
Abstract
Asthma is a chronic disease closely related to airway inflammation. It has been proven that type 2 innate lymphoid cells (ILC2s) play an essential role in airway inflammation in asthma. Furthermore, there is growing evidence that Follistatin-like 1 (FSTL1) can participate in various inflammatory reactions mediated by the JAK/STAT signaling pathway, among others. Therefore, we put forward a new hypothesis: FSTL1 promotes asthmatic airway inflammation by activating ILC2. This study generated an ovalbumin-sensitized asthma model in C57BL/6 and Fstl1+/− mice. The results showed that the absolute number and the proportion of ILC2 in the ovalbumin-challenged Fstl1+/− group were lower than in the ovalbumin-challenged wild-type group. We also measured the levels of Th2-type cytokines in the serum and bronchoalveolar lavage fluid (BALF) of mice and found that the corresponding cytokines in the Fstl1+/− were lower than in the wild-type groups. Finally, we tested whether MEK-JAK-STAT-GATA3 is the specific pathway for FSTL1 to activate ILC2, and further tested our working hypothesis by adding various inhibitors of proteins from this pathway. Overall, these findings reveal that FSTL1 can activate ILC2 through MEK-JAK-STAT-GATA3 to promote airway inflammation and participate in the pathogenesis of asthma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Asthma is a heterogeneous disease usually characterized by chronic airway inflammation and affects 1–22% of the population in different countries [1]. Allergic asthma is a significant phenotype of asthma, and Th2-type cytokines are essential for driving the pathology of allergic diseases, such as allergic asthma and allergic rhinitis [2]. When environmental allergens are encountered by dendritic cells (DCs), they are uptaken, processed into peptides, and presented to naive CD4+ T-cells, inducing the production of Th2-type T-cells and large numbers of Th2-type cytokines such as interleukin-4 (IL-4), IL-5, IL-9, and IL-13, which can activate the effector cells of allergy [3]. Like the Th2-type T-cells, group 2 innate lymphoid cells (ILC2s) can also generate Th2-type cytokines, mainly IL-4, IL-5, and IL-13, but they respond to environmental pathogens and potentially immunogenic triggers at the onset of inflammation, i.e., before the adaptive responses develop [4].
ILC2s are a group of lymphocyte subsets that lack T-cell or B cell diversified antigen receptors [5]. They develop from the common lymphocyte progenitors present in fetal liver and bone marrow and are mainly controlled by the GATA binding protein 3 (GATA3) transcription factor [5, 6]. ILC2 is a Lin− cell group that simultaneously expresses CD45, CD127, spinocerebellar ataxia type 1 (SCA1), CD278, killer cell lectin-like receptor G1 (KLRG1), and CD25, and differentially expresses CD117, IL-33R (ST2), and IL-17BR [5, 7]. In humans and mice, ILC2s are found in the spleen, intestine, liver, lung, and airways [8]. In mice, they are highly specific in the expression of ST2 [7]. In humans, ILC2s specifically express CD294 and CD161 [7]. ILC2s play a key role in maintaining tissue homeostasis, especially in early resistance to infection, pathophysiological mechanisms of the allergic inflammatory response, and tissue injury and repair [9]. There is growing evidence that ILC2 plays an essential role in allergic asthma. Studies have found that the proportion of ILC2s in the peripheral blood of patients with eosinophilic asthma is higher than in non-eosinophilic patients and the healthy control group [10]. A recent epigenetic study showed a strong correlation between gene regulation mechanisms of ILC2s and the genetic basis of asthma, which also confirmed the pathogenic role of ILC2s in asthma patients [11].
Follistatin-like 1 (FSTL1), also known as TGF-β1-stimulated clone 36 (TSC-36), is a glycoprotein that belongs to the secreted protein acidic and rich in cysteine (SPARC) family [12]. FSTL1 is closely associated with the pathogenesis and development of various diseases, such as systemic autoimmune diseases (SADs), tumor growth and metastasis, myocardial infarction, cardiac hypertrophy, and mitral valve disease [13]. FSTL1 acts as a pro-inflammatory cytokine in many SADs, such as arthritis [14, 15]. In rheumatoid arthritis, Ad-mFstl1 can increase the secretion of pro-inflammatory cytokines, interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), IL-1β, and IL-6, inducing synovitis with infiltration of inflammatory cells in the synovium and surrounding tissue [16]. Meanwhile, overexpression of FSTL1 in cultured monocytes, macrophages, or septic shock mouse models can induce caspase-1 and the nod-like receptor family, pyrin domain-containing 3 (NLRP3), confirming that FSTL1 mediates pro-inflammatory events [17]. Moreover, stimulation of nucleus pulposus cells with TNF-α induces FSTL1 secretion, also revealing that FSTL1 has a positive feedback effect on the inflammatory response [18].
Recent studies showed that FSTL1 plays an important role as a pro-inflammatory cytokine in the pathogenesis of bronchial asthma, promoting airway remodeling and airway inflammation in patients with asthma [19]. The FSTL1 levels in asthmatic patients’ serum and bronchoalveolar lavage fluid (BALF) were higher than in healthy controls, and the increasing degree was positively correlated with tracheal airway smooth muscle and reticular basement membrane thickening [19, 20]. Proteomics analysis of the sputum of patients with asthma showed that FSTL1 is one of the most highly expressed proteins [21]. FSTL1 can also promote epithelial autophagy to induce epithelial-mesenchymal transition and airway remodeling in asthma [22].
Our previous study found that serum FSTL1 levels in patients with asthma were higher than in healthy controls and correlated with an ILC2s increase [10]. Many studies have shown that IL-33, IL-25, and thymic stromal lymphopoietin (TSLP) are strong activators of ILC2, but we found the correlation between serum FSTL1 levels and the increasing degree of ILC2s to be even stronger [5]. Thus, we hypothesized that FSTL1 might be a strong activator, able to induce the activation of ILC2s, and promote airway inflammation in asthma. In this study, we tested this hypothesis and revealed the specific mechanism of how FSTL1 activates ILC2.
MATERIALS AND METHODS
Wild-Type Mice Ovalbumin Sensitization and Airway Challenge
Eight-week-old female wild-type C57BL/6 mice were obtained from the Centre of Experimental Animals of Shandong University. Mice were sensitized by intraperitoneal (ip) injection with 100 μg ovalbumin (OVA; grade V; Sigma, St Louis, MO) and 2 mg of aluminum hydroxide (Thermo Scientific Pierce, Rockford, IL) in 200 μl of phosphate-buffered saline (PBS) on days 0 and 14, followed by intranasal administration of 200 μg of OVA in 20 μl of PBS on days 21, 22, and 25. From day 28, mice continued challenges with intranasal OVA twice/week for 4 weeks. Non-OVA-challenged mice were sensitized and challenged with PBS only.
Fstl1+/− Mice
Fstl1flox/+ mice were a generous gift from Xiang Gao (Nanjing University, Nanjing, China) and Xu Zhang (Institute of Neuroscience, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences, Shanghai, China). Fstl1+/− mice, which were generated as described previously, were sensitized and challenged with OVA, as described earlier [23]. All mice (10 mice/group) were euthanized 24 h after the last challenge. Blood, BALF, lung tissue, and spleen tissue were harvested for further experiments.
Mouse BALF Collection
BALF was collected by lavaging the lung four times with 0.5–1 ml of ice-cold PBS via a tracheal catheter. BALF was centrifuged, and the supernatant was collected and stored at − 80 °C before use.
Mouse Serum Collection
After the mice were anesthetized with 10% chloral hydrate, the blood was taken by removing the eyeballs. Approximately 1 ml of blood was taken from each mouse, centrifuged at 5,000 rpm for 15 min, and the supernatant was transferred to a new EP tube. The serum was stored at −80 °C before use.
Western Blot Analysis
Cells were lysed in ice-cold RIPA buffer containing complete protease inhibitor cocktail (Roche) and 1 mM sodium orthovanadate. Protein concentrations were measured using a Protein BCA Assay Kit (Beyotime), and the samples were boiled at 95 °C for 5 min, resolved in sodium dodecyl sulphate–polyacrylamide gel electrophoresis, and transferred to poly (vinylidene fluoride) membranes. Membranes were blocked in 5% skim milk (w/v) in Tris-buffered saline containing 0.05% (v/v) Tween 20 (TBST) for 1 h at room temperature and incubated overnight with the primary antibodies. The following antibodies, acquired from Cell Signaling Technology unless stated otherwise, were used according to the manufacturer’s instructions: p-MEK1/2 (Ser221), p-JAK3 (Tyr980/981), p-STAT3 (Tyr705), p-STAT5 (Tyr694), p-STAT6 (Tyr641), and FSTL1 (ab111969; Abcam). After washing in TBST, the membranes were incubated in mouse anti-rabbit secondary antibody for 1 h at room temperature. Protein bands were visualized using a ChemiDoc XRS Imaging System (Bio-rad), and band intensities were quantified using ImageJ software.
Quantitative Real-Time Polymerase Chain Reaction
Total RNA was extracted using RNAiso Plus (Takara, Japan). cDNA was synthesized using the PrimeScript™ RT reagent Kit (Takara, Japan). Quantification of mRNA levels was performed by real-time PCR using Roche LightCycler480 and SYBR® Premix Ex TaqTM II (Takara, Japan). Gapdh was used as an internal control. Results were calculated using the comparative ΔΔCT method. Primers were designed to span an intron, and the sequences were as follows (5′ to 3′): Gapdh, ACGGCCGCATCTTCTTGTGCA (forward), and AATGGCAGCCCTGGTGACCA (reverse); Fstl1, TCACAGCAGCAATGCCATATCAA (forward) and GATTGGCCAACAGACACTGCAGCTA (reverse). All samples were analyzed in triplicate, and the relative levels were normalized to Gapdh expression.
Hematoxylin–Eosin Staining and Immunohistochemistry
Paraffin-embedded specimens were sectioned at 5-µm thickness with a microtome and stained with hematoxylin–eosin (ZSGB-BIO, China) for inflammatory cell count and morphometric analysis. Other specimens were dewaxed and rehydrated, and antigen retrieval was performed using 10 mM sodium citrate (pH 6.1). The sections were then blocked with 5% bovine serum albumin for 30 min at 37 °C. The slides were incubated with anti-FSTL1 antibody (ab111969; Abcam) overnight at 4 °C and then processed with the corresponding secondary horseradish peroxidase-conjugated antibodies (1:200) for 1 h at room temperature. After washing, 3,3-diaminobenzidine (Zsbio) was applied as the chromogen, resulting in a brown reaction product, and the sections were counter-stained with hematoxylin. PBS was used instead of the primary antibody as a negative control for nonspecific binding.
Enzyme-Linked Immunosorbent Assay
IL-4, IL-5, and IL-13 levels in mice serum and BALF were measured using a standard enzyme-linked immunosorbent assay (ELISA; NRC). All analyses and calibrations were performed in duplicate. Optical densities were determined using an absorbance microplate reader at 450 nm.
Flow Cytometry
Mice were euthanized and systemically perfused by PBS injection into the left heart ventricle. Collagenase IV was dissolved in PBS to prepare a 0.3% digestive solution to digest the whole minced lungs. We used it to digest mice’s lungs and spleens in a 37 °C water bath for 3 h and mixed it up and down every 30 min. Tissues were then filtered with a 70-μm cell strainer and depleted of erythrocytes by adding an RBC lysis solution (Solarbio, Beijing, China) for 10 min. Subsequently, we centrifuged the cells for 10 min at 1,500 rpm, 4 °C, and discarded the supernatant. Finally, the cell pellet was diluted in PBS up to 1 ml. The cells were stained with Mouse Hematopoietic Lineage FITC Cocktail (Ebioscience), anti-mouse ST2 PE, and anti-mouse CD127 PE-Cyanine5 at room temperature in the dark for 15 min to detect ILC2s (Lin−ST2+CD127+). Flow cytometry was performed using a BD FACS machine after the sample was filtered into a single cell suspension. Stained cells were analyzed by flow cytometric analysis using a FAC Scan cytometer equipped with CellQuest software (BD Bioscience, CA, USA).
ILC2 Sorting
The digestion process was the same as previously described. Cells were stained with Mouse Hematopoietic Lineage FITC Cocktail (Ebioscience), mouse CD45 APC, anti-mouse ST2 PE, and anti-mouse CD127 PE-Cyanine5 at room temperature in the dark for 15 min to detect ILC2s (Lin−CD45+ST2+CD127+). Stained cells were analyzed and sorted by a FACS Aria III Flow Cytometer.
ILC2 Culture In Vitro
Cells were cultured in RPMI 1640 (containing 10% fetal bovine serum, 1% penicillin–streptomycin, and 1 mM sodium pyruvate) with added IL-2 (10 ng/ml), IL-7 (10 ng/ml), and IL-33 (10 ng/ml) for ILC2 proliferation. All cells were grown at 37 °C in humidified air with 5% CO2. The cells were seeded at 1 × 105 cells/well in six-well culture plates. Half of the wells were stimulated with FSTL1 100 ng/ml for 16–18 h. The remaining half of the wells were set as the control group without FSTL1 stimulation. Subsequently, we collected the cells and centrifuged them at 1,000 rpm for 5 min. The cells were stained with anti-mouse ST2 PE (Ebioscience) and anti-mouse CD127 PE-Cyanine5 and incubated for 15 min at room temperature in the dark. Finally, we compared the number of ILC2 in these wells.
Statistical Analysis
All experiments in this study were performed at least three times. All statistical analyses were performed using SPSS 19.0 (Abbott Laboratories, USA). Data were presented as mean ± SD. Continuous variables were tested by analysis of Student’s t-test. Differences were considered statistically significant at *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
RESULTS
FSTL1 Is Closely Related to the Asthmatic Airway Inflammation
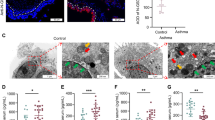
It was reported that FSTL1 is closely associated with the pathogenesis of asthma [19, 21]. To verify this, we performed hematoxylin and eosin staining and immunohistochemistry to stain mouse lung tissue sections, Western blotting, and quantitative real-time polymerase chain reaction (qRT-PCR) to detect the expression of FSTL1 in mouse lung tissue. Hematoxylin and eosin staining results showed plenty of inflammatory cell infiltration around the asthma groups’ airway tissues, accompanied by slight damage to the airway epithelial tissue and mucosal structure. However, the control groups had no obvious inflammatory cell infiltration in the airway, and the epithelial organizational structure was complete. Comparing the OVA-challenged Fstl1+/−model with the OVA-challenged wild-type(WT) model, we found that inflammatory cells around the airway tissue in the OVA-challenged Fstl1+/− mice were significantly fewer than in the OVA-challenged WT mice (Fig. 1a).

The level of FSTL1 is positively correlated with the degree of airway inflammation. a H&E staining of lung sections in four groups of mice (200 × , n = 5 per group). b, c Expression of FSTL1 in the airway epithelium and intercellular lung tissue of mice (immunohistochemistry, 200 × , and 400 × , n = 5 per group) and statistical significance of the differences. d The protein level and statistical difference of FSTL1 in the murine lung tissue (Western blotting). e The mRNA level and statistical difference of FSTL1 in mice lung tissue (qRT-PCR). Data were pooled from at least three independent experiments and presented as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ns, no significance (Student’s t-test).
Immunohistochemistry data demonstrated that the expression of FSTL1 in the airway epithelium and between cells in the WT and OVA-challenged Fstl1+/− groups was higher than that of the control group (Fig. 1b, c). With Western blotting and qRT-PCR, we analyzed the expression level of Fstl1 in each group of mice, and the results were consistent with immunohistochemistry. The expression in lung tissue of OVA-challenged Fstl1+/− mice and OVA-challenged WT mice was both higher than that in their corresponding control group (Fig. 1d, e). These findings indicated that FSTL1 is closely related to the pathogenesis of asthma, especially asthmatic airway inflammation.
The Level of FSTL1 Is Closely Related to the Proportion and Number of ILC2 in the Murine Lung and Spleen
To identify the relationship between FSTL1 and ILC2, we used flow cytometry to analyze the proportion and number of ILC2 in the lung and spleen of four groups of mice (Fig. 2a–d). The results showed that the absolute number and the proportion of ILC2 in the lung tissue of the OVA-challenged WT group were higher than in the control group (Fig. 2e, f). The absolute number and the proportion of ILC2 in the lung tissue of the OVA-challenged Fstl1+/− group were also higher than in the control group (Fig. 2e, f). Moreover, the absolute number and the proportion of ILC2 in the lung tissue of the OVA-challenged Fstl1+/− group were lower than in the OVA-challenged WT group (Fig. 2e, f).

FSTL1 affects ILC2 in the murine lung. a–d The numbers and percentages of ILC2s (Lin−ST2+CD127+) were determined by flow cytometry from the lung tissue of four groups of mice (FO means FSTL1+/− OVA, WO means WT OVA, FC means FSTL1+/− CONTROL, WC means WT CONTROL). e The absolute number of ILC2 in the lung tissue of four groups of mice and statistical differences. f The proportion of ILC2 in the lung tissue of four groups of mice and statistical differences. Data were pooled from at least three independent experiments and were presented as the mean ± SD. *p < 0.05, **p < 0.01 (Student’s t-test).
The same results were obtained in the spleens of WT and Fstl1+/− mice (Fig. 3a–d). The absolute number and the proportion of ILC2 in the spleen of the OVA-challenged WT group were higher than in the control group (Fig. 3e, f). Similarly, the absolute number and the proportion of ILC2 in the spleen of the OVA-challenged Fstl1+/− group were also higher than in the control group (Fig. 3e, f). Meanwhile, the absolute number and the proportion of ILC2 in the spleen of the OVA-challenged Fstl1+/− group were lower than in the OVA-challenged WT group (Fig. 3e, f). These results indicated that ILC2s in the lung and spleen of asthmatic mice were higher than the healthy control group. Simultaneously, the absolute number and the proportion of ILC2s in the lung and spleen of the OVA-challenged Fstl1+/− group were lower than the OVA-challenged WT group. All the differences above were statistically significant, which suggested that FSTL1 can promote the increase in the proportion and number of ILC2 in the lung and spleen of asthmatic mice.

FSTL1 affects ILC2 in the murine spleen. a–d ILC2 (Lin−ST2+CD127+) numbers and percentages were determined by flow cytometry from spleen tissue of four groups of mice (FO means FSTL1+/− OVA, WO means WT OVA, FC means FSTL1+/− CONTROL, WC means WT CONTROL). e The absolute number of ILC2 in the lung tissue of four groups of mice and statistical differences. f The proportion of ILC2 in the spleen tissue of four groups of mice and statistical differences. Data were pooled from at least three independent experiments and are presented as the mean ± SD. *p < 0.05, **p < 0.01 (Student’s t-test).
FSTL1 Promotes the Activation and Proliferation of ILC2
To further clarify the relationship between FSTL1 and ILC2, we used FACS to sort out ILC2 from murine lung (Fig. 4a). We cultured the sorted ILC2 in vitro and added FSTL1 stimulation to observe whether FSTL1 could promote the proliferation and activation of ILC2. After 18 h of FSTL1 stimulation, the proportion of ILC2 (ST2+CD127+) in the FSTL1 stimulation group (21.50 ± 1.35%) was higher than in the control group (10.52 ± 1.15%; Fig. 4b, c). All the results confirmed that FSTL1 induces ILC2 activation and promotes the increase of its number as well as the proportion.

The level of FSTL1 affected the activation of ILC2. a Murine lung ILC2s (Lin−CD45+ST2+CD127+) flow cytometry sorting process. b After sorting, the control group and the FSTL1-stimulated group were set up to culture ILC2, and we stained anti-mouse ST2 PE and anti-mouse CD127 PE-Cyanine5 for flow cytometric analysis to detect ILC2 in each group. c The proportion of ILC2 in the FSTL1 stimulation group and the control group and statistical significance of the differences. Data were pooled from at least three independent experiments and are presented as the mean ± SD. ***p < 0.001 (Student’s t-test).
FSTL1 Promotes the Massive Release of Th2-Type Cytokines by Activating ILC2
Some studies revealed that activated ILC2 produce many Th2-type cytokines, including IL-4, IL-5, and IL-13. These Th2-type cytokines act on eosinophils, mast cells, and basophils, promoting the inflammatory response of airway tissue of asthmatic patients [24, 25]. Therefore, we used ELISA to detect the levels of IL-4, IL-5, and IL-13 in the BALF and serum of the four groups of mice to verify the activation of ILC2 and airway inflammation in each group of mice. ELISA results showed that the concentrations of IL-4, IL-5, and IL-13 in BALF and serum of the OVA-challenged WT group were higher than those of the WT control group (Fig. 5a, b). The same was observed between the OVA-challenged Fstl1+/− and Fstl1+/− control groups. The concentrations of the three kinds of cytokines in BALF and serum of the OVA-challenged Fstl1+/− group were higher than those of the Fstl1+/− control group (Fig. 5a, b). Moreover, a statistical comparison revealed that the concentrations of these cytokines in the OVA-challenged Fstl1+/− group were also significantly lower than those of the OVA-challenged WT group (Fig. 5a, b). These findings indicated that FSTL1 promotes airway inflammation in asthma by inducing the activation of ILC2.

FSTL1 induces ILC2 to produce Th-2 type (IL-4, IL-5, and IL-13) cytokines. a, b The levels of IL-4, IL-5, and IL-13 in the serum and BALF of four groups of mice were detected by ELISA. a The levels of IL-4, IL-5, and IL-13 in each group of mice in BALF and the statistical differences. b The levels of IL-4, IL-5, and IL-13 in each group of mice in serum and the statistical differences. Data were pooled from at least three independent experiments and were presented as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t-test).
FSTL1 Induces the Activation of ILC2 Through the MEK-JAK-STAT-GATA3 Pathway to Promote Airway Inflammation in Asthma
To explore the specific mechanism of ILC2 activation by FSTL1, we reviewed the relevant studies and found that glucocorticoids can inhibit the activation of ILC2 and the production of Th2 cytokines IL-4, IL-5, and IL-13 through the signal transducer and activator of transcription (STAT) signaling pathway [26]. Additionally, the pro-inflammatory effect of FSTL1 is related to JAK-STAT [27]. Consequently, we cultured the ILC2 obtained by sorting in vitro and tested the degree of phosphorylation of STAT protein to determine the best FSTL1 stimulation time. We discovered that the phosphorylation of STAT3, STAT5, and STAT6 in Lin− cells under stimulation markedly increased after 6 h and reached a maximum level in 12 h. After ILC2 was stimulated for 12 h, the changes in the phosphorylation degree of various indicators in the MEK-JAK-STAT-GATA3 pathway were detected (Fig. 6a). According to the Western blotting results, the expression of p-MEK, p-JAK3, p-STAT3, p-STAT5, p-STAT6, and p-GATA3 significantly increased with FSTL1 (Fig. 6b). After adding the mitogen-activated protein kinase kinase (MEK) inhibitor U0126, the expression of p-MEK, p-JAK3, p-STAT3, p-STAT5, p-STAT6, and p-GATA3 decreased (Fig. 6c), and the phosphorylation of pathway proteins JAK3, STATs, and GATA3 was inhibited, indicating that MEK is located upstream of the JAK-STAT-GATA3 signaling pathway. After adding the Janus kinase 3 (JAK3) inhibitor JANEX-1, except for p-MEK, the expression of p-JAK3, p-STAT3, p-STAT5, p-STAT6, and p-GATA3 decreased (Fig. 6d). This variation indicates that JAK is located upstream of the STAT-GATA3 signaling pathway. Based on the above results, we concluded that FSTL1 induces the activation of ILC2 through the MEK-JAK-STAT-GATA3 pathway.

The MEK-JAK-STAT-GATA3 signaling pathway is responsible for the production of IL-4, IL-5, and IL-13 from ILC2 stimulated by FSTL1. a Western blotting analysis of p-STAT3, p-STAT5, p-STAT6, and p-GATA3 levels in sorted Lin− cells in response to IL-2 and IL-7 plus IL-33 at different time points. b–d Western blotting analysis of p-MEK, p-JAK3, p-STAT3, p-STAT5, p-STAT6, and p-GATA3 levels in sorted ILC2s with stimulation of IL-2, IL-7, IL-33, plus FSTL1 or MEK inhibitor (U0126) or JAK3 inhibitor (JANEX-1). Data were pooled from at least three independent experiments and are presented as the mean ± SD. ****p < 0.0001 (Student’s t-test).
MEK-JAK-STAT-GATA3 Pathway Inhibitors Can Inhibit the Activation of ILC2 by FSTL1
Finally, we used various MEK-JAK-STAT-GATA3 pathway inhibitors to measure the levels of IL-4, IL-5, and IL-13 released by ILC2 with ELISA to further confirm the involvement of this pathway. After the JAK3 inhibitor JANEX-1, MEK inhibitor U0126, STAT3 inhibitor S3I-201, STAT5 inhibitor IQDMA, or STAT6 inhibitor AS1517499 was added, the levels of IL-4, IL-5, and IL-13 were significantly lower than those in the control group (Fig. 7). In addition, after stimulation with FSTL1, the contents of IL-4, IL-5, and IL-13 in the ILC2 culture medium were higher than those in the controls (Fig. 7). Collectively, these data confirmed that FSTL1 induces the activation of ILC2 through the MEK-JAK-STAT-GATA3 pathway to promote airway inflammation in asthma.

MEK-JAK-STAT-GATA3 pathway inhibitors can inhibit the activation of ILC2. IL-4, IL-5, and IL-13 levels in ILC2s were measured under STAT3 inhibitor (S3I-201), STAT5 inhibitor (IQDMA), STAT6 inhibitor (AS1517499), JAK3 inhibitor (JANEX-1), MEK inhibitor (U0126), and FSTL1 treatments after the stimulation of IL-2, IL-7, and IL-33. Data were pooled from at least three independent experiments and are presented as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t-test).
DISCUSSION
Asthma affects the health of more than 360 million people worldwide [1]. Recent studies have shown that the prevalence of asthma among people aged 20 years and over in China is 4.2%, with a total of more than 45.7 million patients [28]. The genesis of asthma is closely related to airway inflammation [29, 30]. FSTL1, as a pro-inflammatory factor, is widely involved in the onset and development of many diseases [13]. Studies have shown that ILC2 is related to the pathogenesis of asthma [31]. Therefore, we intended to explore whether FSTL1 and ILC2 are related. To our knowledge, this is the first correlative study to investigate the relationship between ILC2 and FSTL1 in asthma pathogenesis. Our data verified that FSTL1 promotes the proliferation and activation of ILC2, and activated ILC2 can produce large amounts of Th2-type cytokines (IL-4, IL-5, and IL-13), which triggered a series of inflammatory reactions. These observations supported the notion that FSTL1 promotes asthma by activating ILC2.
FSTL1 has been associated with cardiovascular diseases, such as heart failure and acute coronary syndromes [32, 33], and also tumors such as kidney cancer and lung adenocarcinoma [34, 35]. Interestingly, FSTL1 plays a dual role in multiple diseases, which is also confirmed in inflammatory diseases such as asthma and rheumatoid arthritis. FSTL1 can act as an anti-inflammatory factor in the acute phase but has a pro-inflammatory effect in the long term and chronic diseases, probably due to the activation of different signaling pathways [13]. However, it cannot be excluded that additional endogenous or exogenous factors are involved in these regulatory processes [13]. Previous studies in our laboratory proved that FSTL1 promotes asthmatic airway remodeling by affecting autophagy [19]. Airway inflammation, another important mechanism of asthma, is probably also regulated by FSTL1. Therefore, we established asthma models in WT and Fstl1+/− mice. Through staining and Western blotting, we proved that FSTL1 levels were positively correlated with asthma airway inflammation. However, the specific mechanism by which FSTL1 can promote airway inflammation is still unclear; we are determined to continue exploring it.
In 2010, the identification of nuocytes, innate helper type 2 cells, and natural helper cells as major players in type-2 immunity during helminth infections led to a series of investigations into the role of ILC2 in allergic diseases, including asthma [36]. Research has found that ILC2 can be activated in OVA-sensitized mouse asthma models and asthma patients [37]. The signaling pathways related to ILC2 activation have also emerged in large numbers, such as mitogen-activated protein kinase (MAPK) and nuclear factor kappa B (NF-κB) [38, 39]. The JAK-STAT pathway has attracted our attention among the many signaling pathways because FSTL1 can activate it [27]. As far as we know, there is no relevant research on the connection between FSTL1 and ILC2. Thus, we intended to explore the specific connection and internal mechanism between the two. We cultured ILC2 obtained by flow cytometry in vitro, added FSTL1 for stimulation, and found that FSTL1 can promote the proliferation and activation of ILC2. Furthermore, IL-4, IL-5, and IL-13 are recognized as downstream cytokines of ILC2, triggering inflammation, and changes in their levels can support the activation of ILC2. Therefore, we also used ELISA to measure the effects of these cytokines under FSTL1 stimulation. The level changes, and the results agree with our expectations. We have confirmed that FSTL1 can promote the activation of ILC2 and trigger massive production of downstream cytokines, but its related signaling pathways have not yet been clearly established.
To explore the signaling pathway of FSTL1 activating ILC2, we inquired much relevant information. There are several reports about the signaling pathways involved in ILC2s in asthma. Stat6 deficiency would lead to impaired IL-4 and IL-13 receptor expression and proliferation of ILC2s in the mouse, and MEK-JAK-STAT could also be involved [26, 36]. GATA3 is an important transcription factor of ILC2 and a potential therapeutic target for asthma [40,41,42,43,44]. Therefore, we hypothesized that FSTL1 could activate ILC2 through the MEK-JAK-STAT-GATA3 axis. Western blotting found that the addition of FSTL1 could significantly increase the expression of all kinds of proteins in the pathway.
Moreover, when we used the JAK inhibitor JANEX-1, the decline in the expression of STATs and GATA3 proved their downstream position. The application of MEK inhibitors decreased the expression of all related proteins in the pathway and confirmed MEK’s upstream position. After adding multiple pathway-related inhibitors, the ELISA results suggested that FSTL1 can promote the expression of Th2-type cytokines, and the expression levels were significantly lessened after the use of various inhibitors.
Together, our data demonstrated that FSTL1 could activate ILC2 through the MEK-JAK-STAT-GATA3 pathway, and that activated ILC2 produces Th2-type cytokines massively, enhancing airway inflammation and promoting the onset of asthma. There are several monoclonal antibodies against Th-2 type cytokines (e.g., reslizumab and mepolizumab) [45], but the relationship between FSTL1 and ILC2 will open new avenues for the treatment of asthma.
Data Availability
All data and materials are available from the corresponding author upon request.
Abbreviations
- ILC2s:
-
Type 2 innate lymphoid cells
- FSTL1:
-
Follistatin-like 1
- JAK:
-
Janus kinase
- STAT:
-
Signal transducer and activator of transcription
- OVA:
-
Ovalbumin
- WT:
-
Wild-type
- BALF:
-
Bronchoalveolar lavage fluid
- MEK:
-
Mitogen-activated protein kinase kinase
- GATA3:
-
GATA binding protein 3
- DCs:
-
Dendritic cells
- IL-4:
-
Interleukin-4
- SCA1:
-
Spinocerebellar ataxia type 1
- KLRG1:
-
Killer cell lectin-like receptor G1
- TSC-36:
-
TGF-β1-stimulated clone 36
- SPARC:
-
Secreted protein acidic and rich in cysteine
- SADs:
-
Systemic autoimmune diseases
- IFN-γ:
-
Interferon-γ
- TNF-α:
-
Tumor necrosis factor-α
- NLRP3:
-
The nod-like receptor family, pyrin domain-containing 3
- TSLP:
-
Thymic stromal lymphopoietin
- PBS:
-
Phosphate-buffered saline
- TBST:
-
Tris-buffered saline containing 0.05% (v/v) Tween 20
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- ELISA:
-
Enzyme-linked immunosorbent assay
- MAPK:
-
Mitogen-activated protein kinase
- NF-κB:
-
Nuclear factor kappa B
- FO:
-
FSTL1+/− OVA
- WO:
-
WT OVA
- FC:
-
FSTL1+/− CONTROL
- WC:
-
WT CONTROL
References
Global Initiative for Asthma. 2020. Global strategy for asthma management and prevention. Available from:www.ginasthma.org.
Gandhi, N.A., B.L. Bennett, N.M. Graham, G. Pirozzi, N. Stahl, and G.D. Yancopoulos. 2016. Targeting key proximal drivers of type 2 inflammation in disease. Nature reviews. Drug discovery 15 (1): 35–50.
Kucuksezer, U.C., C. Ozdemir, M. Akdis, and C.A. Akdis. 2018. Precision/personalized medicine in allergic diseases and asthma. Archivum immunologiae et therapiae experimentalis 66 (6): 431–442.
Krabbendam, L., S.M. Bal, H. Spits, and K. Golebski. 2018. New insights into the function, development, and plasticity of type 2 innate lymphoid cells. Immunological reviews 286 (1): 74–85.
Vivier, E., D. Artis, M. Colonna, A. Diefenbach, J.P. Di Santo, G. Eberl, S. Koyasu, R.M. Locksley, A. McKenzie, R.E. Mebius, F. Powrie, and H. Spits. 2018. Innate lymphoid cells: 10 years on. Cell 174 (5): 1054–1066.
McKenzie, A.N. 2014. Type-2 innate lymphoid cells in asthma and allergy. Annals of the American Thoracic Society 11 (Suppl 5): S263–S270.
Mjösberg, J.M., S. Trifari, N.K. Crellin, C.P. Peters, C.M. van Drunen, B. Piet, W.J. Fokkens, T. Cupedo, and H. Spits. 2011. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nature immunology 12 (11): 1055–1062.
Puttur, F., L. Denney, L.G. Gregory, J. Vuononvirta, R. Oliver, L.J. Entwistle, S.A. Walker, M.B. Headley, E.J. McGhee, J.E. Pease, M.F. Krummel, L.M. Carlin, and C.M. Lloyd. 2019. Pulmonary environmental cues drive group 2 innate lymphoid cell dynamics in mice and humans. Science immunology 4(36): eaav7638.
Ealey, K.N., and S. Koyasu. 2017. How many subsets of innate lymphoid cells do we need? Immunity 46 (1): 10–13.
Liu, T., J. Wu, J. Zhao, J. Wang, Y. Zhang, L. Liu, L. Cao, Y. Liu, and L. Dong. 2015. Type 2 innate lymphoid cells: A novel biomarker of eosinophilic airway inflammation in patients with mild to moderate asthma. Respiratory medicine 109 (11): 1391–1396.
Stadhouders, R., B. Li, M. de Bruijn, A. Gomez, T.N. Rao, H.J. Fehling, and van IJcken, W., Lim, A. I., Di Santo, J. P., Graf, T., and Hendriks, R. W. 2018. Epigenome analysis links gene regulatory elements in group 2 innate lymphocytes to asthma susceptibility. The Journal of allergy and clinical immunology 142 (6): 1793–1807.
Sundaram, G.M., J.E. Common, F.E. Gopal, S. Srikanta, K. Lakshman, D.P. Lunny, T.C. Lim, V. Tanavde, E.B. Lane, and P. Sampath. 2013. ‘See-saw’ expression of microRNA-198 and FSTL1 from a single transcript in wound healing. Nature 495 (7439): 103–106.
Mattiotti, A., S. Prakash, P. Barnett, and M. van den Hoff. 2018. Follistatin-like 1 in development and human diseases. Cellular and molecular life sciences : CMLS 75 (13): 2339–2354.
Clutter, S.D., D.C. Wilson, A.D. Marinov, and R. Hirsch. 2009. Follistatin-like protein 1 promotes arthritis by up-regulating IFN-gamma. Journal of immunology 182 (1): 234–239.
Li, W., M. Alahdal, Z. Deng, J. Liu, Z. Zhao, X. Cheng, X. Chen, J. Li, J. Yin, Y. Li, G. Wang, D. Wang, K. Tang, and J. Zhang. 2020. Molecular functions of FSTL1 in the osteoarthritis. International immunopharmacology 83: 106465.
Li, D., Y. Wang, N. Xu, Q. Wei, M. Wu, X. Li, P. Zheng, S. Sun, Y. Jin, G. Zhang, R. Liao, and P. Zhang. 2011. Follistatin-like protein 1 is elevated in systemic autoimmune diseases and correlated with disease activity in patients with rheumatoid arthritis. Arthritis research & therapy 13 (1): R17.
Chaly, Y., Y. Fu, A. Marinov, B. Hostager, W. Yan, B. Campfield, J.A. Kellum, D. Bushnell, Y. Wang, J. Vockley, and R. Hirsch. 2014. Follistatin-like protein 1 enhances NLRP3 inflammasome-mediated IL-1β secretion from monocytes and macrophages. European journal of immunology 44 (5): 1467–1479.
Liu, Y., J. Wei, Y. Zhao, Y. Zhang, Y. Han, B. Chen, K. Cheng, J. Jia, L. Nie, and L. Cheng. 2017. Follistatin-like protein 1 promotes inflammatory reactions in nucleus pulposus cells by interacting with the MAPK and NFκB signaling pathways. Oncotarget 8 (26): 43023–43034.
Liu, Y., T. Liu, J. Wu, T. Li, X. Jiao, H. Zhang, J. Zhao, J. Wang, L. Liu, L. Cao, S. Li, J. Xu, J. Xu, X. Ma, L. Yang, and L. Dong. 2017. The correlation between FSTL1 expression and airway remodeling in asthmatics. Mediators of inflammation 2017: 7918472.
Miller, M., A. Beppu, P. Rosenthal, A. Pham, S. Das, M. Karta, D.J. Song, C. Vuong, T. Doherty, M. Croft, B. Zuraw, X. Zhang, X. Gao, S. Aceves, F. Chouiali, Q. Hamid, and D.H. Broide. 2015. Fstl1 promotes asthmatic airway remodeling by inducing oncostatin M. Journal of immunology 195 (8): 3546–3556.
Miller, M., S. Esnault, R.C. Kurten, E.A. Kelly, A. Beppu, S. Das, P. Rosenthal, J. Ramsdell, M. Croft, B. Zuraw, N. Jarjour, Q. Hamid, and D.H. Broide. 2016. Segmental allergen challenge increases levels of airway follistatin-like 1 in patients with asthma. The Journal of allergy and clinical immunology 138 (2): 596-599.e4.
Liu, T., Y. Liu, M. Miller, L. Cao, J. Zhao, J. Wu, J. Wang, L. Liu, S. Li, M. Zou, J. Xu, D.H. Broide, and L. Dong. 2017. Autophagy plays a role in FSTL1-induced epithelial mesenchymal transition and airway remodeling in asthma. American journal of physiology. Lung cellular and molecular physiology 313(1): L27–L40.
Li, K.C., F.X. Zhang, C.L. Li, F. Wang, M.Y. Yu, Y.Q. Zhong, K.H. Zhang, Y.J. Lu, Q. Wang, X.L. Ma, J.R. Yao, J.Y. Wang, L.B. Lin, M. Han, Y.Q. Zhang, R. Kuner, H.S. Xiao, L. Bao, X. Gao, and X. Zhang. 2011. Follistatin-like 1 suppresses sensory afferent transmission by activating Na+, K+-ATPase. Neuron 69 (5): 974–987.
Deo, S.S., K.J. Mistry, A.M. Kakade, and P.V. Niphadkar. 2010. Role played by Th2 type cytokines in IgE mediated allergy and asthma. Lung India: Official organ of Indian Chest Society 27 (2): 66–71.
Kay, A.B. 1996. TH2-type cytokines in asthma. Annals of the New York Academy of Sciences 796: 1–8.
Yu, Q.N., Y.B. Guo, X. Li, C.L. Li, W.P. Tan, X.L. Fan, Z.L. Qin, D. Chen, W.P. Wen, S.G. Zheng, and Q.L. Fu. 2018. ILC2 frequency and activity are inhibited by glucocorticoid treatment via STAT pathway in patients with asthma. Allergy 73 (9): 1860–1870.
Ni, S., C. Li, N. Xu, X. Liu, W. Wang, W. Chen, Y. Wang, and A.J. van Wijnen. 2018. Follistatin-like protein 1 induction of matrix metalloproteinase 1, 3 and 13 gene expression in rheumatoid arthritis synoviocytes requires MAPK, JAK/STAT3 and NF-κB pathways. Journal of cellular physiology 234 (1): 454–463.
Huang, K., T. Yang, J. Xu, L. Yang, J. Zhao, X. Zhang, et al. 2019. Prevalence, risk factors, and management of asthma in China: A national cross-sectional study. Lancet 394 (10196): 407–418.
Busse, W.W., W.F. Calhoun, and J.D. Sedgwick. 1993. Mechanism of airway inflammation in asthma. The American review of respiratory disease 147 (6 Pt 2): S20–S24.
Ma, B., S.S. Athari, E. Mehrabi Nasab, and L. Zhao. 2021. PI3K/AKT/mTOR and TLR4/MyD88/NF-κB signaling inhibitors attenuate pathological mechanisms of allergic asthma. Inflammation. https://doi.org/10.1007/s10753-021-01466-3.
Skevaki, C., and H. Renz. 2018. Advances in mechanisms of allergic disease in 2017. The Journal of allergy and clinical immunology 142 (6): 1730–1739.
Lara-Pezzi, E., L.E. Felkin, E.J. Birks, P. Sarathchandra, K.D. Panse, R. George, J.L. Hall, M.H. Yacoub, N. Rosenthal, and P.J. Barton. 2008. Expression of follistatin-related genes is altered in heart failure. Endocrinology 149 (11): 5822–5827.
Widera, C., R. Horn-Wichmann, T. Kempf, K. Bethmann, B. Fiedler, S. Sharma, R. Lichtinghagen, H. Leitolf, B. Ivandic, H.A. Katus, E. Giannitsis, and K.C. Wollert. 2009. Circulating concentrations of follistatin-like 1 in healthy individuals and patients with acute coronary syndrome as assessed by an immunoluminometric sandwich assay. Clinical chemistry 55 (10): 1794–1800.
Chiou, J., C.Y. Su, Y.H. Jan, C.J. Yang, M.S. Huang, Y.L. Yu, and M. Hsiao. 2017. Decrease of FSTL1-BMP4-Smad signaling predicts poor prognosis in lung adenocarcinoma but not in squamous cell carcinoma. Scientific reports 7 (1): 9830.
Liu, Y., X. Tan, W. Liu, X. Chen, X. Hou, D. Shen, Y. Ding, J. Yin, L. Wang, H. Zhang, Y. Yu, J. Hou, T.C. Thompson, and G. Cao. 2018. Follistatin-like protein 1 plays a tumor suppressor role in clear-cell renal cell carcinoma. Chinese journal of cancer 37 (1): 2.
Lund, S., H.H. Walford, and T.A. Doherty. 2013. Type 2 innate lymphoid cells in allergic disease. Current immunology reviews 9 (4): 214–221.
Lambrecht, B.N., and H. Hammad. 2015. The immunology of asthma. Nature immunology 16 (1): 45–56.
Hu, P.F., C.Y. Ma, F.F. Sun, W.P. Chen, and L.D. Wu. 2019. Follistatin-like protein 1 (FSTL1) promotes chondrocyte expression of matrix metalloproteinase and inflammatory factors via the NF-κB pathway. Journal of cellular and molecular medicine 23 (3): 2230–2237.
Guo, J., W. Liang, J. Li, and J. Long. 2016. Knockdown of FSTL1 inhibits oxLDL-induced inflammation responses through the TLR4/MyD88/NF-κB and MAPK pathway. Biochemical and biophysical research communications 478 (4): 1528–1533.
Nakamura, Y., and M. Hoshino. 2005. TH2 cytokines and associated transcription factors as therapeutic targets in asthma. Current drug targets. Inflammation and allergy 4 (2): 267–270.
Mjösberg, J., J. Bernink, K. Golebski, J.J. Karrich, C.P. Peters, B. Blom, A.A. te Velde, W.J. Fokkens, C.M. van Drunen, and H. Spits. 2012. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity 37 (4): 649–659.
Zhang, D.H., L. Yang, L. Cohn, L. Parkyn, R. Homer, P. Ray, and A. Ray. 1999. Inhibition of allergic inflammation in a murine model of asthma by expression of a dominant-negative mutant of GATA-3. Immunity 11 (4): 473–482.
Wang, J., L. Xiao, L. Zhu, M. Hu, Q. Wang, and T. Yan. 2015. The effect of synthetic salidroside on cytokines and airway inflammation of asthma induced by diisocyanate (TDI) in mice by regulating GATA3/T-bet. Inflammation 38 (2): 697–704.
Chong, L., W. Zhang, Y. Nie, G. Yu, L. Liu, L. Lin, S. Wen, L. Zhu, and C. Li. 2014. Protective effect of curcumin on acute airway inflammation of allergic asthma in mice through Notch1-GATA3 signaling pathway. Inflammation 37 (5): 1476–1485.
Opina, M.T., and W.C. Moore. 2017. Phenotype-driven therapeutics in severe asthma. Current allergy and asthma reports 17 (2): 10.
Acknowledgements
We would like to thank Prof. Xu Zhang and Xiang Gao for generous help with the FSTL1 knockout mice.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81770029).
Author information
Authors and Affiliations
Contributions
Siyuan Huang designed the experiments, analyzed the results, designed the images, and wrote the manuscript. Siyuan Huang performed the experiments with the assistance of Rong Zeng and Xinrui Qiao. Shuo Li and Dong Zhang reviewed the results and the manuscript. Jing Wang and Rong Zeng assisted in solving the problem during the revision process. Liang Dong approved final version of manuscript.
Ethics declarations
Ethics Approval
All mouse experiments were approved by the Institutional Animal Care and Use Committee of Shandong University.
Consent to Participate
Not applicable.
Consent to Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Huang, S., Zeng, R., Wang, J. et al. Follistatin-Like 1 Induces the Activation of Type 2 Innate Lymphoid Cells to Promote Airway Inflammation in Asthma. Inflammation 45, 904–918 (2022). https://doi.org/10.1007/s10753-021-01594-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-021-01594-w