
Abstract
Growth factor independence 1 (Gfi-1) has been widely studied for its anti-inflammatory and anti-apoptotic effects. However, whether Gfi-1 has similar effects on H9c2 cardiomyocytes has not yet been reported. In this study, we explored the effect of Gfi-1 on lipopolysaccharide (LPS)-induced inflammatory responses and apoptosis in H9c2 cells. We found that LPS induced the increased expression of TNF-α and IL-6 in the LPS group. After transfection of the Gfi-1 overexpression plasmid, the expression of TNF-α and IL-6 decreased significantly in the LPS + Gfi-1 group. Gfi-1 clearly blocked LPS-induced NF-κB, TNF-α, TNFR1, cleaved-caspase-3 and cleaved-caspase-8 expression and increased Gfi-1 and Bcl-xL expression in H9c2 cells. Similarly, compared with the LPS group, Gfi-1 significantly decreased the expression of cleaved-caspase3/8 and increased the expression of Bcl-xL in the LPS + Gfi-1 group, as verified by immunocytochemical analysis. Furthermore, Gfi-1 markedly inhibited LPS-induced H9c2 cardiomyocyte apoptosis in the LPS + Gfi-1 group, as determined by TEM, TUNEL and flow cytometry. Taken together, these results demonstrate that Gfi-1 may have protective effects against LPS-induced inflammatory responses and apoptosis in H9c2 cells. Gfi-1 may be a novel molecule for treating septic cardiomyopathy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Sepsis is one of the most challenging issues in intensive care units today. Sepsis is defined as life-threatening organ dysfunction caused by an uncontrolled inflammatory response [1]. Studies have reported that the body’s immune system produces an inflammatory cascade, triggering an inflammatory response that includes myocardial damage, increased vascular permeability and multiple organ dysfunction during the late stage of sepsis [2, 3]. Almost 50% of septic patients present cardiac dysfunction [3]. Furthermore, cardiac dysfunction was associated with higher mortality during sepsis [4]. Many mechanisms have been associated with septic cardiac diseases, such as excessive inflammatory and immune responses, energy metabolic diseases and apoptosis [5,6,7,8]. However, the specific pathogenesis of sepsis-associated cardiac diseases has not yet been clarified.
Endotoxic lipopolysaccharide (LPS) is an important molecule in the pathogenesis of inflammation [8, 9]. Thus, it is used to build an experimental model of sepsis [10]. LPS is one of the key chemical molecules leading to sepsis-associated organ diseases [11]. In addition, LPS induces myocardial diseases, myocardial injury and cardiac dysfunction through an excessive inflammatory response and apoptosis [12]. Growth factor independence 1 (Gfi-1) is a 55-kD nuclear zinc finger protein that contains C-terminal C2H2 zinc finger domains and an N-terminal SNAG domain. Gfi-1 is an important transcription repressor of DNA binding [13]. Gfi-1−/− mice are prone to severe sepsis or septic shock stimulated by LPS [14]. In addition, LPS-induced macrophages from Gfi-1-deficient mice produce several inflammatory factors, such as TNF-α and IL-1β, compared with those from wild-type mice [15].
However, whether Gfi-1 can regulate the LPS-induced inflammatory response and apoptosis of H9c2 cells has not yet been reported. Therefore, the purpose of the present study was to investigate the effects of Gfi-1 on the LPS-induced inflammatory response and apoptosis of H9c2 cells and to explore the related mechanisms.
MATERIAL AND METHODS
Gfi-1 Transfection Assays
H9c2 cardiomyocytes were transfected with the pcDNA-3.1-Rat Gfi-1 plasmid (ELK Biotechnology, Wuhan, China) via Lipofectamine 2000 (Invitrogen, Shanghai, China). The sequences for Gfi-1 were Forward: 5′-GGATCCACTAGTCCAGTGTGGTGGAATTCATGCCGCGCTCATTCCTGGT-3′; Reverse: 5′-GGTTTAAACGGGCCCTCTAGACTCGAGTCATTTGAGTCCATGCTGAGT-3′.
Culture, Treatment and Grouping of H9c2 Cells
H9c2 cells were maintained in DMEM (Dulbecco’s Modified Eagle’s Medium, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% foetal bovine serum (FBS, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and incubated at 37 °C, 5% CO2. The cells were randomly divided into three groups as follows: Control group, in which the cells were only given basic culture medium; LPS group, in which the cells were stimulated with 10 μg/mL LPS for 6 h and 24 h; LPS + Gfi-1 group, after transfection with the Gfi-1 plasmid for 24 h, the cells were stimulated with 10 μg/mL LPS for 6 h and 24 h. The experiments were repeated at least three times.
Cell Viability Assay
The cell viability of H9c2 was measured using a Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Gaithersburg, MD, USA). Briefly, the culture medium was removed and the cells were washed three times with PBS. CCK-8 was added to the culture medium and incubated for 3 h. The assay was performed according to the manufacturer’s instructions. The absorbance was read at 450 nm with a microplate reader (Bio-Rad, Hercules, CA, USA). The OD values (optical density) were used to assess the viability of H9c2 cells. Cell viability was calculated by ((experimental group/control group) × 100).
Reverse Transcription-Quantitative Polymerase Chain Reaction
The mRNA expression levels of TNF-α and IL-6 were detected using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) in H9c2 cells. First, total RNA was extracted by Trizol reagent (Invitrogen Life Technologies, CA, USA). Then, the RNA was reverse transcribed into cDNA using the PrimeScript RT Reagent Kit with oligo(dT) primers and the Transcriptor First-Strand cDNA Synthesis Kit according to the manufacturer’s instructions. The samples were quantified by SYBR® Premix Ex Taq™ (TaKaRa, Japan) and a StepOne™ Real-Time PCR instrument. TNF-α, IL-6 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were amplified with the following primers: TNF-α, forward 5′-AGCATGATCCGAGATGTGGAA-3′; reverse 5′-TAGACAGAAGAG CG TGGTGGC-3′; IL-6, forward 5′-TGGAGTTCCGTTTCTACCTGG-3′ and reverse 5′-GGTCCTTAGCCACTCCTTCTGT-3′; GAPDH, forward 5′-GACATGCCGCCTGGAGAAAC-3′ and reverse 5′-AG CC CAGGATGCCCTTTAGT-3′. The expression levels of TNF-α and IL-6 were normalised to GAPDH. The relative expression of TNF-α and IL-6 was calculated according to the 2−△△Cq method.
Detection of TNF-α and IL-6 Levels by ELISA
The supernatant from each cell group was collected. The expression of TNF-α and IL-6 in H9c2 cells was detected using ELISA kits (Abcam Cambridge, UK) according to the manufacturer’s instructions.
Western Blotting
The protein levels of Gfi-1, NF-κB, TNF-α, cleaved-caspase-3, cleaved-caspase-8 and Bcl-xL were determined by western blotting. First, the total protein from each cell group was extracted, and the concentrations were determined by the bicinchoninic acid (BCA) method. The target protein samples were fractionated according to the molecular weight by SDS-polyacrylamide gel electrophoresis and then transferred to a PVDF membrane (Millipore, USA). Next, the PVDF membranes were incubated with the primary antibodies (all primary antibodies were purchased from Cell Signalling Technology, Inc., USA. 1:1000 dilution) and the secondary antibody (LI-COR, LincoIn, NE, USA. 1:5000 dilution). GAPDH (Santa Cruz Biotechnology, Inc., USA. 1:200 dilution) was used as the loading control. Finally, the membranes were determined using the LI-COR Odyssey Infrared Imaging System (LI-COR Biosciences Inc., USA). The target protein bands were quantified and analysed by Quantity One software (Bio-Rad, USA).
Transmission Electron Microscopy of H9c2 Cells
Each cell line was fixed with 4% paraformaldehyde and 3% glutaraldehyde at room temperature overnight. Next, the cells were incubated with 1% osmium tetroxide for 2 h. The samples were washed three times for 5 min with H2O and dehydrated in graded ethanol concentrations and then embedded in moulds with fresh resin. The sample ultrathin sections were incubated in 2% uranyl acetate for 1 h and stained with lead citrate for 15 min. The ultrathin sections were examined with an H-7100 transmission electron microscope (TEM) (Hitachi Co., Japan) operating at 80 kV.
Immunohistochemistry
The cells were grown on cover slides in cell culture dishes. Next, each cell line was fixed with 4% paraformaldehyde in PBS for 10 min on ice. The cells were perforated with 1% Triton X-100 in PBS for 5 min and then blocked with 3% FBS for 1 h at room temperature. The cells were incubated with primary antibody (cleaved-caspase3, cleaved-caspase8 and Bcl-xL, 1:100 dilution) for 2 h at room temperature. The samples were washed with PBS and incubated with secondary antibody (fluorescein isothiocyanate conjugated goat anti-rabbit lgG antibody) for 1 h in the dark. The samples were washed with PBS and observed by fluorescence microscopy. The expression of cleaved-caspase3, cleaved-caspase8 and Bcl-xL cells was stained brownish as immune-positive cells. The number of immune-positive cells was calculated according to the formula: immune-positive cells/the total cells × 100.
Identification of Apoptosis by Terminal Deoxynucleotidyl Transferase (TdT)-Mediated dUTP Nick End Labeling
The apoptosis of H9c2 cells was determined by terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay using an in situ cell death detection kit (Roche In Situ Cell Death Detection Kit, USA). Briefly, each cell line was fixed with 4% paraformaldehyde in PBS for 15 min at room temperature and then washed with PBS. The cells were blocked with 3% FBS for 1 h at room temperature. Next, the samples were incubated with TUNEL reagent according to the manufacturer’s instructions. The cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 10 min, washed with PBS and observed under a fluorescence microscope. The TUNEL-positive cells were calculated according to the following formula: apoptotic index = TUNEL-positive cells / the total cells × 100.
Flow Cytometry of Apoptosis of H9c2 Cells
The apoptosis rate of H9c2 cells was demined by flow cytometry using a dead cell apoptosis kit with Annexin V Alexa Fluor™ 488 and propidium iodide (Invitrogen). Briefly, 200 μL binding buffer containing 10 μL Annexin V and 5 μL PI was added to the samples for 1 h in the dark. The stained cells were analysed using FACSCaliber™ flow cytometry (BD Biosciences, San Jose, CA, USA) and Cell-Quest software.
Statistical Analysis
SPSS version 20.0 statistical software (SPSS, Inc., USA) was used for statistical analysis. All data were reported as the mean ± standard error of the mean (SEM). One-way analysis of variance (ANOVA), followed by Bonferroni’s post hoc analysis, was performed for comparisons between groups. P values < 0.05 were considered statistically significant.
RESULTS
Effect of Gfi-1 on Cell Viability
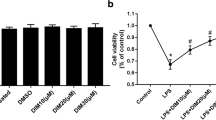
We examined whether Gfi-1 has an effect on H9c2 cardiomyocytes. The results showed that LPS (10 μg/mL) induced a decrease in the viability of H9c2 cells at 6 h and 24 h, but Gfi-1 reduced the toxicity of LPS, especially after Gfi-1 transfection for 24 h (Fig. 1). These results indicated that Gfi-1 attenuated the LPS-induced decrease in cell viability.

Cell viability of H9c2 cells detected with the CCK-8 assay. aP < 0.05 vs. the control group and the vector group, bP < 0.05 vs. the LPS (6 h and 24 h). In the control group, the cells were only given basic culture medium. In the vector group, the cells were transfected with blank plasmid for 24 h. In the LPS group, the cells were stimulated with 10 μg/mL LPS for 6 h and 24 h. In the LPS + Gfi-1 group, the cells were transfected with the Gfi-1 plasmid for 24 h before stimulation with 10 μg/mL LPS for 6 h and 24 h. Each experiment was repeated at least three times. Data are presented as the mean ± SD. LPS, lipopolysaccharide; CCK-8, Cell Counting Kit-8.
Gfi-1 Inhibited the Inflammatory Response Induced by LPS in H9c2 Cells
The effects of Gfi-1 on the production of inflammatory molecules stimulated by LPS in H9c2 cells were detected by qRT-PCR, ELISA and western blotting. TNF-α, IL-6 and NF-κB are key molecules of the inflammatory response. Analysis of the inflammatory response in H9c2 cells is shown in Fig. 2 (A, B and C). RT-PCR revealed that the mRNA levels of TNF-α and IL-6 were significantly elevated in the cells of the LPS group compared with those of the control group (P < 0.01 for all). However, the mRNA levels of TNF-α and IL-6 were significantly lower in the cells of the LPS + Gfi-1 group than in those of the LPS group (P < 0.01 for all, Fig. 2(A)). ELISA showed that the levels of TNF-α and IL-6 were markedly increased in the cells of the LPS group compared with those in the control group. However, transfection with Gfi-1 significantly attenuated the levels of TNF-α and IL-6 in the LPS-Gfi-1 group compared with those in the LPS group (P < 0.01 for all, Fig. 2(B)). Western blotting revealed that stimulation with LPS significantly downregulated the expression level of Gfi-1, and of the expression of NF-κB and TNF-α was significantly upregulated in the H9c2 cells of the LPS group compared with those in the control group (P < 0.05 for all, Fig. 2(C)). Conversely, transfection with Gfi-1 markedly upregulated the expression of Gfi-1, and the expression of NF-κB and TNF-α was markedly downregulated in the H9c2 cells of the LPS + Gfi-1 group compared with those of the LPS group (P < 0.05 for all, Fig. 2(C)).

Effects of Gfi-1 on the LPS-stimulated inflammatory response in H9c2 cells. The LPS group: LPS 10 μg/mL for 6 h and 24 h; the LPS + Gfi-1 group: cells were transfected with Gfi-1 plasmid for 24 h and then induced by 10 μg/mL LPS for 6 h and 24 h. (A) The mRNA levels were determined by RT-qPCR; (B) The TNF-α and IL-6 levels were determined by ELISA. (C) Gfi-1, NF-κB and TNF-α protein expressions were measured by western blotting. A: Control group; B: LPS group; LPS + Gfi-1 group. A: Control group; B: LPS group; C: LPS + Gfi-1 group. Data are presented as the mean ± SD. N = 3. aP < 0.05 vs. the control group, bP < 0.05 vs. the LPS group.
Gfi-1 Inhibited LPS-Induced Apoptotic H9c2 Cells
The effects of Gfi-1 on the apoptosis of H9c2 cells induced by LPS were measured by western blotting, transmission electron microscopy (TEM), immunocytochemistry, TUNEL staining and flow cytometry. Cleaved-caspase-3/8 and Bcl-xL are critical apoptotic and anti-apoptotic molecules, respectively. Evaluation of the apoptosis of H9c2 cells is shown in Fig. 3 (A, B, C, D and E). Western blotting: The data demonstrated that stimulation with LPS markedly upregulated the level of NF-κB, TNF-α, TNFR1 and cleaved-caspase-3/8 and downregulated the expression of Bcl-xL in the H9c2 cells of the LPS group compared with those of the control group (P < 0.05 for all, Fig. 3(A)). Transfection with Gfi-1 downregulated the levels of NF-κB, TNF-α, TNFR1 and cleaved-caspase-3/8 and upregulated the level of Bcl-xL in the H9c2 cells of the LPS + Gfi-1 group compared with those of the LPS group (P < 0.05 for all, Fig. 3(A)). Immunocytochemical analysis: H9c2 cells stained brownish are immune-positive cells expressing the target protein. The expression levels of Cleaved-caspase-3/8 were markedly increased, and the expression level of Bcl-xL was markedly decreased in H9c2 cells after LPS (10 μg/mL) induction for 24 h in the LPS group compared with that in the control group ((P < 0.05, Fig. 3(B)). Interestingly, transfection with Gfi-1 significantly decreased the expression levels of Cleaved-caspase-3/8 and increased the expression level of Bcl-xL in the LPS + Gfi-1 group compared with that in the LPS group ((P < 0.05, Fig. 3(B)). TEM: The images show that in the control group, H9c2 cells had normal nuclei, mitochondria and other organelles and without apoptotic bodies (Fig. 3(C) a, b, c). However, after induction by LPS (10 μg/mL) for 24 h, H9c2 cells showed nuclear shrinkage, cytoplasmic cavitation, mitochondrial disruption and apoptotic bodies, which are characteristics of apoptosis in the LPS group (Fig. 3(C) d, e, f). Compared with the LPS group, Gfi-1 meaningfully reversed the conditions in the LPS + Gfi-1 group (Fig. 3(C) g,h,i). TUNEL staining: the results showed that in the LPS group, in which cells were induced LPS (10 μg/mL) for 24 h, the TUNEL-positive cells were markedly increased compared with those in the control group (P < 0.05, Fig. 3(D)). In contrast, in the LPS + Gfi-1 group, in which H9c2 cells were transfected with Gfi-1, the TUNEL-positive cells were markedly reduced compared with those in the LPS group (P < 0.05, Fig. 3 (D)). Flow cytometry: H9c2 cells in the upper right quadrant and lower right quadrant are apoptotic cells. The data demonstrated that the apoptotic rate of H9c2 cells in the LPS group was 27.5 ± 0.56%, which was significantly increased compared with the control group (P < 0.05, Fig. 3(E)). Furthermore in the LPS + Gfi-1 group, after transfection with Gfi-1, the apoptotic rate of H9c2 cells induced by LPS was decreased (12.3 ± 0.37%), which was significantly decreased compared with the apoptotic rate in the LPS group (P < 0.05, Fig. 3(E)). These data strongly indicated that Gfi-1 attenuated the apoptosis induced by LPS in H9c2 cells.



Gfi-1 could suppress the apoptosis induced by LPS in H9c2 cells. (A) The apoptosis-associated protein levels of NF-κB, TNF-α, TNFR1, cleaved-caspase-3/8 and Bcl-xL were analysed by western blotting. A: Control group; B: LPS group; LPS + Gfi-1 group. (B) Cleaved-caspase3/8 and Bcl-xL were detected by immunocytochemistry. Representative images (× 400) are shown. The immune-positive cells were stained brownish (black arrow). (C) Detection of apoptosis in H9c2 cardiomyocytes by TEM. N, nucleus; m, mitochondrion; white arrow, cytoplasmic cavitation; black arrow, apoptotic bodies. (D) TUNEL staining results show apoptosis in H9c2 cells. Representative images (× 400) are shown. (E) The apoptotic rate of H9c2 cells was detected by flow cytometry. Representative apoptotic images. Data are presented as the mean ± SD. N = 3. aP < 0.05 vs. the control group, bP < 0.05 vs. the LPS group.
DISCUSSION
Recent studies have reported that the inflammatory response and apoptosis play an important role in cardiac dysfunction during sepsis [16]. Therefore, the prevention and treatment of the myocardial inflammatory response and apoptosis may have clinical value for patients with sepsis. H9C2 cardiomyocytes are an embryonic rat heart-derived cell line and have similar biological functions as primary cardiomyocytes [17]. However, it is not clear whether Gfi-1 can regulate the damage to H9c2 cells induced by LPS. In this study, we assessed the inflammatory response and apoptosis of H9c2 cells induced by LPS and whether Gfi-1 has protective effects on H9c2 cardiomyocytes. Furthermore, we studied the underlying mechanisms of this protection.
The lung tissue of Gfi-1 knockout mice can cause the massive production of inflammatory cytokines, such as TNF-α and IL-1β, after stimulation by LPS [18]. Recent studies have shown that Gfi-1 is a negative regulator of LPS-induced immune responses, such as septic shock and other severe inflammatory diseases [19]. In the present study, we found that LPS induced an increase in TNF-α and IL-6 levels and upregulated the expression of NF-κB and TNF-α in H9c2 cells at 6 h and 24 h after LPS induction in the LPS group (Fig. 2A–C). Interestingly, we also found that transfection of H9c2 cells with a Gfi-1 overexpression plasmid reduced TNF-α and IL-6 levels and downregulated the expression of NF-κB and TNF-α in H9c2 cells at 6 h and 24 h after LPS induction in the LPS + Gfi-1 group (Fig. 2A–C). These data strongly demonstrate that Gfi-1 plays a protective role in LPS-induced inflammatory injury in experimental cells. This effect is associated with a reduction in the level of inflammatory factors, such as NF-κB, TNF-α and IL-6, during sepsis.
In addition, in Gfi-1−/− mice, Gfi-1 protects against the apoptosis of haematopoietic cells [20]. Consistently, compared with wild-type mice, Gfi-1 gene knockout mice show the severe apoptosis of pancreatic acinar cells [21]. In this study, our results demonstrated that LPS downregulated the expression of anti-apoptotic Bcl-xL and upregulated and proapoptotic cleaved-caspase-3/8 in H9c2 cells in the LPS group (Fig. 3(A)). In contrast, we found that the transfection of H9c2 cells with Gfi-1 overexpression plasmid upregulated anti-apoptotic Bcl-xL and downregulated proapoptotic cleaved-caspase-3/8 in H9c2 cells after LPS induction in the LPS + Gfi-1 group (Fig. 3(A)). Similarly, our data showed that transfection of H9c2 cells with Gfi-1 overexpression plasmid could significantly increase the nuclear staining of anti-apoptotic Bcl-xL and decrease proapoptotic cleaved-caspase-3/8 expression in the LPS + Gfi-1 group, as confirmed by immunocytochemical analysis (Fig. 3(B)). TEM, TUNEL and flow cytometry revealed that LPS could significantly increase the apoptosis of H9c2 cells in the LPS group (Fig. 3(C–E)). Conversely, Gfi-1 attenuated the apoptosis of H9c2 cells induced by LPS in the LPS + Gfi-1 group (Fig. 3(C–E)). The above results strongly indicate that Gfi-1 plays a protective role in LPS-induced apoptotic H9c2 cells in vitro. Furthermore, the study of mouse macrophages after LPS stimulation showed that Gfi-1 can directly antagonise NF-κB through the TLR pathway to inhibit the production of TNF-α [22]. Numerous studies have reported that sepsis induces the significant production of proapoptotic molecules [23].
In these molecules, TNF-α and NF-κB are considered to be one of the links regulating apoptosis [24]. Nuclear factor-κB (NF-κB) belongs to transcription factors, including NF-κB1 (p50), NF-κB2 (p52), cREL, RELB and RELA (p65) [25]. Additionally, some authors have demonstrated that NF-κB regulates inflammation, immunity, thrombosis and apoptosis [26]. Much data have also shown that cell damage caused by lipopolysaccharides (LPS), such as the inflammatory response and apoptosis, is closely related to NF-κB [27]. NF-κB induced apoptosis by upregulating the expression of the proapoptotic genes Bax in the rat model of cerebral ischaemia [28, 29]. Previous studies have confirmed that NF-κB induced the upregulation of TNF-α; the NF-κB inhibitor, PDTC, could inhibit TNF-α production in H9c2 cardiomyocytes [30]. Evidence has indicated that the binding of TNF-α and tumour necrosis factor receptor 1 (TNF-R1) can lead to the activation of caspase-8 and eventually cause myocardial apoptosis [31]. The present study shows that NF-κB/TNF-α expression is inhibited by Gfi-1. The expression of NF-κB/TNF-α may be related to inflammatory response and apoptosis of H9c2 cells during sepsis.
CONCLUSION
In summary, the data of our study suggest that Gfi-1 is an effective anti-inflammatory and anti-apoptotic molecule that acts primarily by inhibiting the NF-κB/TNF-α pathway during sepsis (Fig. 4). These findings may help in the development of new therapeutic strategy for prevention and treatment sepsis-associated cardiac disease.

Schematic representation depicting the potential molecular mechanisms underlying the protective effects of Gfi-1 on lipopolysaccharide-induced inflammatory responses and apoptosis by inhibition of the NF-κB/TNF-α pathway in H9c2 cells. Gfi-1, growth factor independence 1; LPS, lipopolysaccharide; TLR4, Toll like receptor 4; TNF-R1, tumour necrosis factor receptor 1.
References
Seymour, C.W., V.X. Liu, T.J. Iwashyna, F.M. Brunkhorst, T.D. Rea, A. Scherag, G. Rubenfeld, J.M. Kahn, M. Shankar-Hari, M. Singer, C.S. Deutschman, G.J. Escobar, and D.C. Angus. 2016. Assessment of clinical criteria for sepsis: for the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3)[J]. Jama 315 (8): 762–774.
Perner, A., M. Joannidis, S. Opal, et al. 2017. Sepsis: frontiers in supportive care, organisation and research[J]. Intensive Care Medicine 43 (4): 496–508.
Charpentier, J., C.E. Luyt, Y. Fulla, C. Vinsonneau, A. Cariou, S. Grabar, J.F. Dhainaut, J.P. Mira, and J.D. Chiche. 2004. Brain natriuretic peptide: a marker of myocardial dysfunction and prognosis during severe sepsis.[J]. Critical Care Medicine 32 (3): 660–665.
Sanfilippo, F., C. Corredor, N. Fletcher, G. Landesberg, U. Benedetto, P. Foex, and M. Cecconi. 2015. Diastolic dysfunction and mortality in septic patients: a systematic review and meta-analysis. Intensive Care Medicine 41: 1004–1013.
Havaldar, Amarja Ashok. 2018. Evaluation of sepsis induced cardiac dysfunction as a predictor of mortality[J]. Cardiovascular Ultrasound 16 (1): 31–42.
Zhang, L., Y.L. Zheng, R.H. Hu, L. Zhu, C.C. Hu, F. Cheng, S. Li, and J.G. Li. 2018. Annexin A1 mimetic peptide AC2-26 inhibits sepsis-induced cardiomyocyte apoptosis through LXA4/PI3K/AKT signaling pathway[J]. Current Medical Science 38 (6): 997–1004.
Wang, Y.Y., Y. Wang, D. Yang, X. Yu, H. Li, X. Lv, D. Lu, and H. Wang. 2015. β1 -adrenoceptor stimulation promotes LPS-induced cardiomyocyte apoptosis through activating PKA and enhancing CaMKII and IκBα phosphorylation[J]. Critical Care 19 (1): 76–82.
Tupchong, K., A. Koyfman, and M. Foran. 2015. Sepsis, severe sepsis, and septic shock: a review of the literature[J]. African Journal of Emergency Medicine 5: 127–135.
Lv, H., Q. Liu, Z. Wen, H. Feng, X. Deng, and X. Ci. 2017. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3β-Nrf2 signal axis[J]. Redox Biology 12 (C): 311–324.
Thomas, R.C., M.F. Bath, C.M. Stover, D.G. Lambert, and J.P. Thompson. 2014. Exploring LPS-induced sepsis in rats and mice as a model to study potential protective effects of the nociceptin/orphanin FQ system[J]. Peptides 61: 56–60.
Sato, R., and M. Nasu. 2015. A review of sepsis-induced cardiomyopathy. Journal of Intensive Care 3: 1–7.
Balija, T.M., and S.F. Lowry. 2011. Lipopolysaccharide and sepsis-associated myocardial dysfunction[J]. Current Opinion in Infectious Diseases 24 (3): 248–253.
Hongbing, C., Z. Fan, and L. Zhen. 2018. Gfi-1 promotes proliferation of human cervical carcinoma via targeting of FBW7 ubiquitin ligase expression[J]. Cancer Management and Research 10: 2849–2857.
Jin, J., H. Zeng, K.W. Schmid, M. Toetsch, S. Uhlig, and T. Möröy. 2006. The zinc finger protein Gfi1 acts upstream of TNF to attenuate endotoxin-mediated inflammatory responses in the lung[J]. European Journal of Immunology 36 (2): 421–430.
Holger, K., Z. Hui, S. Thorsten, et al. 2002. Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1.[J]. Nature Genetics 30 (3): 295–300.
Liu, R., H. Jiang, Y. Tian, W. Zhao, and X. Wu. 2016. Astragaloside IV protects against polymicrobial sepsis through inhibiting inflammatory response and apoptosis of lymphocytes[J]. Journal of Surgical Research 200 (1): 315–323.
Watkins, S.J., G.M. Borthwick, and H.M. Arthur. 2011. The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro[J]. In Vitro Cellular & Developmental Biology Animal 47 (2): 125–131.
Jin, J., H. Zeng, K.W. Schmid, M. Toetsch, S. Uhlig, and T. Mo¨ro¨y. 2006. The zinc fifinger protein Gfi-1 acts upstream of TNF to attenuate endotoxin-mediated inflammatory responses in the lung. European Journal of Immunology 36: 421–430.
Sharif-Askari, E., L. Vassen, C. Kosan, C. Khandanpour, M.C. Gaudreau, F. Heyd, T. Okayama, J. Jin, M.E.B. Rojas, H.L. Grimes, H. Zeng, and T. Moroy. 2010. Zinc finger protein Gfi1 controls the endotoxin-mediated Toll-like receptor inflammatory response by antagonizing NF- B p65[J]. Molecular and Cellular Biology 30 (16): 3929–3942.
Khandanpour, C., C. Kosan, M.C. Gaudreau, U. Dührsen, J. Hébert, H. Zeng, and T. Möröy. 2011. Growth factor independence 1 protects hematopoietic stem cells against apoptosis but also prevents the development of a myeloproliferative-like disease[J]. Stem Cells (Dayton, Ohio) 29 (2): 376–385.
Qu, Xiaoling, Pia Nyeng, Fan Xiao, Jorge Dorantes, and Jan Jensen. 2015 Mar. Growth factor independence-1 (Gfi1) is required for pancreatic acinar unit formation and centroacinar cell differentiation[J]. Cellular and Molecular Gastroenterology and Hepatology 1 (2): 233–247.
Geissler, S., M. Textor, S. Stumpp, S. Seitz, A. Lekaj, S. Brunk, S. Klaassen, T. Schinke, C. Klein, S. Mundlos, U. Kornak, and J. Kühnisch. 2018. Loss of murine Gfi1 causes neutropenia and induces osteoporosis depending on the pathogen load and systemic inflammation[J]. PLoS One 13 (6): e0198510.
Ma, H., X. Wang, T. Ha, M. Gao, L. Liu, R. Wang, K. Yu, J.H. Kalbfleisch, R.L. Kao, D.L. Williams, and C. Li. 2016. MicroRNA-125b prevents cardiac dysfunction in polymicrobial sepsis by targeting TRAF6-mediated nuclear factor κB activation and p53-mediated apoptotic signaling[J]. Journal of Infectious Diseases 214 (11): 1773–1783.
Zhao, G., Y.M. Yu, M. Kaneki, et al. 2014. Simvastatin reduces burn injury-induced splenic apoptosis via downregulation of the TNF-α/NF-кB pathway[J]. Annals of Surgery 261 (5): 1006–1012.
Hayden, M.S., and S. Ghosh. 2008. Shared principles in NF-kappaB signaling.[J]. Cell 132 (3): 344–362.
Mussbacher, M., M. Salzmann, C. Brostjan, B. Hoesel, C. Schoergenhofer, H. Datler, P. Hohensinner, J. Basílio, P. Petzelbauer, A. Assinger, and J.A. Schmid. 2019. Cell type-specific roles of NF-κB linking inflammation and thrombosis. Frontiers in Immunology 10: 85.
Chand, H.S., J.F. Harris, and Y. Tesfaigzi. 2018. IL-13 in LPS-induced inflammation causes Bcl-2 expression to sustain hyperplastic mucous cells[J]. Scientific Reports 8 (1): 436.
Mckenna, S., B. Butler, L. Jatana, et al. 2017. Inhibition of IκBβ/NFκB signaling prevents LPS-induced IL1β expression without increasing apoptosis in the developing mouse lung:[J]. Pediatric Research 82 (6): 1064–1072.
Shou, Y., N. Li, L. Li, J.L. Borowitz, and G.E. Isom. 2002. NF-κB-mediated up-regulation of Bcl-XS and Bax contributes to cytochrome c release in cyanide-induced apoptosis[J]. Journal of Neurochemistry 81 (4): 842–852.
Zhong, L., X.L. Zhou, Y.S. Liu, et al. 2015. Estrogen receptor α mediates the effects of notoginsenoside R1 on endotoxin-induced inflammatory and apoptotic responses in H9c2 cardiomyocytes. Molecular Medicine Reports 12 (1): 119–126.
Liu, C., J. Lo, C. Kuo, et al. 2009. Akt mediates 17β-estradiol and/or estrogen receptor-α inhibition of LPS-induced tumor necresis factor-α expression and myocardial cell apoptosis by suppressing the JNK1/2-NFκB pathway. Journal of Cellular and Molecular Medicine 13 (9B): 3655–3667.
Funding
This work was financially supported by the Natural Science Foundation of Hubei Province [grant number 2018CFC847].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zheng, Y., Li, S., Hu, R. et al. GFI-1 Protects Against Lipopolysaccharide-Induced Inflammatory Responses and Apoptosis by Inhibition of the NF-κB/TNF-α Pathway in H9c2 Cells. Inflammation 43, 74–84 (2020). https://doi.org/10.1007/s10753-019-01095-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-019-01095-x