
Abstract
The present study aimed to investigate the role of Forkhead box protein C2 (Foxc2) in oxidized low-density lipoprotein (ox-LDL)-induced macrophages and identify the potential mechanisms. RAW264.7 cells, the murine macrophage cell line, were stimulated by ox-LDL, and cell proliferation was examined. The levels of inflammation- and oxidative stress-related markers were detected using kits after induction with ox-LDL. Subsequently, the expression of Foxc2 was measured using Western blotting. After transfection with Foxc2 pcDNA3.1, intracellular lipid droplets were examined using oil red O staining. The levels of total cholesterol (TC), free cholesterol (FC), inflammatory cytokines, and oxidative stress markers were determined. Moreover, apoptosis of RAW264.7 cells was detected using flow cytometry, and apoptosis-related proteins were measured using Western blotting. Angiopoietin-like protein 2 (Angptl2) was predicted as a target gene of Foxc2. Therefore, the expression of Angptl2 was examined after Foxc2 overexpression in ox-LDL-induced RAW264.7 cells. Then, the changes of intracellular lipid droplets, TC, FC, inflammatory cytokines, oxidative stress factors, and cell apoptosis were detected after Angptl2 overexpression or co-transfection with Foxc2 and Angptl2 pcDNA3.1. The results revealed that ox-LDL induction inhibited proliferation of RAW264.7 cells and promoted the release of inflammatory factors. Importantly, the expression of Foxc2 was obviously decreased after stimulation by ox-LDL. Foxc2 overexpression suppressed lipid accumulation, TC, FC levels, inflammation, oxidative stress, and apoptosis induced by ox-LDL, whereas these inhibitory effects were relieved after co-transfection with Angptl2 pcDNA3.1. These findings demonstrated that Foxc2 can alleviate ox-LDL-induced lipid accumulation, inflammation, and apoptosis of macrophage via regulating the expression of Angptl2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Atherosclerosis (AS) is a chronic degenerative disease of the arterial wall among aged people around the world, which contributes to the development and progression of multiple cardiovascular and cerebral vascular diseases [15, 22]. Statistical analysis has revealed that AS is a leading health threat with high morbidity and mortality to aged people [8, 19]. Recently, although a great deal of basic research and clinical therapy studies about AS have been carried out, the treatment status is still unsatisfactory [21, 35]. Therefore, investigation and development of effective strategies to treat AS is urgent and imperative.
The pathogenesis of AS is closely related to endothelial lesions, lipid deposition, inflammation, and apoptosis of macrophages [23]. Numerous studies have unveiled that oxidized low-density lipoprotein (ox-LDL) is a key atherogenic risk factor for the genesis and progression of AS [17, 36]. Ox-LDL can induce the dysfunction of endothelial cells, activate macrophages, and promote the formation of foam cells [7]. It has been well reported that macrophages are major contributors to all pathological changes during AS [16]. Macrophages can swallow normal or oxidative lipoproteins and subsequently transform themselves into foam cells, which results in the formation of the necrotic core in atheromatous plaques [19]. In addition, these foam cells initiate a cascade of inflammation by overwhelming release of pro-inflammatory cytokines that accelerates the progression of AS [3, 37]. Mounting evidence has supported that the apoptosis of macrophages in atheromatous plaques promotes inflammation and plaque instability [29, 34]. Therefore, inhibition of lipid metabolism, inflammation, and apoptosis in ox-LDL-induced macrophages might be promising for the treatment of AS.
Forkhead box protein C2 (Foxc2), a member of the forkhead/winged helix transcription factor family, plays significant roles in lots of crucial functions during development [14, 32]. Compelling evidence indicates that Foxc2 can suppress inflammation and accelerate browning of white adipose tissue via regulating leptin signal [11]. Moreover, emerging evidence supports the notion that the expression of Foxc2 was decreased in lipopolysaccharide-induced human umbilical vein endothelial cells, and Foxc2 overexpression reduced the release of inflammatory factors and cell adhesion [32]. However, the functions of Foxc2 in AS remain to be elucidated.
In the present study, murine macrophage cell line RAW264.7 cells were stimulated by ox-LDL. And then, the functions of Foxc2 in lipid accumulation, inflammation, and apoptosis of macrophage were investigated, with the hope of identifying the underlying regulatory mechanisms of Foxc2 in AS. Our study may provide a novel therapeutic strategy for AS.
MATERIALS AND METHODS
Cell Culture and Treatment
Murine macrophage cell line RAW264.7 cells were purchased from Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum in a humidified atmosphere with 5% CO2 at 37 °C. When the cells were confluent, they were exposed to 20, 40, 60, and 100 μg/ml ox-LDL (Yiyuan Biotechnology, Guangzhou, China) for 24 h. Cells untreated were used as control group.
Cell Transfection
Foxc2-overexpressing plasmid (pcDNA-Foxc2), angiopoietin like-2 (Angptl2), overexpressing plasmid (pcDNA-Angptl2) and the empty vector plasmid (pcDNA-NC) were synthesized by GenePharma (Shanghai, China). RAW264.7 cells were plated in a 6-well plate and cultured at 37 °C until 80% confluence. Subsequently, transfection with abovementioned plasmids was performed using Lipofectamine 2000 (Invitrogen, CA, USA) following the product manual. At 24 h after post-transfection, successful transfections were evaluated by RNA extraction, reverse transcription-quantitative polymerase chain reaction (RT-qPCR), and Western blot assay, respectively.
Cell Counting Kit-8 (CCK-8) Assay
The ability of cell proliferation was determined using CCK-8 kit (Beyotime, Shanghai, China). Cells were seeded in a 96-well plate (5 × 103 cells per well). Cells were stimulated with different concentrations of ox-LDL and cultured at 37 °C for 24 h. Subsequently, 100 μl CCK-8 solution was added into each well. Following incubation for further 4 h, absorbance was detected at 450 nm using a microplate reader (Bio-Tek, Winooski, VT).
Enzyme-Linked Immunosorbent Assay (ELISA) for Inflammatory Cytokines
The contents of inflammatory cytokines including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6 in culture medium were detected using commercially available ELISA Kits (Shanghai Xitang Biotechnology Co., Ltd., Shanghai, China) according to the manufacturer’s recommendations. Absorbance at 405 nm was detected using a microplate reader (Bio-Tek, Winooski, VT).
Test for the Contents of Total Cholesterol (TC) and Free Cholesterol (FC)
The levels of TC and FC in culture medium were measured using commercial kits provided by Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Evaluation of Oxidative Stress
The levels of oxidative stress-related biomarkers including reactive oxygen species (ROS), malondialdehyde (MDA), and glutathione peroxidase (GSH-Px) were examined using commercial kits following the manufacturer’s guidelines. These kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Oil Red O Staining
The changes of intracellular lipid droplets were examined using oil red O staining. After transfection, RAW264.7 cells were stimulated by ox-LDL for 24 h. Subsequently, cells were washed with PBS and fixed with 4% paraformaldehyde solution. Oil red O working solution was used to stain cells for 15 min. Then, cells were lightly counterstained with hematoxylin for 5 min. The cells’ morphology was observed by a light microscope (Olympus, Tokyo, Japan). Intracellular lipid was red and the nucleus was blue.
Cell Apoptosis Assay
Apoptosis was detected by using a cell apoptosis detection kit (KeyGEN BioTECH, China). Following transfection for 24 h, RAW264.7 cells were stained with Annexin V-PE/7AAD in the dark at room temperature. Then, cell apoptosis was analyzed by flow cytometry (BD Biosciences, NJ, USA). Finally, the data were analyzed using the FlowJo software (Becton-Dickinson-San Jose CA, USA).
RT-qPCR
Total RNA from macrophages were extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA). Then, RNA was reverse-transcribed to cDNA by a Reverse Transcription kit (Takara, Japan). qPCR was performed using SYBR Green I (Takara, Japan) on an ABI 7500 system (Applied Biosystems, Foster, CA). Specific primers were designed by RiboBio (Guangzhou, China). The expression of target genes was normalized against GAPDH. The relative fold change of target gene expression was calculated using the 2−ΔΔCt method.
Western Blotting
Total proteins were extracted from RAW264.7 cells and quantified using a BCA protein assay kit (Bio-rad, China). Then, 40 μg protein in each group was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Proteins on gels were subsequently transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, USA), which were blocked in 5% skimmed milk. These blots were incubated with diluted primary antibodies for the target proteins. Following incubation with secondary antibodies (Santa Cruz Biotechnology, CA, USA), the bands were visualized using an Odyssey Infrared Imaging Scanner (LI-COR Biosciences). Intensity of bands was examined using the Image-J software (National Institutes of Health, Bethesda, MA, USA). The protein expression was normalized to GAPDH levels to correct for loading.
Statistical Analysis
All data were expressed as the mean ± standard deviation (SD). Statistical analysis was conducted by GraphPad Prism version 6.0 (GraphPad Software, Inc.). Statistical comparisons between two groups were analyzed using Student’s t test, and the comparisons among multiple groups were performed using analysis of variance (ANOVA) followed by Turkey’s post hoc test. A value of P < 0.05 was considered statistically significant.
RESULTS
The Expression of Foxc2 Was Downregulated in Ox-LDL-Induced RAW264.7 Cells
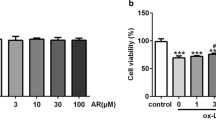
As presented in Fig. 1a, the viability of RAW264.7 cells was gradually reduced with the increasing concentration of ox-LDL (0–100 μg/mL). When cells were treated with 60 μg/mL ox-LDL, cell viability was decreased by about 50%. Based on the abovementioned result, 60 μg/mL ox-LDL was selected for the subsequent experiments. Then, the levels of inflammation-related factors were tested using ELISA. Results from Fig. 1b–d demonstrated that ox-LDL treatment enhanced the levels of TNF-α, IL-1β, and IL-6 in a concentration-dependent manner compared with the control. In addition, the expression of Foxc2 was examined by RT-qPCR, and Western blotting after RAW264.7 cells were induced by ox-LDL. Obvious downregulation of Foxc2 expression was observed in Fig. 1e, f. The results above indicated that Foxc2 was low expressed in RAW264.7 cells stimulated by ox-LDL.

The expression of Foxc2 was downregulated in ox-LDL-induced RAW264.7 cells. a Cell viability was tested using CCK-8 assay after RAW264.7 cells were treated with a series of different concentrations of ox-LDL. The contents of b TNF-α, c IL-1β, and d IL-6 were determined using ELISA kits. e RT-qPCR analysis and f Western blotting were employed to examine the expression of Foxc2 at transcription level and protein level, respectively. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
Foxc2 Overexpression Decreased the Lipid Accumulation in Ox-LDL-Induced RAW264.7 Cells
To investigate the function of Foxc2 in macrophages induced by ox-LDL, Foxc2-overexpressed plasmid was transfected into RAW264.7 cells. Results from Fig. 2a, b suggested that the expression of Foxc2 was remarkably upregulated after transfection with Foxc2 pcDNA3.1 relative to pcDNA-NC. Subsequently, the effect of Foxc2 overexpression on lipid accumulation was evaluated using oil red O staining. The result from Fig. 2c revealed that lipid droplets stained by oil red O in ox-LDL-induced macrophages were significantly increased compared with the control, while decreased after Foxc2 overexpression. Consistently, the levels of TC and FC in Foxc2 overexpression group were notably reduced relative to the ox-LDL+pcDNA-NC group (Fig. 2d, e). In light of the above findings, we proved that overexpression of Foxc2 suppressed the lipid accumulation in ox-LDL-induced RAW264.7 cells.

Foxc2 overexpression decreased the lipid accumulation in ox-LDL-induced RAW264.7 cells. The expression of Foxc2 was evaluated by a RT-qPCR and b Western blotting after transfection with pcDNA-Foxc2 in ox-LDL-induced RAW264.7 cells. c Oil red O staining was used to examine the level of lipid accumulation. The relative concentration of d TC and e FC was measured using commercial kits. ***P < 0.001 vs. control; ##P < 0.01, ###P < 0.001 vs. ox-LDL-pcDNA-NC.
Foxc2 Overexpression Inhibited Inflammation and Oxidative Stress in Ox-LDL-Induced RAW264.7 Cells
The effects of Foxc2 overexpression on inflammation and oxidative stress were assessed in the current study. Results from Fig. 3a–c uncovered that Foxc2 overexpression dramatically reduced the levels of inflammatory factors (TNF-α, IL-1β, and IL-6). Additionally, the contents of ROS and MDA were notably enhanced coupled with obviously decrease in GSH-Px activity after treatment with ox-LDL in RAW264.7 cells (Fig. 3d, f). The visualized and quantized results from Western blotting suggested that ox-LDL stimulation significantly increased the expression of related proteins of NOD-like receptor protein 3 (NLRP3) inflammasome, including NLRP3, apoptosis-associated speck-like protein (ASC), and caspase-1, whereas these trends have been reversed following transfection with Foxc2 pcDNA3.1 (Fig. 3g). These findings confirmed the proposal that Foxc2 overexpression could restrain inflammation and oxidative stress in RAW264.7 cells stimulated by ox-LDL.

Foxc2 overexpression inhibited inflammation and oxidative stress in ox-LDL-induced RAW264.7 cells. The contents of a TNF-α, b IL-1β, c IL-6, d ROS, e MDA, and the activity of f GSH-Px were detected by commercial kits. g Western blotting was utilized to assess the expression of NLRP3 inflammasome signaling proteins. ***P < 0.001 vs. control; ##P < 0.01, ###P < 0.001 vs. ox-LDL-pcDNA-NC.
Foxc2 Overexpression Suppressed Apoptosis in Ox-LDL-Induced RAW264.7 Cells
To understand the functions of Foxc2 overexpression in apoptosis of ox-LDL-stimulated RAW264.7 cells, cell apoptosis was detected using flow cytometry. As exhibited in Fig. 4a, b, the number of apoptotic cells was greatly reduced in ox-LDL-treated RAW264.7 cells compared with the control, while overexpression of Foxc2 reversed this trend. Consistent with the alteration of apoptotic rate, the level of anti-apoptotic protein Bcl-2 was downregulated, accompanied by upregulated expression of pro-apoptotic proteins Bax and cleaved-caspase-3 following ox-LDL induction, and the subsequent Foxc2 overexpression attenuated the effects of ox-LDL on apoptosis (Fig. 4c). To sum up, these data demonstrated that Foxc2 overexpression inhibited apoptosis of ox-LDL-induced RAW264.7 cells.

Foxc2 overexpression suppressed apoptosis in ox-LDL-induced RAW264.7 cells. a Apoptosis of RAW264.7 cells was examined by flow cytometry. b Apoptotic rate was qualified. c The expression of apoptosis-related proteins including Bcl-2, Bax, and cleaved caspase-3 was tested using Western blotting. ***P < 0.001 vs. control; ###P < 0.001 vs. ox-LDL-pcDNA-NC.
Angptl2 Overexpression Reversed the Effects of Foxc2 Overexpression on Lipid Accumulation in Ox-LDL-Induced RAW264.7 Cells
To determine the potential mechanisms of Foxc2 in AS, TRRUST database (https://www.grnpedia.org/trrust) was employed to predict the target regulator of Foxc2, and the result demonstrated that Angptl2 could be the target of Foxc2. Therefore, the expression of Angptl2 in ox-LDL-stimulated RAW264.7 cells was examined. As displayed in Fig. 5a, ox-LDL treatment evidently increased the expression of Angptl2 at protein level, whereas subsequent Foxc2 overexpression reduced that of it in ox-LDL-stimulated RAW264.7 cells. To further illuminate whether Foxc2 played a part in the lipid accumulation of ox-LDL-induced RAW264.7 cells via regulating Angptl2 expression, Angptl2 was overexpressed by transfection with Angptl2 plasmid (Fig. 5b, c). As illuminated in Fig. 6a, Angptl2 overexpression appeared the highest level of lipid accumulation in ox-LDL-stimulated RAW264.7 cells. Conversely, the lowest level of lipid accumulation was observed after Foxc2 overexpression in ox-LDL-stimulated RAW264.7 cells, whereas this inhibitory effect was relieved following Angptl2 overexpression. As expected, the contents of TC and FC exhibited the same alteration with abovementioned oil red O staining (Fig. 6b, c). These observations revealed that Angptl2 overexpression reversed the effects of Foxc2 overexpression on lipid accumulation in ox-LDL-induced RAW264.7 cells.

Foxc2 overexpression suppressed the expression of Angptl2 in ox-LDL-induced RAW264.7 cells. a Western blot analysis was exploited for testing the level of Angptl2. ***P < 0.001 vs. control; #P < 0.05 vs. ox-LDL-pcDNA-NC. The expression of Angptl2 was measured by b RT-qPCR and c Western blotting after transfection with pcDNA-Angptl2. **P < 0.01, ***P < 0.001 vs. control; ##P < 0.01, ###P < 0.001 vs. ox-LDL-pcDNA-NC.

Angptl2 overexpression reversed the effects of Foxc2 overexpression on lipid accumulation in ox-LDL-induced RAW264.7 cells. a Oil red O staining was applied to estimate the level of lipid accumulation. The levels of b TC and c FC were measured using commercial kits. ***P < 0.001 vs. control; ##P < 0.01, ###P < 0.001 vs. ox-LDL-pcDNA-NC; △△P < 0.01 vs. ox-LDL-pcDNA-Foxc2.
Angptl2 Overexpression Attenuated the Effects of Foxc2 Overexpression on Inflammation and Oxidative Stress in Ox-LDL-Induced RAW264.7 Cells
Results from Fig. 7a–c revealed that transfection with pcDNA-Angptl2 aggravated the inflammatory responses via increasing the production of TNF-α, IL-1β, and IL-6, and Angptl2 overexpression alleviated the inhibitory effects of Foxc2 overexpression in ox-LDL-stimulated RAW264.7 cells. Meanwhile, the contents of ROS and MDA showed the same alteration tendency with the above inflammation-related factors, while the activity of GSH-Px presented the opposite change (Fig. 7d–f). Moreover, the expression of NLRP3, ASC, and caspase-1 was remarkably upregulated in the ox-LDL+pcDNA-Angptl2 group, and the significantly downregulated expression of abovementioned proteins in ox-LDL+pcDNA-Foxc2 group was reversed after Angptl2 overexpression (Fig. 7g). These results implicated that Angptl2 overexpression mitigated the effects of Foxc2 overexpression on inflammation and oxidative stress in ox-LDL-induced RAW264.7 cells.

Angptl2 overexpression attenuated the effects of Foxc2 overexpression on inflammation and oxidative stress in ox-LDL-induced RAW264.7 cells. The levels of a TNF-α, b IL-1β, c IL-6, d ROS, e MDA, and the activity of f GSH-Px were examined using commercial kits. g Western blotting was employed to determine the expression of NLRP3 inflammasome signaling proteins. **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. ox-LDL-pcDNA-NC; △P < 0.05, △△P < 0.01, △△△P < 0.001 vs. ox-LDL-pcDNA-Foxc2.
Angptl2 Overexpression Relieved the Effects of Foxc2 Overexpression on Apoptosis in Ox-LDL-Induced RAW264.7 Cells
As displayed in Fig. 8a–c, the apoptotic rate presented the highest level after Angptl2 overexpression alone, accompanied by decreased expression of Bcl-2 and increased expression of Bax, cleaved-caspase-3 in ox-LDL-treated RAW264.7 cells. The effects of Foxc2 overexpression on apoptosis exhibited the opposite results with Angptl2 overexpression alone, whereas these effects were restored following transfection with both pcDNA-Foxc2 and pcDNA-Angptl2. Taken together, the conclusion might be drawn as Angptl2 overexpression attenuated the effects of Foxc2 overexpression on apoptosis in ox-LDL-treated RAW264.7 cells.

Angptl2 overexpression relieved the effects of Foxc2 overexpression on apoptosis in ox-LDL-induced RAW264.7 cells. a Apoptosis of RAW264.7 cells was examined by flow cytometry. b Qualification of apoptotic rate. c The expression of apoptosis-related proteins including Bcl-2, Bax and cleaved caspase-3 was tested using Western blotting. ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. ox-LDL-pcDNA-NC; △P < 0.05, △△P < 0.01, △△△P < 0.001 vs. ox-LDL-pcDNA-Foxc2.
DISCUSSION
AS is recognized as a complex metabolic disease characterized by lipid deposition, inflammation, and apoptosis [31]. As a major component of AS lesions, foam cells play an important role in the development and progression of AS [4]. In the current study, RAW264.7 cells stimulated by ox-LDL were applied to mimic a foam cell model in vitro. The headmost finding was that ox-LDL stimulation dramatically reduced the level of Foxc2. We confirmed that Foxc2 overexpression suppressed lipid accumulation, inflammation, and apoptosis of macrophage via regulating Angptl2 expression, suggesting a novel insight into the mechanisms of AS.
It is well known that the risk of AS is increased by elevated ox-LDL levels in cells [27]. Uptake of ox-LDL by macrophages plays essential roles in the progression of AS by enhancing intracellular lipid accumulation and foam cell formation [24]. However, the relationship between Foxc2 and lipid accumulation of macrophages cells remains ambiguous. Significant increase of lipid accumulation could be found by ox-LDL induction in RAW264.7 cells, which was in agreement with the previous findings [13, 25]. Consistent with the alteration of lipid droplets stained by oil red O, the contents of TC and FC were evidently enhanced after ox-LDL stimulation. Foxc2 overexpression showed reversal functions on the regulation of lipid accumulation, revealing a vital role of Foxc2 in AS.
Reports have demonstrated previously that ox-LDL stimulates the production of pro-inflammatory factors and ROS, which contribute to AS development [2, 5]. Macrophages serve as key participants during AS via secreting inflammatory factors including TNF-α, IL-1β, and IL-6, which are closely implicated in future cardiovascular risk [26]. Emerging evidence supports that Foxc2 can suppress inflammation and promote browning of white adipose tissue; therefore, the authors speculate that Foxc2 may be of potential effects to treat metabolic syndrome induced by obese [11]. Foxc2 overexpression decreased the release of inflammatory factors in lipopolysaccharide-induced human umbilical vein endothelial cells [32]. Recent research by celebrated scholars have confirmed that NLRP3 inflammasome had a positive relation with AS-associated inflammation, which was recognized as a novel correlation between lipid metabolism and inflammation [9]. Importantly, the decreased inflammation and lesions of AS were observed in the tissues with suppressed NLRP3 inflammasome signaling [1]. According to the aforementioned evidence, we tested on the expression of NLRP3 inflammasome-associated genes at protein level. In line with the results of inflammation markers, Foxc2 overexpression decreased the expression of NLRP3, ASC, and caspase-1. To sum up, our findings demonstrated that Foxc2 overexpression inhibited inflammation and oxidative stress in ox-LDL-induced RAW264.7 cells via suppression of NLRP3 inflammasome signaling.
Excessive accumulation of inflammatory factors and ROS contributes to apoptosis of macrophages via the detrimental modification of DNA, proteins, and lipids [39]. Therefore, interventions in the apoptosis of macrophages are crucial for AS treatment [18, 33]. Accumulating evidence shows that Foxc2 suppresses apoptosis of mouse preadipocytes via activation of Akt/mTORC1 pathway [12]. The present study revealed that overexpression of Foxc2 restrained apoptosis of ox-LDL-induced RAW264.7 cells coupled with the alteration of apoptosis-related proteins expression, which was in line with the previous studies [20, 38]. The data above suggested that Foxc2 inhibited apoptosis of macrophages and showed protective effects against AS.
To determine the potential mechanisms of Foxc2 in AS, TRRUST database was utilized to predict the target regulator of Foxc2, and the result demonstrated that Angptl2 could be the target of Foxc2. Therefore, we focused on the expression of Angptl2 in ox-LDL-stimulated RAW264.7 cells. Angptl2 is a chronic inflammatory mediator and is detectable in most organs of adult mice [30]. Studies have shown that the expression of Angptl2 is increased in the serum of patients with AS and can promote the formation of AS in mice [10]. Knockdown of Angptl2 can slow atherogenesis in mice through inhibiting inflammation and apoptosis of vascular endothelial senescent cells [6]. It is also worth noting that robust evidence suggests that Angptl2 overexpression enhanced the mRNA or protein expression of pro-inflammatory genes in human macrophage-like cell line, and treatment with adenovirus mediated Angptl2 leads to lipid accumulation and increased fatty acid synthesis [28]. In the current study, Angptl2 overexpression promoted lipid accumulation, inflammation, and apoptosis in ox-LDL-stimulated RAW264.7 cells, which was in line with the abovementioned evidence. The expression of Angptl2 was inhibited after Foxc2 overexpression, and Angptl2 overexpression restored the effects of Foxc2 overexpression on lipid accumulation, inflammation, and apoptosis in ox-LDL-induced RAW264.7 cells. Collectively, the conclusion might be drawn as Foxc2 alleviated ox-LDL-induced lipid accumulation, inflammation, and apoptosis of macrophage via regulating Angptl2 expression.
CONCLUSION
Taken together, our findings above demonstrated that Foxc2 overexpression meliorated lipid accumulation, inflammation, and apoptosis in ox-LDL-stimulated RAW264.7 cells through inhibiting the expression of Angptl2. This study provides insights into the pathogenesis of AS, which potentially serve as a novel direction for exploring therapeutic strategies for AS.
References
Abderrazak, A., D. Couchie, D.F. Mahmood, R. Elhage, C. Vindis, M. Laffargue, V. Mateo, et al. 2015. Anti-inflammatory and antiatherogenic effects of the NLRP3 inflammasome inhibitor arglabin in ApoE2.Ki mice fed a high-fat diet. Circulation 131 (12): 1061–1070. https://doi.org/10.1161/CIRCULATIONAHA.114.013730.
Aviram, M. 2011. Atherosclerosis: Cell biology and lipoproteins--inflammation and oxidative stress in atherogenesis: Protective role for paraoxonases. Current Opinion in Lipidology 22 (3): 243–244. https://doi.org/10.1097/MOL.0b013e3283474beb.
Bhansali, S., S. Khatri, and V. Dhawan. 2019. Terminalia Arjuna bark extract impedes foam cell formation and promotes apoptosis in ox-LDL-stimulated macrophages by enhancing UPR-CHOP pathway. Lipids in Health and Disease 18 (1): 195. https://doi.org/10.1186/s12944-019-1119-z.
Bhaskar, S., P.R. Sudhakaran, and A. Helen. 2016. Quercetin attenuates atherosclerotic inflammation and adhesion molecule expression by modulating TLR-NF-kappaB signaling pathway. Cellular Immunology 310: 131–140. https://doi.org/10.1016/j.cellimm.2016.08.011.
Bryk, D., W. Olejarz, and D. Zapolska-Downar. 2017. The role of oxidative stress and NADPH oxidase in the pathogenesis of atherosclerosis. Postȩpy Higieny i Medycyny Doświadczalnej (Online) 71 (0): 57–68. https://doi.org/10.5604/17322693.1229823.
Caland, L., P. Labbe, M. Mamarbachi, L. Villeneuve, G. Ferbeyre, P.E. Noly, M. Carrier, N. Thorin-Trescases, and E. Thorin. 2019. Knockdown of angiopoietin-like 2 induces clearance of vascular endothelial senescent cells by apoptosis, promotes endothelial repair and slows atherogenesis in mice. Aging (Albany NY) 11 (11): 3832–3850. https://doi.org/10.18632/aging.102020.
Chen, D.D., L.L. Hui, X.C. Zhang, and Q. Chang. 2018. NEAT1 contributes to ox-LDL-induced inflammation and oxidative stress in macrophages through inhibiting miR-128. Journal of Cellular Biochemistry. https://doi.org/10.1002/jcb.27541.
Chen, K.C., and S.H. Juo. 2012. MicroRNAs in atherosclerosis. The Kaohsiung Journal of Medical Sciences 28 (12): 631–640. https://doi.org/10.1016/j.kjms.2012.04.001.
Duewell, P., H. Kono, K.J. Rayner, C.M. Sirois, G. Vladimer, F.G. Bauernfeind, G.S. Abela, L. Franchi, G. Nuñez, M. Schnurr, T. Espevik, E. Lien, K.A. Fitzgerald, K.L. Rock, K.J. Moore, S.D. Wright, V. Hornung, and E. Latz. 2010. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464 (7293): 1357–1361. https://doi.org/10.1038/nature08938.
Farhat, N., N. Thorin-Trescases, M. Mamarbachi, L. Villeneuve, C. Yu, C. Martel, N. Duquette, M. Gayda, A. Nigam, M. Juneau, B.G. Allen, and E. Thorin. 2013. Angiopoietin-like 2 promotes Atherogenesis in mice. Journal of the American Heart Association 2 (3): 13. https://doi.org/10.1161/jaha.113.000201.
Gan, L., Z. Liu, F. Feng, T. Wu, D. Luo, C. Hu, and C. Sun. 2018. Foxc2 coordinates inflammation and browning of white adipose by leptin-STAT3-PRDM16 signal in mice. International Journal of Obesity 42 (2): 252–259. https://doi.org/10.1038/ijo.2017.208.
Gan, L., Z.J. Liu, W. Jin, Z.J. Zhou, and C. Sun. 2015. Foxc2 enhances proliferation and inhibits apoptosis through activating Akt/mTORC1 signaling pathway in mouse preadipocytes. Journal of Lipid Research 56 (8): 1471–1480. https://doi.org/10.1194/jlr.M057679.
Guo, C.X., R. Ma, X.Y. Liu, T. Chen, Y. Li, Y. Yu, J.C. Duan, X.Q. Zhou, Y.B. Li, and Z.W. Sun. 2018. Silica nanoparticles promote oxLDL-induced macrophage lipid accumulation and apoptosis via endoplasmic reticulum stress signaling. Science of the Total Environment 631-632: 570–579. https://doi.org/10.1016/j.scitotenv.2018.02.312.
Iida, K., H. Koseki, H. Kakinuma, N. Kato, Y. Mizutani-Koseki, H. Ohuchi, H. Yoshioka, S. Noji, K. Kawamura, Y. Kataoka, F. Ueno, M. Taniguchi, N. Yoshida, T. Sugiyama, and N. Miura. 1997. Essential roles of the winged helix transcription factor MFH-1 in aortic arch patterning and skeletogenesis. Development 124 (22): 4627–4638.
Jia, H., H. Li, Y. Zhang, C. Li, Y. Hu, and C. Xia. 2015. Association between red blood cell distribution width (RDW) and carotid artery atherosclerosis (CAS) in patients with primary ischemic stroke. Archives of Gerontology and Geriatrics 61 (1): 72–75. https://doi.org/10.1016/j.archger.2015.04.005.
Jia, S.J., K.Q. Gao, and M. Zhao. 2017. Epigenetic regulation in monocyte/macrophage: A key player during atherosclerosis. Cardiovascular Therapeutics 35 (3). https://doi.org/10.1111/1755-5922.12262.
Li, D., and Y. Tan. 2019. TIPE2 suppresses atherosclerosis by exerting a protective effect on macrophages via the inhibition of the Akt signaling pathway. Experimental and Therapeutic Medicine 17 (4): 2937–2944. https://doi.org/10.3892/etm.2019.7316.
Li, E., T. Wang, F. Wang, T. Wang, L.Q. Sun, L. Li, S.H. Niu, and J.Y. Zhang. 2015. FGF21 protects against ox-LDL induced apoptosis through suppressing CHOP expression in THP1 macrophage derived foam cells. BMC Cardiovascular Disorders 15: 80. https://doi.org/10.1186/s12872-015-0077-2.
Libby, P., P.M. Ridker, and G.K. Hansson. 2011. Progress and challenges in translating the biology of atherosclerosis. Nature 473 (7347): 317–325. https://doi.org/10.1038/nature10146.
Liu, J.Y., S. Liang, Z. Du, J.Y. Zhang, B.Y. Sun, T. Zhao, X.Z. Yang, Y.F. Shi, J.C. Duan, and Z.W. Sun. 2019. PM2.5 aggravates the lipid accumulation, mitochondrial damage and apoptosis in macrophage foam cells. Environmental Pollution 249: 482–490. https://doi.org/10.1016/j.envpol.2019.03.045.
Long, L., and Y. Song. 2018. Dietary ellagic acid is protective for atherosclerosis. International Journal of Cardiology 256: 12. https://doi.org/10.1016/j.ijcard.2017.12.094.
Meng, F., J. Yan, Q. Ma, Y. Jiao, L. Han, J. Xu, F. Yang, and J. Liu. 2018. Expression status and clinical significance of lncRNA APPAT in the progression of atherosclerosis. PeerJ 6: e4246. https://doi.org/10.7717/peerj.4246.
Moore, K.J., and I. Tabas. 2011. Macrophages in the pathogenesis of atherosclerosis. Cell 145 (3): 341–355. https://doi.org/10.1016/j.cell.2011.04.005.
Pan, M.S., Y.J. Huo, C.T. Wang, Y.H. Zhang, Z.Y. Dai, and B. Li. 2019. Positively charged peptides from casein hydrolysate show strong inhibitory effects on LDL oxidation and cellular lipid accumulation in Raw264.7 cells. International Dairy Journal 91: 119–128. https://doi.org/10.1016/j.idairyj.2018.09.011.
Peng, S., L.W. Xu, X.Y. Che, Q.Q. Xiao, J. Pu, Q. Shao, and B. He. 2018. Atorvastatin inhibits inflammatory response, attenuates lipid deposition, and improves the stability of vulnerable atherosclerotic plaques by modulating autophagy. Frontiers in Pharmacology 9: 17. https://doi.org/10.3389/fphar.2018.00438.
Rahman, K., Y. Vengrenyuk, S.A. Ramsey, N.R. Vila, N.M. Girgis, J. Liu, V. Gusarova, J. Gromada, A. Weinstock, K.J. Moore, P. Loke, and E.A. Fisher. 2017. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. The Journal of Clinical Investigation 127 (8): 2904–2915. https://doi.org/10.1172/JCI75005.
Rios, F.J., M. Gidlund, and S. Jancar. 2011. Pivotal role for platelet-activating factor receptor in CD36 expression and oxLDL uptake by human monocytes/macrophages. Cellular Physiology and Biochemistry 27 (3–4): 363–372. https://doi.org/10.1159/000327962.
Sasaki, Y., M. Ohta, D. Desai, J.L. Figueiredo, M.C. Whelan, T. Sugano, M. Yamabi, W. Yano, T. Faits, K. Yabusaki, H. Zhang, A.K. Mlynarchik, K. Inoue, K. Mizuno, and M. Aikawa. 2015. Angiopoietin like protein 2 (ANGPTL2) promotes adipose tissue macrophage and T lymphocyte accumulation and leads to insulin resistance. PLoS One 10 (7): 18. https://doi.org/10.1371/journal.pone.0131176.
Seimon, T., and I. Tabas. 2009. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. Journal of Lipid Research 50 (Suppl): S382–S387. https://doi.org/10.1194/jlr.R800032-JLR200.
Tabata, M., T. Kadomatsu, S. Fukuhara, K. Miyata, Y. Ito, M. Endo, T. Urano, H.J. Zhu, H. Tsukano, H. Tazume, K. Kaikita, K. Miyashita, T. Iwawaki, M. Shimabukuro, K. Sakaguchi, T. Ito, N. Nakagata, T. Yamada, H. Katagiri, M. Kasuga, Y. Ando, H. Ogawa, N. Mochizuki, H. Itoh, T. Suda, and Y. Oike. 2009. Angiopoietin-like protein 2 promotes chronic adipose tissue inflammation and obesity-related systemic insulin resistance. Cell Metabolism 10 (3): 178–188. https://doi.org/10.1016/j.cmet.2009.08.003.
Wang, J.C., and M. Bennett. 2012. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circulation Research 111 (2): 245–259. https://doi.org/10.1161/CIRCRESAHA.111.261388.
Xu, Y.J., P. Li, L. Zheng, F.X. Guo, C.M. Kang, L. Ding, B.M. Xu, et al. 2019. Forkhead box C2 attenuates lipopolysaccharide-induced cell adhesion via suppression of intercellular adhesion Molecule-1 expression in human umbilical vein endothelial cells. DNA and Cell Biology 38 (6): 583–591. https://doi.org/10.1089/dna.2019.4663.
Yan, L., Z. Liu, H. Yin, Z. Guo, and Q. Luo. 2019. Silencing of MEG3 inhibited ox-LDL-induced inflammation and apoptosis in macrophages via modulation of the MEG3/miR-204/CDKN2A regulatory axis. Cell Biology International 43 (4): 409–420. https://doi.org/10.1002/cbin.11105.
Zahid, M. K., M. Rogowski, C. Ponce, M. Choudhury, N. Moustaid-Moussa, and S. M. Rahman. CCAAT/enhancer-binding protein beta (C/EBP beta) knockdown reduces inflammation, ER stress, and apoptosis, and promotes autophagy in oxLDL-treated RAW264.7 macrophage cells. Molecular and Cellular Biochemistry:13. https://doi.org/10.1007/s11010-019-03642-4.
Zhang, C., J. Chen, Y. Liu, and D. Xu. 2019. Sialic acid metabolism as a potential therapeutic target of atherosclerosis. Lipids in Health and Disease 18 (1): 173. https://doi.org/10.1186/s12944-019-1113-5.
Zhang, E., and Y. Wu. 2013. MicroRNAs: Important modulators of oxLDL-mediated signaling in atherosclerosis. Journal of Atherosclerosis and Thrombosis 20 (3): 215–227. https://doi.org/10.5551/jat.15180.
Zhang, Q., J. Hu, Y. Wu, H. Luo, W. Meng, B. Xiao, X. Xiao, Z. Zhou, and F. Liu. 2019. Rheb (Ras homolog enriched in brain 1) deficiency in mature macrophages prevents atherosclerosis by repressing macrophage proliferation, inflammation, and lipid uptake. Arteriosclerosis, Thrombosis, and Vascular Biology 39 (9): 1787–1801. https://doi.org/10.1161/ATVBAHA.119.312870.
Zheng, G.L., H.Z. Li, T. Zhang, L.B. Yang, S.T. Yao, S.H. Chen, M.C. Zheng, Q. Zhao, and H. Tian. 2018. Irisin protects macrophages from oxidized low density lipoprotein-induced apoptosis by inhibiting the endoplasmic reticulum stress pathway. Saudi Journal of Biological Sciences 25 (5): 849–857. https://doi.org/10.1016/j.sjbs.2017.08.018.
Zhou, P.L., M. Li, X.W. Han, Y.H. Bi, W.G. Zhang, Z.Y. Wu, and G. Wu. 2019. Perilipin 5 deficiency promotes atherosclerosis progression through accelerating inflammation, apoptosis, and oxidative stress. Journal of Cellular Biochemistry 120 (11): 19107–19123. https://doi.org/10.1002/jcb.29238.
Funding
The present study was supported by the Natural Science Foundation of Hunan Province, China (Grant No. 14JJ7006), the Science and Technology Innovation Planning Project of Hunan Province, China (Grant No. 2017SK50104), and the Scientific Research Project of Hunan Health Commission (Grant No. 20201228).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yang, L., Li, T. & Zha, L. Foxc2 Alleviates Ox-LDL-Induced Lipid Accumulation, Inflammation, and Apoptosis of Macrophage via Regulating the Expression of Angptl2. Inflammation 43, 1397–1410 (2020). https://doi.org/10.1007/s10753-020-01217-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-020-01217-w