
Abstract
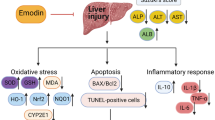
Gastrodin is a major active phenolic glycoside extract from Gastrodia elata, an important herb used in traditional medicine. Previous research has reported that gastrodin possesses anti-inflammatory and anti-oxidant properties. Therefore, we aimed to investigate its hepatoprotective effects and mechanisms on acetaminophen (APAP)-induced liver injury in a mouse model. Mice included in this study were intraperitoneally administered with a hepatotoxic APAP dose (300 mg/kg). At 30 min after APAP administration, gastrodin was intraperitoneally injected at concentrations of 0, 15, 30, and 45 mg/kg. Then, all mice were sacrificed at 16 h after APAP injection for further analysis. The results showed that gastrodin treatment ameliorated acute liver injury caused by APAP, as indicated by serum alanine aminotransferase level, hepatic myeloperoxidase activity, and cytokine (TNF-α, IL-1β, and IL-6) production. It also significantly decreased hepatic malondialdehyde activity but increased superoxide dismutase activity. In addition, gastrodin decreased ERK/JNK MAPK expression but promoted Nrf2 expression. These results demonstrated that gastrodin may be a potential therapeutic target for the prevention of APAP-induced hepatotoxicity via amelioration of the inflammatory response and oxidative stress, inhibition of ERK/JNK MAPK signaling pathways, and activation of Nrf2 expression levels.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Acute liver injury poses a serious threat to human health. One of the most common factors that cause acute liver injury is drug overdose. Acetaminophen (APAP) is a frequently used antipyretic and analgesic drug. Previous research has reported that APAP overdose results in liver damage, which is the most common cause of drug-induced liver failure in the world, despite it being relatively safe at therapeutic doses [1]. APAP-induced hepatotoxicity usually occurs within 48 h of ingestion. Clinically, the standard treatment options for emergent APAP-induced liver damage are limited. Although N-acetylcysteine is used as a first-line treatment, it is associated with various side effects, and its effectiveness is restricted to the early stages [2, 3].
At therapeutic doses, APAP is metabolized into a highly toxic, reactive metabolite called N-acetyl-p-benzoquinone imine (NAPQI), which is depleted by the hepatic glutathione (GSH) antioxidant system [4]. However, under toxic APAP doses, excess toxic metabolite accumulation causes oxidant stress generation and mitochondrial dysfunction, which subsequently results in centrilobular necrosis and hepatocyte death [5, 6]. APAP-induced hepatotoxicity is associated with innate immunity and oxidative stress. After initial liver damage to parenchymal hepatocytes, resident phagocytic macrophages called Kupffer cells trigger increased release of pro-inflammatory mediators, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6, as well as oxidative stress. Then, a number of other activating inflammatory cells, including macrophages and neutrophils, are recruited into the liver vasculature, which worsens the progression of APAP-induced liver damage [7, 8].
The inflammatory response and reactive oxygen species (ROS) accumulation are two of the main aspects associated with APAP-induced liver injury. Previous studies have reported that mitogen-activated protein kinase (MAPK) activation by oxidative stress is a key element of the intracellular signal transduction pathway [9]. Activation of c-Jun N-terminal kinase (JNK), one of the MAPK pathways, by ROS causes increased mitochondrial permeabilization and cytochrome c release [10]. Extracellular signal-regulated kinase (ERK) also participates in the regulation of inflammation and oxidative stress [11]. Moreover, recent studies have indicated that nuclear factor erythroid-2-related factor 2 (Nrf2) activates related gene regulation in APAP-induced hepatotoxicity [12]. Nrf2 can regulate antioxidant defense genes and enzymes to protect against cell injury [13, 14]. In animal studies, Nrf2-deficient mice were susceptible to drug-induced liver hepatotoxicity due to decreased antioxidant protection [15]. Thus, Nrf2 signaling pathways may be a potential target in the protection against APAP-induced liver injury.
Gastrodin is a major active phenolic glycoside extract from the rhizome of Gastrodia elata, commonly known as Tianma. It is used in traditional medicine and has a wide range of properties, such as antioxidation, anti-inflammation, and modulating neurotransmitters [16]. Previous studies have shown that gastrodin can attenuate the expression of inflammatory cytokines via inhibition of the MAPK signaling pathway in lipopolysaccharide-stimulated microglia [17]. It also has protective effects on organs in ischemia/reperfusion-induced spine and heart injury [18, 19]. In addition, gastrodin exerts hepatoprotective effects against alcohol-induced liver injury by suppressing the inflammatory response and oxidative stress [20]. However, the pharmacological effects of gastrodin in APAP-induced liver injury have rarely been reported and remain to be elucidated. In this study, the detailed mechanisms of gastrodin were investigated following APAP-induced hepatotoxicity in a mouse model.
MATERIALS AND METHODS
Animals
Adult C57BL/6 (B6) mice were purchased from BioLASCO Taiwan Co., Ltd. (Taipei, Taiwan). All procedures carried out in this study were approved by the Institutional Animal Care and Use Committee of Chang Gung Memorial Hospital (Taoyuan, Taiwan). All animal experiments were performed according to the guidelines of the Animal Welfare Act and the Guide for Care and Use of Laboratory Animals from the National Institutes of Health.
Experimental Protocols and Drug Treatment
All mice were housed in a room with a controlled environment and made to fast overnight before the procedure. After 1 week of adaptive breeding, 36 male mice were randomly divided into 6 groups (n = 6 mice/group) as follows: control, gastrodin (GA) only (45 mg/kg), APAP only (300 mg/kg), APAP + GA (15 mg/kg), APAP + GA (30 mg/kg), and APAP + GA (45 mg/kg). APAP (Sigma Chemical Co., St. Louis, MO, USA) was dissolved in 0.9% sterile normal saline at a concentration of 20 mg/mL. The treatment mice received an intraperitoneal hepatotoxic injection of APAP (300 mg/kg), whereas the control mice were injected with an equal volume of normal saline. At 30 min after APAP administration, the mice were intraperitoneally injected with gastrodin (Sigma Chemical Co.) at a concentration of 0, 15, 30, or 45 mg/kg. Then, all mice were sacrificed via cervical dislocation under isoflurane anesthesia at 16 h after APAP exposure. Blood samples from the vena cava were collected using heparinized syringes. Immediately after collecting the blood, the livers were harvested for further analysis.
Measurement of Hepatic Injury
Blood samples were collected and centrifuged at 15,000 × g for 15 min. The separated serum was collected for liver function tests. Liver enzyme alanine aminotransferase (ALT), aspartate aminotransferase (AST), and γ-glutamyltransferase (GGT) were measured to determine liver damage using a Vitros DT60 II Chemistry System (Ortho-Clinical Diagnostics; Johnson & Johnson, New York, NY, USA). All serum sample processing and procedures were performed according to the manufacturer’s protocols.
Liver Histological Examination
For histopathological assessment of tissue damage, liver samples were harvested, fixed with 4% paraformaldehyde over 24 h, and embedded in paraffin. Sections of 4 μm thickness stained with standard hematoxylin and eosin (H&E) were observed to evaluate liver damage using a light microscope (Olympus BX60, Tokyo, Japan).
Measurement of Liver Tissue Myeloperoxidase (MPO) Activity
Liver MPO activity is an indicator of neutrophil accumulation and acts a biomarker of oxidative stress and inflammation. Mouse liver tissues were homogenized and centrifuged at 15,000 × g and 4 °C for 15 min. The resulting pellet was washed twice, resuspended in 10 volumes of room temperature 50 mM KPO4 buffer, incubated at 60 °C for 2 h and then sonicated for 10 s using a sonicator. The suspension was centrifuged at 15,000 × g and 4 °C for 15 min. Subsequently, the supernatant was added to phosphate buffer consisting of o-dianisidine, 0.3% hydrogen peroxide, and 50 mM KPO4. Light absorbance was measured at 460 nm over an interval of 5 min.
Immunohistochemical Analysis of Liver Tissues
For immunohistochemical analysis, hepatic sections were blocked with buffer for 30 min and incubation with anti-Ly6G and anti-Nrf2 antibodies (Abcam, Cambridge, UK) at 37 °C for 2 h. After washing for 5 min, the samples were incubated with a biotinylated secondary antibody for 1 h at room temperature. Then, the slides underwent 3,3′-diaminobenzidine staining and hematoxylin counter staining according to the manufacturer’s instructions (Millipore IHC select kit; Burlington, MA, USA). The reaction times for all experimental mice were identical. A brownish-yellow color in the nuclei or cytoplasm of liver cells was considered to be positive using light microscopy (Olympus BX60, Tokyo, Japan).
Measurement of Liver Tissue Cytokine Levels by ELISA
To determine the inflammatory response in liver injury, we evaluated the inflammatory profiles (TNF-α, IL-1β, and IL-6) of liver tissues. The liver tissues were homogenized and centrifuged for 10 min (12,000 × g, 4 °C). The supernatants were assayed using an eBiosciences ELISA Kit (San Diego, CA, USA) according to the manufacturer’s protocols. After adding the stopping solution, the absorbance at 450 nm was measured using an ELISA reader (TECAN infinite 200).
Measurement of Malondialdehyde (MDA) and Superoxide Dismutase (SOD) Oxidative Markers in Liver Homogenates
Liver tissues were homogenized in 10 volumes of ice-cold 10% trichloroacetic acid and centrifuged at 1000 × g and 4 °C for 15 min. The supernatant was used to evaluate the MDA and SOD levels. We measured MDA production as an indicator of lipid peroxidation using a Bioxytech MDA-586 Kit (OxisResearch, Portland, OR, USA). SOD was determined using a spectrophotometric method, which is a modification of the Ellman procedure. All manipulations were performed according to the manufacturer’s protocols.
Western Blot Analysis
The cytoplasmic and nuclear proteins were extracted using cytoplasmic and nuclear protein extraction kits (BIOTOOLS Co., Ltd., New Taipei City, Taiwan) according to the manufacturer’s protocol for Nrf2. Equal quantities of proteins from each group were separated using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Schleicher & Schuell, Middlesex, UK). Subsequently, the membrane was blocked with 5% fat-free dry milk for 2 h and rinsed 3 times using tris-buffered saline with 1% Tween 20 (TBST). The membrane was incubated with primary antibodies for ERK, phospho-ERK, JNK, phospho-JNK, p38, phospho-p38, and Nrf2 (Cell Signaling Technology, Danvers, MA, USA) overnight at 4 °C. After washing thoroughly with TBST for 5 min, the membranes were incubated with a horseradish peroxidase-conjugated secondary antibody for 1 h (Cell Signaling Technology, MA, USA). Then, the proteins were detected using an enhanced chemiluminescence system (Amersham, Piscataway, NJ, USA). All western blot analyses were performed at least three times.
Statistical Analysis
All values are expressed as means ± standard errors of the means. Calculations were performed with GraphPad Prism 6.0 software (GraphPad Software Inc., San Diego, CA, USA). The data were analyzed using a one-way analysis of variance, followed by post hoc Tukey–Kramer multiple comparison tests. The significance level was set at p < 0.05 for all tests.
RESULTS
Gastrodin Decreases APAP-Induced Liver Injury
Mice liver ALT levels were increased after a toxic dose of APAP (300 mg/kg) injection compared to the control group (Fig. 1a). At 30 min after exposure to APAP, treatment with gastrodin significantly decreased ALT enzyme activity. ALT levels were significantly lower in the 3 different gastrodin treatment groups (15, 30, and 45 mg/kg) than those in the APAP-only group. AST and GGT are another two enzymes associated with a hallmark of liver function. Levels of AST and GGT were significantly increased in the APAP-only group compared with the control group, and after treatment with gastrodin, these enzymes were improved compared with the APAP-only group (Fig. 1b and c). Effects of gastrodin appear to be more obvious when administered at a dose of 30 or 45 mg/kg. H&E staining analysis of the liver parenchyma from the APAP group showed extensive liver damage with centrilobular necrosis and fatty infiltration (Fig. 1e). Gastrodin treatment (30 or 45 mg/kg) clearly alleviated liver injury with reduced hepatocyte degeneration and decreased necrotic area. The extent of liver necrotic area also confirmed these findings (Fig. 1d). The percentage of liver tissue with necrotic damage was markedly increased in APAP-only group. After gastrodin treatment, the mice showed similar trend and decreased liver necrotic areas compared with the APAP-only mice.

Gastrodin protects against APAP-induced liver injury. Mice were administered with APAP (300 mg/kg, i.p.) alone or with different concentrations of gastrodin (15, 30, and 45 mg/kg) 30 min after APAP injection. Serum was obtained 16 h after APAP injection. The serum a ALT, b AST, and c GGT are expressed as means ± SEM (n = 6 mice/group). **p < 0.01, ***p < 0.005 vs. control; ##p < 0.01, ###p < 0.005 vs. APAP alone. d Bar graph showed quantitative analysis of the percentage of necrotic area. Each value is mean ± SEM of 6 mice per group. ***p < 0.05 vs. control; ###p < 0.005 vs. APAP alone. e H&E staining of the liver tissues from six groups. Typical histological images were chosen from each experimental group (50 × and 100 × magnifications). Black arrows indicate hepatocyte necrosis. Abbreviations: CV, central vein.
Gastrodin Decreases Neutrophil Recruitment in APAP-Induced Liver Injury
To further investigate which cells contribute to inflammation, we used liver MPO expression as a biomarker of neutrophil accumulation and identified neutrophils via immunohistochemistry detection of the liver tissue with a granulocyte-specific marker, Ly6G antibody. Liver MPO expression was significantly increased at 16 h post-APAP (300 mg/kg) injection compared to that of the control group (p < 0.005, Fig. 2a), which indicated that neutrophil infiltration into the liver was induced by APAP. At 30 min after APAP administration, the gastrodin-treated groups (15, 30, and 45 mg/kg) showed significantly reduced liver MPO activity compared to the APAP-only group (p < 0.01 or 0.005). Moreover, we investigated the degree of neutrophil infiltration in the liver tissues via immunohistochemistry staining (Fig. 2c). As expected, the number of Ly6G-positive cells was significantly higher in the APAP-only mice compared to the control mice (Fig. 2b) and demonstrated dominant infiltrated neutrophils in the necrotic area. The gastrodin-treated mice (15, 30, and 45 mg/kg) showed significantly reduced liver neutrophil accumulation in the liver parenchyma relative to APAP-only mice.

Effects of gastrodin on liver MPO activity and neutrophil infiltration after APAP overdose. Mice were administered with APAP (300 mg/kg, i.p.) alone or with different concentrations of gastrodin (15, 30, and 45 mg/kg) 30 min after APAP injection. Liver tissues were obtained 16 h after APAP injection. a The liver MPO activity data are expressed as means ± SEM (n = 6 mice/group). *p < 0.05, ***p < 0.005 vs. control; ##p < 0.01, ###p < 0.005 vs. APAP alone. b and c Immunohistochemistry staining of the liver tissues with anti-Ly6G antibody (black arrows) from six groups. Typical images were chosen from each experimental group (200 × magnifications). Bar graph showed quantitative analysis of Ly6G-positive neutrophils under high power field in the injury liver. Each value is mean ± SEM of 6 mice per group. *p < 0.05, ***p < 0.05 vs. control; ###p < 0.005 vs. APAP alone. Abbreviations: CV, central vein.
Gastrodin Decreases the APAP-Induced Inflammatory Response in Liver Tissues
Essential pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, are involved in the progression of APAP-induced liver injury. In our study, as shown in Fig. 3, APAP (300 mg/kg) injection caused a significant increase in these cytokines after 16 h compared to those in the normal group (p < 0.01). However, gastrodin treatment (15, 30, and 45 mg/kg) significantly decreased these cytokine levels, which were induced by a toxic dose of APAP after 30 min. These results indicate that gastrodin treatment attenuates the production and release of these cytokines in APAP-induced hepatotoxicity.

Effects of gastrodin on the inflammatory cytokine expression levels of a TNF-α, b IL-1β, and c IL-6 in APAP-treated mice. Mice were administered with APAP (300 mg/kg, i.p.) alone or with different concentrations of gastrodin (15, 30, and 45 mg/kg) 30 min after APAP injection. Liver tissues were obtained 16 h after APAP injection. The liver inflammatory cytokine data are expressed as means ± SEM (n = 6 mice/group). **p < 0.01 vs. control; #p < 0.05, ##p < 0.01, ###p < 0.005 vs. APAP alone.
Gastrodin Decreases APAP-Induced Oxidative Stress
APAP overdose results in oxidative stress injury. In order to investigate the protective mechanisms of gastrodin, we used MDA and SOD as indicators to evaluate the expression of oxidative stress in liver tissues. MDA, a lipid peroxidation product, is frequently utilized to evaluate the degree of oxidative stress. Compared to the control group, MDA concentrations were significantly increased in the APAP group (p < 0.01) (Fig. 4a). After treatment with gastrodin (15 mg/kg), MDA levels were not significantly decreased relative to those in the APAP-only mice. However, treatment with higher doses of gastrodin (30 and 45 mg/kg) significantly decreased the MDA levels in mice exposed to APAP (p < 0.05). On the other hand, oxidative stress caused by excess ROS formation decreased the activity of SOD, a liver antioxidant defense enzyme. SOD contents were significantly decreased after APAP overdose compared to the control group (p < 0.05) (Fig. 4b). Treatment with gastrodin (30 and 45 mg/kg) restored hepatic SOD activity (p < 0.01, p < 0.005). These results suggest that gastrodin may suppress the oxidative stress injury caused by APAP.

Gastrodin treatment inhibits oxidative stress in APAP-induced liver injury. Mice were administered with APAP (300 mg/kg, i.p.) alone or with different concentrations of gastrodin (15, 30, and 45 mg/kg) 30 min after APAP injection. Liver tissues were obtained 16 h after APAP injection. The liver tissue a MDA and b SOD data are expressed as means ± SEM (n = 6 mice/group). *p < 0.05, **p < 0.01 vs. control; #p < 0.05, ##p < 0.01, ###p < 0.005 vs. APAP alone.
Gastrodin Regulates the ERK and JNK Signaling Pathways in APAP-Induced Liver Injury
To explore the protective roles of gastrodin in APAP-induced hepatotoxicity, we measured the activity of the hepatic MAPK protein family, including ERK, JNK, and p38 kinase. The ERK, JNK, and p38 expression levels, as determined by their phosphorylation levels, were analyzed by western blot analysis. The results revealed that APAP significantly increased ERK and JNK phosphorylation compared to the control group (Fig. 5a and b), but the p38 protein expression levels displayed no significant differences (data not shown). Gastrodin treatment at 30 min after APAP overdose was observed to reverse these phosphorylated ERK and JNK expression levels. These results show that gastrodin exerts a potential protective effect against APAP-induced liver damage through downregulation of the ERK/JNK MAPK signaling pathways.

Western blot analysis of the effects of gastrodin on hepatic a ERK and b JNK expression levels after APAP overdose. Mice were administered with APAP (300 mg/kg, i.p.) alone or with different concentrations of gastrodin (15, 30, and 45 mg/kg) 30 min after APAP injection. Liver tissues were obtained 16 h after APAP injection. The expression levels of ERK and JNK were detected by western blot analysis. Equal protein loading is illustrated by the β-actin bands in all lanes. The bands were analyzed using densitometry, and each value represents the mean ± SEM (n = 6 mice/group). **p < 0.01, ***p < 0.005 vs. control; #p < 0.05, ##p < 0.01, ###p < 0.005 vs. APAP alone.
Hepatic Expression of Nrf2 in APAP-Induced Liver Injury
To investigate the possible anti-oxidant mechanism of gastrodin in APAP-induced hepatotoxicity, we performed immunohistochemistry staining and western blot analysis with the Nrf2 antibody in liver tissues. These results unveiled that gastrodin treatment (15, 30, and 45 mg/kg) led to increased Nrf2 accumulation in the nuclei of hepatocytes compared to the APAP-only treatment group (Fig. 6a and c) (p < 0.05). In addition, western blot analysis showed that the gastrodin (30 and 45 mg/kg)-treated groups had significantly lower Nrf2 expression levels in the cytoplasm compared to the APAP group (p < 0.05) (Fig. 6b). These results indicate that gastrodin treatment exerts protective effects against oxidative stress due to APAP overdose, which is attributable to Nrf2 nuclear translocation.

Effects of gastrodin treatment on Nrf2 activation in APAP-induced liver injury. Mice were administered with APAP (300 mg/kg, i.p.) alone or with different concentrations of gastrodin (15, 30, and 45 mg/kg) 30 min after APAP injection. Liver tissues were obtained 16 h after APAP injection. a Immunohistochemical staining of liver Nrf2 expression (brown) from six groups. Typical images were chosen from each experimental group (400 × magnification; black arrows indicate nucleus-positive cells). The expression levels of b cytoplasm and c nucleus Nrf2 were detected by western blot analysis. GAPDH and histone H3 were used as the internal control for normalization of cytoplasmic and nuclear protein loading. The bands were analyzed using densitometry, and each value represents the mean ± SEM (n = 6 mice/group). **p < 0.01 vs. control; #p < 0.05 vs. APAP alone. Abbreviations: CV, central vein.
DISCUSSION
Acute overdose of APAP-induced hepatotoxicity is the most common cause of liver damage and acute liver failure. However, the therapeutic options for APAP-induced acute liver failure remain limited. The aim of our study was to investigate the protective effects of gastrodin in APAP-induced hepatotoxicity using a mouse model. Our results showed that at 16 h post-APAP overdose liver injury, serum ALT, hepatic MPO, oxidative stress, and inflammatory cytokine expression levels were increased. Gastrodin treatment significantly improved these hepatic parameters. In addition, gastrodin treatment also increased nuclear Nrf2 expression and antioxidant enzyme levels, such as SOD, as well as decreased ERK and JNK phosphorylation.
The innate immune response plays a key role in APAP-induced liver injury. When excess electrophilic APAP metabolic product NAPQI conjugates with the cellular GSH antioxidant system, the GSH content causes a sharp depletion, which eventually results in mitochondrial dysfunction and hepatocyte necrosis [21]. Then, activated resident macrophages release inflammatory cytokines and chemokines to recruit infiltrating macrophages and neutrophils into the hepatic vasculature [22]. Previous studies have shown that these inflammatory mediators, such as TNF-α, IL-1β, and IL-6, are associated with many inflammatory diseases and mediated the maturation of infiltrating macrophages [23,24,25]. The formation of cytokines significantly increases in drug-induced liver injury models [26, 27]. Previous evidence has also illustrated that gastrodin exerts an anti-neuroinflammatory injury effect after cerebral ischemia via downregulation of macrophage activation [28]. In addition, neutrophil-driven hepatocytes injury has also been shown in ischemia–reperfusion [29] and alcoholic hepatitis [30] liver injury. Past studies have demonstrated that the severity of APAP-induced hepatotoxicity is amplified by neutrophil accumulation [31]. Hepatocyte injury after APAP overdose is accompanied by increased release of cytokines and neutrophil activation as well as transmigration into the hepatic parenchyma. In our study, early treatment with gastrodin after APAP-induced damage effectively reduced inflammatory cytokine expression levels (TNF-α, IL-1β, and IL-6), macrophage and neutrophil infiltration, and hepatic MPO activity. These results suggest that gastrodin treatment after APAP-induced hepatotoxicity alleviates the progression and severity of inflammation.
Oxidative stress can elicit MAPK activation in APAP-induced liver injury. The intracellular signaling pathways can further control oxidative stress response and cytokine production. The MAPK family, including ERK and JNK, is a serine-threonine protein kinase family that is associated with regulating inflammatory mediators and cell death. JNK is an important modulator of mitochondrial permeabilization. JNK activation causes mitochondrial dysfunction and promotes ROS formation in APAP overdose [32]. Knockdown of JNK gene expression protects against APAP toxicity [33]. Previous studies have reported that oxidative stress can also activate the JNK signaling pathway through inhibition of JNK phosphatase in APAP treatment [34]. ERK is another important member of the MAPK family that is related to oxidative stress formation and the apoptotic pathway [35]. The protective effects of APAP-induced hepatotoxicity have been observed to be mediated by decreased ROS production and suppression of the ERK signaling pathway [36]. A previous study showed that gastrodin exerts anti-inflammatory responses in cardiomyocytes through inhibition of the MAPK signaling pathways [37]. In our study, phospho-ERK and phospho-JNK expression levels significantly increased after APAP administration based on western blot analysis. Gastrodin treatment effectively alleviated this phosphorylation, thus decreasing the ERK and JNK expression levels. These data suggest that gastrodin exerts protective effects against APAP-induced hepatotoxicity by inhibiting the ERK/JNK MAPK pathways.
Oxidative stress is considered to be the major cellular event in APAP-induced liver injury. In APAP overdose, excessive NAPQI depletes cellular GSH and generates free radicals and superoxide anion, which results in oxidative stress [6]. Oxidative stress can activate pro-inflammatory gene expression, triggering inflammatory cells to produce more ROS, which forms a vicious cycle in the development of liver damage [38]. Previous reports have demonstrated that gastrodin may exert a hepatoprotective effect against H2O2-induced oxidative stress in liver sinusoidal endothelial cells [39]. Pretreatment of gastrodin can protect against mouse liver ischemia–reperfusion injury through anti-apoptotic and anti-oxidant mechanisms [40]. A previous study also reported that Gastrodia elata extracts have organ protective effects against APAP-induced liver and kidney toxic injury via anti-inflammatory and anti-oxidative activities [41]. In our study, excessive APAP caused increased MDA formation, indicating accumulation of lipid peroxidation and ROS production. Gastrodin treatment significantly reduced this APAP-induced change and increased the levels of antioxidant proteins, such as SOD. These results suggest that gastrodin may relieve APAP-induced liver injury via its anti-oxidant effects.
APAP toxicity can cause ROS formation and deterioration of anti-oxidant capacity. Nrf2 is a major transcription factor and regulator of oxidative stress. It plays essential roles in regulating the expression of antioxidant genes to eliminate ROS. In its inactivated form, Nrf2 is normally sequestered in the cytoplasm. After stimulation by oxidative stress, it can translocate from the cytoplasm into the nucleus and bind with the antioxidant response element to induce the transcription of anti-oxidant genes, such as SOD and heme oxygenase-1 (HO-1) [12]. HO-1 functions as a suppressor of the TNF-α-mediated signaling pathway by decreasing intracellular ROS production in airway inflammation in humans and mice [42]. Previous studies have shown that in Nrf2-knockout mice, APAP-induced liver injury is more serious compared to that in wild-type mice. The protective effects of APAP-induced hepatotoxicity occur via attenuation of liver oxidative stress and activation of Nrf2 [43, 44]. Previous evidence has also demonstrated that APAP inhibits Nrf2 nuclear translocation, making liver cells highly susceptible to oxidative stress [45]. In our study, we investigated the importance of Nrf-2 in gastrodin against oxidative stress in APAP-induced hepatotoxicity. Previous reports have demonstrated that in a model of acute lung injury, gastrodin exerts anti-inflammatory effects via upregulation of Nrf2 and HO-1 expression levels [46]. Another study also found that gastrodin reduces the inflammatory response and oxidative stress via activation of the Nrf2 signaling pathway in a model of nonalcoholic fatty liver disease [47]. In this study, we showed that gastrodin can increase the expression of Nrf2 and facilitate translocation into the nucleus. These results suggest that activation of Nrf2 protein by gastrodin may contribute to its protective effects against APAP-induced liver injury.
CONCLUSIONS
This study demonstrated that gastrodin treatment attenuates APAP-induced liver injury in a mouse model. Its anti-inflammatory and anti-oxidant mechanisms involve suppressing ERK/JNK MAPK signaling pathways while activating Nrf2 signaling pathways. Our findings suggest that gastrodin may develop as an effective preventive and treatment method in the clinical management of APAP-induced hepatotoxicity in the future.
Change history
10 May 2022
A Correction to this paper has been published: https://doi.org/10.1007/s10753-022-01679-0
References
Hinson, J.A., D.W. Roberts, and L.P. James. 2010. Mechanisms of acetaminophen-induced liver necrosis. Handbook of Experimental Pharmacology 196: 369–405. https://doi.org/10.1007/978-3-642-00663-0_12.
Kao, L.W., M.A. Kirk, R.B. Furbee, N.H. Mehta, J.R. Skinner, and E.J. Brizendine. 2003. What is the rate of adverse events after oral N-acetylcysteine administered by the intravenous route to patients with suspected acetaminophen poisoning? Annals of Emergency Medicine 42 (6): 741–750. https://doi.org/10.1016/s0196-0644(03)00508-0.
Singh, D., W.C. Cho, and G. Upadhyay. 2015. Drug-induced liver toxicity and prevention by herbal antioxidants: An overview. Frontiers in Physiology 6: 363. https://doi.org/10.3389/fphys.2015.00363.
Bunchorntavakul, C., and K.R. Reddy. 2013. Acetaminophen-related hepatotoxicity. Clinical Liver Disease 17 (4):587–607, viii. doi:https://doi.org/10.1016/j.cld.2013.07.005
Jaeschke, H., M.R. McGill, C.D. Williams, and A. Ramachandran. 2011. Current issues with acetaminophen hepatotoxicity—a clinically relevant model to test the efficacy of natural products. Life Sciences 88 (17–18): 737–745. https://doi.org/10.1016/j.lfs.2011.01.025.
McGill, M.R., M.R. Sharpe, C.D. Williams, M. Taha, S.C. Curry, and H. Jaeschke. 2012. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. The Journal of Clinical Investigation 122 (4): 1574–1583. https://doi.org/10.1172/JCI59755.
Jaeschke, H., C.D. Williams, A. Ramachandran, and M.L. Bajt. 2012. Acetaminophen hepatotoxicity and repair: The role of sterile inflammation and innate immunity. Liver International 32 (1): 8–20. https://doi.org/10.1111/j.1478-3231.2011.02501.x.
Krenkel, O., J.C. Mossanen, and F. Tacke. 2014. Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surgery and Nutrition 3 (6): 331–343. https://doi.org/10.3978/j.issn.2304-3881.2014.11.01.
Wang, A.Y., L.H. Lian, Y.Z. Jiang, Y.L. Wu, and J.X. Nan. 2010. Gentiana manshurica Kitagawa prevents acetaminophen-induced acute hepatic injury in mice via inhibiting JNK/ERK MAPK pathway. World Journal of Gastroenterology 16 (3): 384–391. https://doi.org/10.3748/wjg.v16.i3.384.
Latchoumycandane, C., C.W. Goh, M.M. Ong, and U.A. Boelsterli. 2007. Mitochondrial protection by the JNK inhibitor leflunomide rescues mice from acetaminophen-induced liver injury. Hepatology 45 (2): 412–421. https://doi.org/10.1002/hep.21475.
Yu, S.M., and S.J. Kim. 2015. The thymoquinone-induced production of reactive oxygen species promotes dedifferentiation through the ERK pathway and inflammation through the p38 and PI3K pathways in rabbit articular chondrocytes. International Journal of Molecular Medicine 35 (2): 325–332. https://doi.org/10.3892/ijmm.2014.2014.
Gum, S.I., and M.K. Cho. 2013. Recent updates on acetaminophen hepatotoxicity: The role of nrf2 in hepatoprotection. Toxicological Research 29 (3): 165–172. https://doi.org/10.5487/TR.2013.29.3.165.
Liu, X.M., K.J. Peyton, A.R. Shebib, H. Wang, and W. Durante. 2011. Compound C stimulates heme oxygenase-1 gene expression via the Nrf2-ARE pathway to preserve human endothelial cell survival. Biochemical Pharmacology 82 (4): 371–379. https://doi.org/10.1016/j.bcp.2011.05.016.
Donovan, E.L., J.M. McCord, D.J. Reuland, B.F. Miller, and K.L. Hamilton. 2012. Phytochemical activation of Nrf2 protects human coronary artery endothelial cells against an oxidative challenge. Oxidative Medicine and Cellular Longevity 2012: 132931. https://doi.org/10.1155/2012/132931.
Lv, H., Q. Xiao, J. Zhou, H. Feng, G. Liu, and X. Ci. 2018. Licochalcone A upregulates Nrf2 antioxidant pathway and thereby alleviates acetaminophen-induced hepatotoxicity. Frontiers in Pharmacology 9: 147. https://doi.org/10.3389/fphar.2018.00147.
Liu, Y., J. Gao, M. Peng, H. Meng, H. Ma, P. Cai, Y. Xu, Q. Zhao, and G. Si. 2018. A review on central nervous system effects of gastrodin. Frontiers in Pharmacology 9: 24. https://doi.org/10.3389/fphar.2018.00024.
Dai, J.N., Y. Zong, L.M. Zhong, Y.M. Li, W. Zhang, L.G. Bian, Q.L. Ai, Y.D. Liu, J. Sun, and D. Lu. 2011. Gastrodin inhibits expression of inducible NO synthase, cyclooxygenase-2 and proinflammatory cytokines in cultured LPS-stimulated microglia via MAPK pathways. PLoS ONE 6 (7): e21891. https://doi.org/10.1371/journal.pone.0021891.
Fang, H., J.C. Zhang, M. Yang, H.F. Li, J.P. Zhang, F.X. Zhang, Q.Y. Wang, R.R. Wang, and J. Liu. 2016. Perfusion of gastrodin in abdominal aorta for alleviating spinal cord ischemia reperfusion injury. Asian Pacific Journal of Tropical Medicine 9 (7): 688–693. https://doi.org/10.1016/j.apjtm.2016.05.007.
Fu, S., L. Chen, Y. Wu, Y. Tang, L. Tang, Y. Zhong, S. Wang, H. Liu, X. Wang, and A. Chen. 2018. Gastrodin pretreatment alleviates myocardial ischemia/reperfusion injury through promoting autophagic flux. Biochemical and Biophysical Research Communications 503 (4): 2421–2428. https://doi.org/10.1016/j.bbrc.2018.06.171.
Li, X.X., Z.H. Jiang, B. Zhou, C. Chen, and X.Y. Zhang. 2019. Hepatoprotective effect of gastrodin against alcohol-induced liver injury in mice. Journal of Physiology and Biochemistry 75 (1): 29–37. https://doi.org/10.1007/s13105-018-0647-8.
McGill, M.R., M. Lebofsky, H.R. Norris, M.H. Slawson, M.L. Bajt, Y. Xie, C.D. Williams, D.G. Wilkins, D.E. Rollins, and H. Jaeschke. 2013. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: Dose-response, mechanisms, and clinical implications. Toxicology and Applied Pharmacology 269 (3): 240–249. https://doi.org/10.1016/j.taap.2013.03.026.
Holt, M.P., L. Cheng, and C. Ju. 2008. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. Journal of Leukocyte Biology 84 (6): 1410–1421. https://doi.org/10.1189/jlb.0308173.
Bradley, J.R. 2008. TNF-mediated inflammatory disease. The Journal of Pathology 214 (2): 149–160. https://doi.org/10.1002/path.2287.
Sierra-Filardi, E., C. Nieto, A. Dominguez-Soto, R. Barroso, P. Sanchez-Mateos, A. Puig-Kroger, M. Lopez-Bravo, J. Joven, C. Ardavin, J.L. Rodriguez-Fernandez, C. Sanchez-Torres, M. Mellado, and A.L. Corbi. 2014. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: Identification of CCL2/CCR2-dependent gene expression profile. The Journal of Immunology 192 (8): 3858–3867. https://doi.org/10.4049/jimmunol.1302821.
Dinarello, C.A. 2018. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunological Reviews 281 (1): 8–27. https://doi.org/10.1111/imr.12621.
Mizrahi, M., T. Adar, G. Lalazar, D. Nachman, M. El Haj, A. Ben Ya’acov, Y. Lichtenstein, Y. Shabat, D. Kanovich, L. Zolotarov, and Y. Ilan. 2018. Glycosphingolipids prevent APAP and HMG-CoA reductase inhibitors-mediated liver damage: A novel method for “safer drug” formulation that prevents drug-induced liver injury. Journal of Clinical and Translational Hepatology 6 (2): 127–134. https://doi.org/10.14218/JCTH.2017.00071.
Wang, Z., W. Hao, J. Hu, X. Mi, Y. Han, S. Ren, S. Jiang, Y. Wang, X. Li, W. Li. 2019. Maltol improves APAP-induced hepatotoxicity by inhibiting oxidative stress and inflammation response via NF-kappaB and PI3K/Akt signal pathways. Antioxidants (Basel) 8 (9). doi:https://doi.org/10.3390/antiox8090395
Mao, X.N., H.J. Zhou, X.J. Yang, L.X. Zhao, X. Kuang, C. Chen, D.L. Liu, and J.R. Du. 2017. Neuroprotective effect of a novel gastrodin derivative against ischemic brain injury: Involvement of peroxiredoxin and TLR4 signaling inhibition. Oncotarget 8 (53): 90979–90995. https://doi.org/10.18632/oncotarget.18773.
Jaeschke H. 2006. Mechanisms of liver injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. American Journal of Physiology-Gastrointestinal and Liver Physiology 290 (6):G1083–1088. doi:https://doi.org/10.1152/ajpgi.00568.2005
Bertola, A., O. Park, and B. Gao. 2013. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: A critical role for E-selectin. Hepatology 58 (5): 1814–1823. https://doi.org/10.1002/hep.26419.
Marques, P.E., S.S. Amaral, D.A. Pires, L.L. Nogueira, F.M. Soriani, B.H. Lima, G.A. Lopes, R.C. Russo, T.V. Avila, J.G. Melgaco, A.G. Oliveira, M.A. Pinto, C.X. Lima, A.M. De Paula, D.C. Cara, M.F. Leite, M.M. Teixeira, and G.B. Menezes. 2012. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 56 (5): 1971–1982. https://doi.org/10.1002/hep.25801.
Saito, C., J.J. Lemasters, and H. Jaeschke. 2010. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicology and Applied Pharmacology 246 (1–2): 8–17. https://doi.org/10.1016/j.taap.2010.04.015.
Gunawan, B.K., Z.X. Liu, D. Han, N. Hanawa, W.A. Gaarde, and N. Kaplowitz. 2006. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 131 (1): 165–178. https://doi.org/10.1053/j.gastro.2006.03.045.
Bourdi, M., M.C. Korrapati, M. Chakraborty, S.B. Yee, and L.R. Pohl. 2008. Protective role of c-Jun N-terminal kinase 2 in acetaminophen-induced liver injury. Biochemical and Biophysical Research Communications 374 (1): 6–10. https://doi.org/10.1016/j.bbrc.2008.06.065.
Nowak, G. 2002. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. Journal of Biological Chemistry 277 (45): 43377–43388. https://doi.org/10.1074/jbc.M206373200.
Noh, J.R., Y.H. Kim, J.H. Hwang, G.T. Gang, K.S. Kim, I.K. Lee, B.S. Yun, and C.H. Lee. 2013. Davallialactone protects against acetaminophen overdose-induced liver injuries in mice. Food and Chemical Toxicology 58: 14–21. https://doi.org/10.1016/j.fct.2013.04.005.
Yang, P., Y. Han, L. Gui, J. Sun, Y.L. Chen, R. Song, J.Z. Guo, Y.N. Xie, D. Lu, and L. Sun. 2013. Gastrodin attenuation of the inflammatory response in H9c2 cardiomyocytes involves inhibition of NF-kappaB and MAPKs activation via the phosphatidylinositol 3-kinase signaling. Biochemical Pharmacology 85 (8): 1124–1133. https://doi.org/10.1016/j.bcp.2013.01.020.
Jaeschke, H. 2011. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. Journal of Gastroenterology and Hepatology 26 (Suppl 1): 173–179. https://doi.org/10.1111/j.1440-1746.2010.06592.x.
Zhang, H., B. Yuan, H. Huang, S. Qu, S. Yang, and Z. Zeng. 2018. Gastrodin induced HO-1 and Nrf2 up-regulation to alleviate H2O2-induced oxidative stress in mouse liver sinusoidal endothelial cells through p38 MAPK phosphorylation. Brazilian Journal of Medical and Biological Research 51 (10): e7439. https://doi.org/10.1590/1414-431X20187439.
Sun, B., J. Jiang, X. Zhu, D. Yang, Z. Cui, Y. Zhang, M. Zhang, Y. Qian, R. Liu, and W. Yang. 2021. Protective effects of gastrodin pretreatment on mouse hepatic ischemia-reperfusion occurring through antioxidant and anti-apoptotic mechanisms. Experimental and Therapeutic Medicine 21 (5): 471. https://doi.org/10.3892/etm.2021.9902.
Seok, P.R., J.H. Kim, H.R. Kwon, J.S. Heo, J.R. Choi, and J.H. Shin. 2018. Protective effects of Gastrodia elata Blume on acetaminophen-induced liver and kidney toxicity in rats. Food Sci Biotechnol 27 (5): 1445–1454. https://doi.org/10.1007/s10068-018-0374-5.
Lee, I.T., S.F. Luo, C.W. Lee, S.W. Wang, C.C. Lin, C.C. Chang, Y.L. Chen, L.Y. Chau, and C.M. Yang. 2009. Overexpression of HO-1 protects against TNF-alpha-mediated airway inflammation by down-regulation of TNFR1-dependent oxidative stress. American Journal of Pathology 175 (2): 519–532. https://doi.org/10.2353/ajpath.2009.090016.
Yan, H., Z. Huang, Q. Bai, Y. Sheng, Z. Hao, Z. Wang, and L. Ji. 2018. Natural product andrographolide alleviated APAP-induced liver fibrosis by activating Nrf2 antioxidant pathway. Toxicology 396–397: 1–12. https://doi.org/10.1016/j.tox.2018.01.007.
Wang, L., W. Wei, Q. Xiao, H. Yang, and X. Ci. 2019. Farrerol ameliorates APAP-induced hepatotoxicity via activation of Nrf2 and autophagy. International Journal of Biological Sciences 15 (4): 788–799. https://doi.org/10.7150/ijbs.30677.
Gao, Y., S. Chu, Z. Zhang, W. Zuo, C. Xia, Q. Ai, P. Luo, P. Cao, and N. Chen. 2017. Early stage functions of mitochondrial autophagy and oxidative stress in acetaminophen-induced liver injury. Journal of Cellular Biochemistry 118 (10): 3130–3141. https://doi.org/10.1002/jcb.25788.
Zhang, Z., J. Zhou, D. Song, Y. Sun, C. Liao, and X. Jiang. 2017. Gastrodin protects against LPS-induced acute lung injury by activating Nrf2 signaling pathway. Oncotarget 8 (19): 32147–32156. https://doi.org/10.18632/oncotarget.16740.
Qu, L.L., B. Yu, Z. Li, W.X. Jiang, J.D. Jiang, and W.J. Kong. 2016. Gastrodin ameliorates oxidative stress and proinflammatory response in nonalcoholic fatty liver disease through the AMPK/Nrf2 pathway. Phytotherapy Research 30 (3): 402–411. https://doi.org/10.1002/ptr.5541.
Funding
This work was supported in part by grants from the National Science Council (MOST 108–2314-B-182A-059-MY2) and Chang Gung Memorial Hospital (CMRPG3J1561-2) to Fu-Chao Liu and Chang Gung Memorial Hospital (CORPG3G0601) to Chia-Chih Liao.
Author information
Authors and Affiliations
Contributions
C.C.L., H.P.Y., and F.C.L. conceived and designed this study. A.H.C. and H.C.L. carried out experiments. C.C.L. and L.M.H. collected and analyzed data. L.M.H. performed statistical analysis. C.C.L. and H.P.Y. wrote the manuscript, which was critically reviewed and revised by H.C.L. and F.C.L. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
All procedures carried out in this study were approved by the Institutional Animal Care and Use Committee of Chang Gung Memorial Hospital (Taoyuan, Taiwan) (No. 2019091102). All animal experiments were performed according to the guidelines of the Animal Welfare Act and the Guide for Care and Use of Laboratory Animals from the National Institutes of Health.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Gastrodin Decreases Hepatotoxicity via MAPK and Nrf2 Signaling Pathways
Chia-Chih Liao and Huang-Ping Yu were equal contributors and co-first authors.
The original online version of this article was revised: The originally published version of this article contained mistakes. The marks "*" and "#" were missing in Figures 3, 4, 5 and 6.
Rights and permissions
About this article

Cite this article
Liao, CC., Yu, HP., Chou, AH. et al. Gastrodin Alleviates Acetaminophen-Induced Liver Injury in a Mouse Model Through Inhibiting MAPK and Enhancing Nrf2 Pathways. Inflammation 45, 1450–1462 (2022). https://doi.org/10.1007/s10753-021-01557-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-021-01557-1