (HS), CPI-0610 (CPI), or lenalidomide treated ex vivo HS skin models from A-L (A), Bio (B), L-S (C) platforms.
Reconstructing a heat map from these model cultures was achieved by dividing the heatmap of HS day 0 skin detailing the expression profile of inflammatory genes in eight clusters, as shown in (Fig. 4A (iii)). Thereafter, we searched the expression profiles of all three cultures at day 3 and day 14 and identified the segments where we did not find any alteration in the expression of the clusters compared to day 0 profile. These unaltered segments were sutured to make an artificial heatmap, as shown in Fig. 4A (iii). This heatmap could be utilized to predict the impact of drug treatment on the profile of inflammatory genes in HS skin (day 0). In sum, this reconstruction of the heatmap was necessary for drug evaluation, as our culture methods provide a dynamic environment for the gene expression and many of these genes are continuously altered.
Overall, various ex vivo cultures together provide a powerful system to assess the drug effects as exemplified by demonstrating the effect of two distinct classes of mechanistically anti-inflammatory drugs.
DISCUSSION
In this study, we established three different ex vivo culture model systems for healthy and HS skin and assessed suitability of these ex vivo models to screen drugs which can affect inflammatory signaling and progression of inflammation in HS skin. Previously, Vossen and colleagues used Transwell ex vivo skin culture from healthy and HS skin [26]. Using these cultures authors showed that this methodology could be used for the ex vivo evaluation of drugs against HS. In this Transwell culture, HS skin tissue was exposed to various anti-inflammatory drugs, including prednisolone or biologics targeting TNF-α (adalimumab and infliximab), interleukin (IL)-17A (secukinumab), IL-12/23p40 (ustekinumab), or CD20 (rituximab). Of these, TNF-α inhibitors and prednisolone were found to be the most powerful inhibitors of proinflammatory cytokines. In comparison to the previous efforts to establish skin tissue cultures, our three different types of culture platforms, namely, air–liquid interface, liquid submersion, and a more controlled perfusion bioreactor, together provide a more powerful and rigorous tool to establish ex vivo cultures of healthy and HS skin. In an attempt to demonstrate the suitability of these cultures to aid in understanding the molecular pathogenesis of disease progression, particularly with respect to the dynamic regulation of inflammatory cascade, we characterized various cell populations including keratinocytes and immune cells in these cultures. Our initial findings demonstrated that these ex vivo cultures were able to maintain a significant level of the tissue architecture and integrity at least up to day 3 of culture. Furthermore, when cultures were extended to day 14, the tissue architecture and integrity were maintained in HS skin culture, but not in healthy skin culture. To avoid any complexity in group comparisons, the longer duration culture conditions were not modified for healthy skin.
These results are primarily based on H&E staining of the cultured skin. However, a further detailed analysis is needed to assess each cell type and their ability to maintain the molecular signatures, for example, using single-cell analyses. The observed differences in healthy and HS skin cells may be due to multiple factors, particularly due to differences in metabolic signatures. In this regard, vitamin D metabolism, steroid metabolism, biological processes associated with collagen and extracellular matrix remodeling, growth and tissue factors such as tissue factor 3 (T3), growth and differentiation factor 2 (GDF2), midkine, and clusterin which are known to play important roles in vital cellular and physiological processes and are dysregulated in HS skin [27,28,29,30]. The compromised metabolic environment of HS skin is considered to be associated with HS disease progression [4, 31].
Next, evaluation of various cell populations in these ex vivo cultures showed that Bio culture at day 3 was able to best represent the CD4+ T cell populations seen in the starting HS tissue (day 0). However, with further evaluation of CD4+ T cells, TRM, Treg, and central memory T cells were found to expand in all culture platforms in HS specimen when compared to the starting (day 0) tissue. CD8+ T cell populations remained unaltered when compared to the starting tissue across all culture platforms; however, small increases in Treg and central memory T cells, as well as decreases in effector memory cells were seen. When activated T cell populations were evaluated, an increase in activated CD4+ T and CD8+ T cells was observed in all culture platforms, but statistical significance could only be achieved for CD4+ T cells in ex vivo cultures of healthy skin. Moreover, proliferating CD4+ T cells were increased with all the platforms, but significance could only be achieved with Bio culture in HS tissue. The ratio of proliferating CD8 + T cells was higher in Bio culture of HS skin (Fig. 3). Evaluation of keratinocytes and proliferating keratinocytes showed that these cultures only support the healthy growth of epidermal cells up to day 3 of evaluation but hyper-proliferation of keratinocytes was maintained in these ex vivo cultures of HS skin out to day 14. The proportions of macrophages and proliferating macrophages were small within both healthy and HS skin specimen, and these cell populations were found to increase with bio culture in healthy skin. However, the proliferating M2 population was found to be decreased in A-L and L-S culture platforms of HS skin. Overall, these data demonstrate that these ex vivo models were not only able to maintain various cells populations but were also able to preserve the disease tissue system physiology at least partially. Further details and mechanistic aspects remain to be investigated and are beyond the scope of this study.
Molecular profiling of inflammatory genes in these cultures showed that there are dynamic expression profiles and that a single culture platform cannot precisely represent the inflammatory status of HS skin at day 0. However, using all three ex vivo culture platforms at day 3 and day 14 could recapitulate most of the inflammatory pattern seen in HS skin. Molecular expression data from these cultures also indicate that these cultures could serve as useful models to screen anti-inflammatory drugs. In this regard, our data employing the immunomodulatory drug, lenalidomide, and the anti-inflammatory/epigenetic modulatory drug, CPI-0610 (a BET inhibitor) in these ex vivo HS models showed that many inflammatory cytokines/chemokines were downregulated by these treatments. Although, as described earlier, a single culture method was not as powerful as the results obtained from the three culture methods together.
Thus, our combined ex vivo culture models provide a unique opportunity to employ these systems to understand the pathobiology of HS as well as screening and development of various therapeutic agents, particularly those which are already FDA approved for other inflammatory skin conditions and could be repurposed for HS treatment.
CONCLUSION
We believe that these three ex vivo culture systems collectively provide a powerful tool for defining the pathobiology of HS and assessing drugs for suppressing inflammatory responses.
Availability of Data and Materials
Data generated/analyzed during this study are included in this published article (and its supplementary information files).
References
Garg, A., J.S. Kirby, J. Lavian, G. Lin, and A. Strunk. 2017. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA dermatology 153: 760–764.
Phan, K., O. Charlton, and S.D. Smith. 2020. Global prevalence of hidradenitis suppurativa and geographical variation—Systematic review and meta-analysis. Biomedical Dermatology 4: 2.
Frew, J.W., K. Navrazhina, M. Marohn, P.C. Lu, and J.G. Krueger. 2019. Contribution of fibroblasts to tunnel formation and inflammation in hidradenitis suppurativa/acne inversa. Experimental dermatology 28: 886–891.
Guenin-Mace, L., J.D. Morel, J.M. Doisne, A. Schiavo, L. Boulet, V. Mayau, P. Goncalves, S. Duchatelet, A. Hovnanian, V. Bondet, et al. 2020. Dysregulation of tryptophan catabolism at the host-skin microbiota interface in hidradenitis suppurativa. JCI Insight 5: e140598.
Byrd, A.S., C. Carmona-Rivera, L.J. O’Neil, P.M. Carlucci, C. Cisar, A.Z. Rosenberg, M.L. Kerns, J.A. Caffrey, S.M. Milner, J.M. Sacks, et al. 2019. Neutrophil extracellular traps B cells, and type I interferons contribute to immune dysregulation in hidradenitis suppurativa. Science translational medicine 11: 508.
van der Zee, H.H., L. de Ruiter, J. Boer, D.G. van den Broecke, J.C. den Hollander, J.D. Laman, and E.P. Prens. 2012. Alterations in leucocyte subsets and histomorphology in normal-appearing perilesional skin and early and chronic hidradenitis suppurativa lesions. British Journal of Dermatology 166: 98–106.
Shah, N. 2005. Hidradenitis suppurativa: A treatment challenge. American family physician 72: 1547–1552.
Hotz, C., M. Boniotto, A. Guguin, M. Surenaud, F. Jean-Louis, P. Tisserand, N. Ortonne, B. Hersant, R. Bosc, F. Poli, et al. 2016. Intrinsic defect in keratinocyte function leads to inflammation in hidradenitis suppurativa. Journal of Investigative Dermatology 136: 1768–1780.
Navrazhina, K., J.W. Frew, P. Gilleaudeau, M. Sullivan-Whalen, S. Garcet, and J.G. Krueger. 2021. Epithelialized tunnels are a source of inflammation in hidradenitis suppurativa. Journal of Allergy and Clinical Immunology 147: 2213–2224.
Sbidian, E., C. Hotz, J. Seneschal, A. Maruani, F. Amelot, F. Aubin, C. Paul, M. Beylot-Barry, P. Humbert, A. Dupuy, et al. 2016. Antitumour necrosis factor-alpha therapy for hidradenitis suppurativa: Results from a national cohort study between 2000 and 2013. British Journal of Dermatology 174: 667–670.
Theut-Riis, P., L.R. Thorlacius, and G.B. Jemec. 2018. Investigational drugs in clinical trials for hidradenitis suppurativa. Expert opinion on investigational drugs 27: 43–53.
van der Zee, H.H., L. de Ruiter, D.G. van den Broecke, W.A. Dik, J.D. Laman, and E.P. Prens. 2011. Elevated levels of tumour necrosis factor (TNF)-alpha, interleukin (IL)-1beta and IL-10 in hidradenitis suppurativa skin: A rationale for targeting TNF-alpha and IL-1beta. British Journal of Dermatology 164: 1292–1298.
Lowe, M.M., H.B. Naik, S. Clancy, M. Pauli, K.M. Smith, Y. Bi, R., Dunstan, J.E. Gudjonsson, M. Paul, H. Harris, et al. 2020. Immunopathogenesis of hidradenitis suppurativa and response to anti-TNF-alpha therapy. JCI Insight 5: e139932.
Folkes, A.S., F.Z. Hawatmeh, A. Wong, and F.A. Kerdel. 2020. Emerging drugs for the treatment of hidradenitis suppurativa. Expert Opinion on Emerging Drugs 25: 201–211.
Marzano, A.V., G. Genovese, G. Casazza, C. Moltrasio, P. Dapavo, G. Micali, R. Sirna, P. Gisondi, A. Patrizi, V. Dini, et al. 2021. Evidence for a “window of opportunity” in hidradenitis suppurativa treated with adalimumab: A retrospective real-life multicentre cohort study. British Journal of Dermatology 184: 133–140.
Frew, J.W., J.E. Hawkes, and J.G. Krueger. 2018. A systematic review and critical evaluation of inflammatory cytokine associations in hidradenitis suppurativa. F1000Research 7: 1930.
Goliwas, K.F., J.R. Richter, H.C. Pruitt, L.M. Araysi, N.R. Anderson, R.S. Samant, S.M. Lobo-Ruppert, J.L. Berry, and A.R. Frost. 2017. Methods to evaluate cell growth, viability, and response to treatment in a tissue engineered breast cancer model. Scientific reports 7: 14167.
Kashyap, M., N. Kawamorita, V. Tyagi, Y. Sugino, M. Chancellor, N. Yoshimura, and P. Tyagi. 2013. Down-regulation of nerve growth factor expression in the bladder by antisense oligonucleotides as new treatment for overactive bladder. The Journal of urology 190: 757–764.
Goliwas, K.F., C.S. Simmons, S.A. Khan, A.M. Wood, Y. Wang, J.L. Berry, M. Athar, J.A. Mobley, Y.I. Kim, V.J. Thannickal, et al. 2021. Local SARS-CoV-2 peptide-specific immune responses in convalescent and uninfected human lung tissue models. medRxiv.
Bao, Y., X. Wu, J. Chen, X. Hu, F. Zeng, J. Cheng, H. Jin, and X., Lin, and L.F. Chen. 2017. Brd4 modulates the innate immune response through Mnk2-eIF4E pathway-dependent translational control of IkappaBalpha. Proceedings of the National Academy of Sciences 114: E3993–E4001.
Fink, E.C., and B.L. Ebert. 2015. The novel mechanism of lenalidomide activity. Blood 126: 2366–2369.
Banerjee, A., S. McNish, and V.K. Shanmugam. 2017. Interferon-gamma (IFN-gamma) is elevated in wound exudate from hidradenitis suppurativa. Immunological investigations 46: 149–158.
Hessam, S., M. Sand, T. Gambichler, M. Skrygan, I. Ruddel, and F.G. Bechara. 2018. Interleukin-36 in hidradenitis suppurativa: Evidence for a distinctive proinflammatory role and a key factor in the development of an inflammatory loop. British Journal of Dermatology 178: 761–767.
Jimenez-Gallo, D., R. de la Varga-Martinez, L. Ossorio-Garcia, C. Albarran-Planelles, C. Rodriguez, and M. Linares-Barrios. 2017. The clinical significance of increased serum proinflammatory cytokines C-reactive protein and erythrocyte sedimentation rate in patients with hidradenitis suppurativa. Mediators of inflammation 2017: 2450401.
Kanni, T., V. Tzanetakou, A. Savva, B. Kersten, A. Pistiki, F.L. van de Veerdonk, M.G. Netea, J.W. van der Meer, and E.J. Giamarellos-Bourboulis. 2015. Compartmentalized cytokine responses in hidradenitis suppurativa. PLoS One 10: e0130522.
Vossen, A., C.B. Ardon, H.H. van der Zee, E. Lubberts, and E.P. Prens. 2019. The anti-inflammatory potency of biologics targeting tumour necrosis factor-alpha, interleukin (IL)-17A, IL-12/23 and CD20 in hidradenitis suppurativa: An ex vivo study. British Journal of Dermatology 181: 314–323.
Brandao, L., R. Moura, P.M. Tricarico, R. Gratton, G. Genovese, C. Moltrasio, S. Garcovich, M. Boniotto, S. Crovella, and A.V. Marzano. 2020. Altered keratinization and vitamin D metabolism may be key pathogenetic pathways in syndromic hidradenitis suppurativa: A novel whole exome sequencing approach. Journal of Dermatological Science 99: 17–22.
Filippou, P.S., G.S. Karagiannis, and A. Constantinidou. 2020. Midkine (MDK) growth factor: A key player in cancer progression and a promising therapeutic target. Oncogene 39: 2040–2054.
Hoffman, L.K., L.E. Tomalin, G. Schultz, M.D. Howell, N. Anandasabapathy, A. Alavi, M. Suarez-Farinas, and M.A. Lowes. 2018. Integrating the skin and blood transcriptomes and serum proteome in hidradenitis suppurativa reveals complement dysregulation and a plasma cell signature. PLoS One 13: e0203672.
Jones, S.E., and C. Jomary. 2002. Clusterin. The international journal of biochemistry & cell biology 34: 427–431.
Sabat, R., A. Chanwangpong, S. Schneider-Burrus, D. Metternich, G. Kokolakis, A. Kurek, S. Philipp, D. Uribe, K. Wolk, and W. Sterry. 2012. Increased prevalence of metabolic syndrome in patients with acne inversa. PLoS One 7: e31810.
Funding
NIH grant RO1 ES026219 and NCI grant 5P01CA210946 and by intramural UAB funds to M.A.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: J.S.D., C.R., M.S.M., M.A. Performed the experiments: K.F.G., M.P.K., J.K., R.S., Z.W., A.O., L.J., V.A., M.B.L. Analyzed the data: K.F.G., M.P.K., J.K., R.S., Z.W., A.O., L.J., V.A., M.B.L., C.A.E., M.S.M., C.R., J.S.D., M.A. Contributed reagents/materials/analysis tools: C.A.E., M.S.M., C.R., J.S.D., M.A. Wrote the paper: K.F.G., M.P.K., J.S.D., and M.A.
Corresponding authors
Ethics declarations
Ethical Approval and Consent to Participate
The institutional Review Board of the University of Alabama at Birmingham approved the protocol (IRB-300005214) for obtaining surgically removed discarded skin tissues from breast/tummy reduction from otherwise healthy individuals and HS patients.
Consent of Publication
All authors agree to submit this manuscript.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Goliwas, K., Kashyap, M.P., Khan, J. et al. Ex Vivo Culture Models of Hidradenitis Suppurativa for Defining Molecular Pathogenesis and Treatment Efficacy of Novel Drugs. Inflammation 45, 1388–1401 (2022). https://doi.org/10.1007/s10753-022-01629-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-022-01629-w