
Abstract
Inflammation and reactive oxygen species (ROS) play crucial roles in the progression of chronic kidney diseases. Vitamin D has been shown anti-inflammatory effects, but the underlying mechanism is not fully understood. Here, we investigated whether calcitriol exerts protective effects via upregulating A20 in angiotensinII (AngII)-induced renal injury. Male C57BL/6 mice were infused with vehicle or AngII for 10 days. Calcitriol reduced infiltration of T lymphocytes and macrophages. This reduction of inflammatory cells was accompanied by elevated A20 and decreased pro-inflammatory cytokines (PICs) and reactive oxygen species (ROS). Calcitriol could inhibit NF-κB activation and necroptotic pathway. Induction of A20 was located primarily to the tubular epithelial cells. In rat proximal tubular epithelial cells (NRK-52E), calcitriol stably upregulated A20 and reduced the PICs and ROS. Inhibitory effect of A20 on PICs and ROS depended on suppressing NF-κB pathway and necroptotic pathway, respectively. A20 knockdown diminished the effect of calcitriol on suppressing NF-κB and necroptotic pathways. However, A20 deficiency could not abrogate the inhibitory effect of calcitriol on NF-κB and necroptotic pathways. Our results established that A20 is involved in the renoprotective effect by calcitriol via negatively modulating the NF-κB pathway and necroptotic pathway in AngII-induced renal injury.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Chronic kidney disease (CKD) progression to end stage renal disease (ESRD) has become a worldwide health problem. Inflammation and oxidative stress are regarded as underlying mechanisms in the progression of chronic kidney disease (CKD) [1, 2]. AngiotensinII (AngII)-induced pro-inflammatory cytokines (PICs) and reactive oxygen species (ROS) in renal cells cause injuries in an autocrine or paracrine way [3, 4].
Zinc finger protein A20, the product of TNFAIP3 gene, is a potent NF-κB pathway negative regulator and necroptosis inhibitor [5, 6]. Studies have exhibited A20 can function as both a ubiquitin ligase and a deubiquitinating enzyme [7].The dual roles in ubiquitination and deubiquitination enable A20 to disrupt the necroptotic pathway activation and NF-κB activation including phosphorylation of IKKβ [6, 8, 9]. Overexpressed or upregulated A20 achieves protection of renal cells against inflammation by suppressing NF-κB activation and necroptotic pathway in ischemia/reperfusion model, cytokine-induced inflammation, and LPS-mediated sepsis [10,11,12,13].
Active vitamin D is widely used to counteract loss of bone mass caused by glucocorticoids in clinical nephrology and in the correction of secondary hyperparathyroidism in CKD patients [14]. For a long time, vitamin D has been regarded as a pivotal bone mineral metabolism regulator. However, a number of studies showed vitamin D or its analogs reduced albuminuria independent of PTH suppression or impact on hemodynamics. The renoprotective effects of vitamin D are due to its pleiotropic functions such as downregulating renin gene expression [15], suppressing inflammation [16], and reducing ROS [17]. Moreover, active vitamin D was showed to exert cytoprotection against cytokine-induced apoptosis via upregulating zinc finger protein A20 [18].
Although vitamin D has been shown remarkable role in alleviating renal injury, the underlying mechanism is not fully understood. Considering the inhibitory effects of A20 on NF-κB activation and necroptotic pathway in acute organ injury and organ transplantation, we aimed to identify whether calcitriol ameliorates inflammation via A20 in AngII-induced chronic renal injury.
MATERIALS AND METHODS
Animals
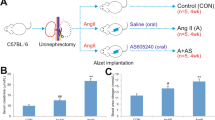
Male C57BL/6 mice aged 7–8 weeks were purchased from the Experimental Animal Center of Chongqing Medical University. This study was approved by the ethics committee of Chongqing Medical University. The mice underwent uninephrectomy (left) under chloralic hydras anesthesia and were allowed to recover for 1 week. The mice were assigned randomly into three groups (control, AngII, and AngII + calcitriol, n = 5 per group). Osmotic minipumps (Alzet model 2002; USA) were implanted subcutaneously in the backs to deliver AngII (Abcam, China) or 0.9% sterile saline at rate of 1500 ng/min/kg. The control group was given vehicle by gavage. The AngII-infused mice were orally given vehicle or calcitriol (5 μg/kg, three times per week; Selleck, China) [19]. All mice were sacrificed after 10 days.
Cell Culture
The rat renal tubular epithelial cell line NRK-52E was provided by Department of Nephrology of the First Affiliated Hospital of Chongqing Medical University. The cells were cultured in DMEM high glucose medium (Corning, USA) supplemented with 5% FBS (Bovogen, Australia) at 37 °C in a humidified incubator with 5% CO2. Cells were subcultured when confluence reached 80%.
Cell Lentivirus Transfection
An appropriate amount of lentivirus (Genechem Co. Ltd., China) containing negative control (shCTRL) and shRNA targeting rat TNFAIP3 (shA20) (multiplicity of infection: 60) was added into the medium with polybrene at 5 μg/ml when confluence reached 35–45%. The cells were again cultured in normal medium after 12 h of incubation. Enhanced-green fluorescent protein (eGFP)-tagged gene expression was observed under a fluorescence microscope 3–4 days after transfection. Cells with transfection efficiency >70% were selected for subsequent experiments.
Cell Treatment
A pilot experiment was performed to choose the best time-point for 10−6 M AngII (Sigma-Aldrich, USA) [20] to stimulated cells because AngII can induce compensatory upregulation of A20 [21]. 10−7 M calcitriol (Sigma-Aldrich, USA) was selected to treat NRK-52E [22]. ShCTRL cells or A20-knockdown (shA20) cells were pretreated with 10−7 M calcitriol 0.5 h before AngII exposure for the following 24 h. All treatments were done after 12 h of starvation with serum-free medium.
Histopathologic Analysis
The right kidneys were harvested, fixed in 4% paraformaldehyde then embedded in paraffin and cut at 4 μm. Hematoxylin and eosin (HE) staining was performed. Immunohistochemistry was performed according to PV-9000 kit instructions (ZSGB-BIO, China).The primary antibodies were anti-A20 (Novus, USA), anti-CD3 (Abcam, China), anti-F4/80 (Abcam, China), and anti-p65 (Proteintech, China). CD3+ cells and F4/80+ cells were counted under ×400 magnification observing ten consecutive nonoverlapping fields per animal.
Enzyme-Linked Immune Sorbent Assay (ELISA)
After treatments, the kidney tissue and cell medium were harvested. The supernatant was collected and preserved at −80 °C after being spun at 2000 RPM for 10 min. TNF-α and IL-6 in kidney homogenate and supernatant were determined according to the manufacturer’s instructions (Cloud-Clone Corp, USA).
Immunofluorescence
Cells were seeded on round glass coverslips. After treatments, cells were harvested, fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.5% TritonX-100 for 15 min, and blocked with goat serum for 30 min. The cells were incubated with mouse anti-A20 (Novus, USA), rabbit anti-NF-κB p65 (Proteintech, China) antibodies at 4 °C overnight, then incubated with goat anti-rabbit 549-labeled (Abbkine, USA) and goat anti-mouse FITC-labeled (Proteintech, China) secondary antibodies at room temperature for 1 h. After being counterstained with DAPI, the cells were visualized with laser confocal microscope (Leica TCP SP2, Germany).
Quantitative Real-time PCR
Total RNA was extracted from kidneys and NRK-52E cells with TRIzol Reagent (Invitrogen, China) according to the manufacturer’s instructions. Approximately 1000 ng of total RNA was reverse transcribed with the Prime Script® ™ RT Reagent Kit with gDNA Eraser (TaKaRa Biotechnology Inc., Japan) and amplified using SYBR Green (SYBR® Premix Ex TaqTM II, TaKaRa Biotechnology Inc., Japan). RT-qPCR was performed on an iQ5 Gradient Real-Time PCR detection system (Bio-Rad, USA). The primer sequences in Table 1.were used. Target gene expressions normalized to the β-actin gene were determined using the 2-∆∆Ct method.
ROS Level
The ROS level in kidney tissue was tested as previously described [23]. ROS production in NRK-52E was detected with a Reactive Oxygen Species assay kit (dihydroethidium, DHE, Beyotime, China) according to the manufacturer’s instructions.
Renal Function
Blood urea nitrogen and creatinine were determined according to the manufacturer’s instructions (Leagene, China).
Albuminuria
Mice were placed into metabolic cages to collect urine. Albumin and creatinine in the urine were determined with commercially available kits (Leagene, China) according to the manufacturer’s instructions.
Western Blotting
Total protein was extracted using RIPA buffer containing proteinase and phosphatase inhibitors. Cytoplasmic and nuclear proteins were extracted using a Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, China). Immunoblotting was performed according to standard procedures with mouse monoclonal anti-A20 (Novus, USA), rabbit anti-IKKβ (Proteintech, China), rabbit anti-IκBα (Proteintech, China), rabbit anti-phospho-IKKβ (Abcam, China), rabbit anti-phospho-IκBα (Abcam, China), rabbit anti-RIP1 (Cell Signaling Technology, China), rabbit anti-phospho-RIP3 (Abcam, China), rabbit anti-RIP3 (Abcam, China), rabbit anti-phospho-MLKL (Abcam, China), goat anti-MLKL (Santa Cruz, China), and β-actin (ZSGB-BIO, China) antibodies at 4 °C overnight. Conventional methods were used to perform the subsequent Western blotting procedures.
Statistical Analysis
All data were expressed as mean ± SD. Statistical analysis was performed with SPSS 22.0 using a t test and a one-way ANOVA test. P < 0.05 was considered statistically significant.
RESULTS
Upregulation of A20 Accompanied Suppression of Calcitriol on Inflammation In Vivo
In vivo, we observed that calcitriol induced A20 after 10-day administration of calcitriol primarily in tubular epithelial cells. In control and AngII groups, immunoblotting and qPCR exhibited A20 expression was low. Upregulated A20 was observed in calcitriol group. A20 immunohistochemical staining confirmed the immunoblotting and qPCR results (Fig. 1a).

Calcitriol alleviates inflammation in vivo accompanied by upregulated A20. a Expression of A20 in vivo after 10 days treatment. b mRNA and protein expression of TNF-α and IL-6 in vivo. c HE staining, CD3+ lymphocyte and F4/80+ macrophage staining. Values are shown as mean ± SD, n = 5 per group. **P < 0.01 among groups. ΔΔ P < 0.01 versus control.
TNF-α and IL-6 were drastically elevated by AngII treatment. Calcitriol decreased TNF-α and IL-6 (Fig. 1b). Meanwhile, tubulointerstitial inflammation was ameliorated by calcitriol, which was confirmed by HE staining, CD3+ lymphocyte and F4/80+ macrophage staining (Fig. 1c).
Decline of serum urea nitrogen, serum creatinine, and urine albumin/creatinine ratio confirmed calcitrol’s renoprotection biochemically. Serum calcium cation was improved by calcitriol than AngII group (Table 2.).
Calcitriol Suppressed NF-κB and Necroptotic Pathway In Vivo
Immunoblotting showed AngII significantly promoted phosphorylation of IKKβ and IκBα and translocation of p65 into nucleus. Administration of calcitriol reduced phosphorylated IKKβ and IκBα and prevented p65 translocation (Fig. 2a). Immunohistochemical staining of p65 showed that almost all p65 translocated into nucleus in AngII group. Calcitriol sequestered p65 in cytoplasm in some tubular epithelial cells. (Fig. 2b).

Calcitriol suppresses NF-κB signaling and necroptotic pathway in vivo. a Effect of calcitriol on NF-κB pathway. b Immunohistochemical staining of p65. c Effect of calcitriol on necroptotic pathway. d DHE analysis for the ROS level in the kidney tissue. Values are shown as mean ± SD, n = 5 per group. **P < 0.01 among groups.
We also observed that administration of AngII increased protein level of RIP1 and promoted phosphorylation of RIP3 and MLKL. Calcitriol decreased protein level of RIP1. Meanwhile, phosphorylation of RIP3 and MLKL was disrupted by calcitriol as well (Fig. 2c). The results showed elevated ROS levels in the AngII group tissue and noticeable reduction by calcitriol (Fig. 2d).
AngII Induced Transient Upregulation of A20 but Calcitriol Stably Upregulated A20
In vitro, we investigated the role of A20 in calcitriol’s renoprotection. In a pilot experiment, we treated NRK-52E with AngII or calcitriol for 24 h and examined the mRNA level of A20. We also determined the mRNA of TNF-α and IL-6 induced by AngII at different time points. A20 in AngII-treated cells was elevated after 6 h of exposure and then decreased. At 24 h, the mRNA level of A20 was not significantly different from the basal level. Meanwhile, A20 in calcitriol-treated cells gradually increased after 24-h exposure. Accordingly, upregulation of A20 after 24 h was due to calcitriol. There was no significant difference in the expression of TNF-α and IL-6 between the 6-h groups and 24-h groups. Thus, 24 h was selected as the exposure time for the following experiments (Fig. 3a). We successfully developed A20-knockdown NRK-52E with approximately 85% knock-down efficacy (Fig. 3b).

AngII induces transient upregulation of A20 and calcitriol stably upregulates A20. a Time-dependent response of A20 to AngII or calcitriol, time-dependent response of TNF-α and IL-6 to AngII. b A20 knockdown efficacy. Values are shown as mean ± SD. **P < 0.01 versus control or wild type, ***P < 0.001 versus control or wild type.
Role of A20 in Calcitriol’s Negative Modulation on NF-κB and Necroptotic Pathway in NRK-52E Cells
We used shCTRL and shA20 cells to investigate the role of A20 in calcitriol’s renoprotection. The secreted TNF-α and IL-6 were elevated in AngII-treated cells in shCTRL NRK-52E. Calcitriol could decrease TNF-α and IL-6. However, A20-knockdown cells showed higher expression levels of TNF-α and IL-6 than shCTRL. In terms of NF-κB signaling, calcitriol could reduce phosphorylated IKKβ, phosphorylated IκBα, and translocated p65 in shCTRL cells. Whereas, in A20-knockdown cells, Western blotting detected more phosphorylated IKKβ, phosphorylated IκBα, and more p65 in the nucleus than those in shCTRL (Fig. 4a).

Role of A20 in calcitriol’s negative modulation on NF-κB signaling and necroptotic pathway in NRK-52E cells. a Effect of calcitriol on TNF-α, IL-6, A20 and NF-κB pathway in shCTRL and shA20 NRK-52E. b Immunofluorescence of A20 and p65 after treatment in shCTRL and shA20 NRK-52E. c Effect of calcitriol on necroptotic pathway in shCTRL and shA20 NRK-52E. d Immunofluorescence of ROS indicated by DHE after treatment in shCTRL and shA20 NRK-52E. Data are representative of three independent experiments. Values are shown as mean ± SD. **P < 0.05, **P < 0.01 among groups.
Immunofluorescence showed A20 expression levels were low in control and AngII groups in shCTRL NRK-52E. Calcitriol could significantly upregulate A20 in shCTRL cells. We could observe that p65 translocation was triggered by AngII; however, upregulated A20 by calcitriol sequestered p65 translocation. In the shA20 NRK-52E, due to A20 deficiency, AngII stimulated more p65 into nucleus (Fig. 4b).
Similar to NF-κB pathway, AngII induced RIP1 protein expression and promoted phosphorylation of RIP3 and MLKL. Calcitriol downregulated RIP1 expression and impaired phosphorylation of RIP3 and MLKL. A20 deficiency diminished calcitirol’s inhibitory effects on necroptotic pathway (Fig. 4c). Downstream ROS generation indicated by DHE was decreased by calcitriol. Deficiency of A20 weakened calcitriol’s capability to reduce ROS (Fig. 4d).
DISCUSSION
PICs and ROS induced by overproduced AngII are deleterious factors to renal cells in the self-sustained progression of CKD. The results presented in our study demonstrated that calcitriol displayed a potent anti-inflammatory effect evidenced by inhibited inflammatory cells infiltration, reduced pro-inflammatory cytokines, and alleviated ROS production in an AngII-infused mouse model. Calcitriol induced A20 to disrupt the activation of IKKβ, thereby suppressing translocation of p65 into nucleus. Necroptotic pathway was also disrupted by upregulated A20 in vivo and in vitro, leading to reduction of ROS. A20 knockdown did not abrogate inhibitory effect of calcitriol on NF-κB and necroptotic pathway. Nonetheless, A20 knockdown diminished the negative modulation of calcitriol on NF-κB signaling and necroptotic pathway.
In vivo, we found calcitriol ameliorated renal inflammation and renal dysfunction, which was evidenced by alleviated CD3(+) lymphocyte and F4/80(+) macrophage infiltration, downregulated TNF-α and IL-6, and reduced proteinuria. The underlying mechanism of vitamin D or analogs in renoprotection is quite complex. Evidence has shown that several pathways contributing to kidney injury, such as NF-κB [16], RAAS [15], and slit diaphragm proteins [24] are engaged in the protection mechanisms of calcitriol. However, the viewpoints on how calcitriol or its analogs disrupts NF-κB activation are discrepant or even contradictory. Tan et al. showed paricalcitol induced the formation of a complex containing VDR and NF-κB p65 components thus reducing p65 binding to target genes [16], but IκB phosphorylation and p65 translocation were not disrupted by paricalcitol. Nonetheless, stabilization of IκB by calcitriol leading to an inhibition of p65 translocation to the nucleus was also considered to be one mechanism of downregulation of NF-κB signaling pathway [25]. Chen et al. [26] reported VDR-IKKβ interaction blocks IKK complex formation and IKKβ phosphorylation. They also pointed out calcitriol enhances the interaction. Similar to Chen’s finding, we observed suppression of the canonical NF-κB pathway involving phosphorylation of IKKβ and IκBα by calcitriol in this study. Unlike Tan’s finding, in our study, administration of calcitriol disrupted IκB phosphorylation and sequestered p65 in cytoplasm in vivo and in vitro.
Although the expression of A20 is low in most cells under basal conditions, it can be induced by a number of stimuli including AngII and calcitriol [18, 21, 27].We found A20 in tubular epithelial cells was stably upregulated by calcitriol in vivo and in vitro. AngII can affect the transcription of multiple genes via activating NF-κB including transiently upregulating A20 gene TNFAIP3. A20 in turn inhibits activation-induced NF-κB to restrict its overactivation [28]. Thus, finding some means to induce A20 without initiating an inflammatory response to struggle against inappropriate inflammation is quite significant in CKD. In our study, Ang II and calcitriol could both induce A20. However, upregulation of A20 by Ang II was compensatory because, after the peak at approximately 3–6 h, the expression of A20 decreased gradually to the basal level. Twenty-four hours was selected for the treatment time course to investigate the effect of calcitriol on A20.Consistent with Riachy’s study [18], we found calcitriol could still prominently upregulate A20 at 24 h.
We developed A20-knockdown NRK-52E to determine the role of A20 in the protection by calcitriol against PICs and ROS production in NRK-52E. Calcitriol reduced TNF-α and IL-6 production induced by Ang II in both shCTRL and shA20 NRK-52E cells. This indicates that A20 knockdown could not abrogate renoprotection of calcitriol. Similar to the TNF-α and IL-6, phosphorylations of IKKβ and IκBα were disrupted by calcitriol. Meanwhile, the same occurred in A20-knockdown cells. However, the significant differences between shCTRL and shA20 groups revealed that A20 knockdown indeed weakened calcitriol’s negative role in regulating NF-κB signaling, but A20 was not the only mediator through which calcitriol suppresses NF-κB pathway. A20 maintains the balance of inflammatory responses and prevents overactive or prolonged inflammation in the canonical NF-κB signaling pathway. A20 has been shown to have a renoprotective effect in various renal injury models, such as ischemia/reperfusion injury [13], and pristine-induced lupus nephritis [29]. Nonetheless, A20’s precise inhibitory mechanism still remains poorly understood. Initially, A20 was observed to ubiquitinate or deubiquitinate upstream adaptors such as RIP1, TRAF6, and TRADD to negatively regulate the NF-κB pathway. However, Skaug recently showed A20 even directly inhibits IKK activity independently of its deubiquitinase activity in a manner that depends upon binding to NEMO [30]. In support of Skaug’s study, Zilberman-Rudenko demonstrated that failure to recruit A20 due to C-terminal mutations in NEMO enhanced IKK kinase activity and targeting this molecular interaction by enhancing A20 expression may be a therapeutic strategy [31].
In addition to PICs, we observed administering calcitriol to mice and NRK-52E reduced ROS production. A20 participated in the reduction of ROS by calcitriol via inhibiting necroptotic pathway. RIP1 expression was downregulated and phosphorylation of RIP3 and MLKL were impaired in our study. RIP1, RIP3, and MLKL are core proximal components of necroptotic pathway although downstream molecular componentry of the pathway is poorly understood. Activation of necroptotic pathway results in ROS generation. In necroptotic pathway, multiple proteins such as glutamate–ammonia ligase (GLUL), PGAM5, Drp1, and even RIP1/RIP3 themselves were reported to be engaged in ROS generation; however, RIP1, RIP3, and MLKL play pivotal role [32,33,34]. ROS production via necroptotic pathway has been revealed in CKD as well as in acute kidney injury [23]. A20 inhibits necroptotic pathway by adding K48-linked polyubiquitin chains to RIP1 and targeting it for proteasomal degradation. A recent study has shown that A20 suppresses activation of necroptotic pathway by restricting RIP3 ubiquitination at lysine 5 (K5) [6]. The differences between AngII + shCTRL and AngII + shA20 + calcitriol groups suggest that other mechanisms may contribute to inhibitory effects of calcitriol on necroptotic pathway in addition to A20.
Unveiling the role of A20 in calcitriol’s renoprotection, we will carry on investigating the role of calcitriol-upregulated A20 in different CKD model based on our previous work. Taken together, the current findings suggest the involvement of zinc finger protein A20 in the renoprotection of calcitriol against inflammation and ROS generation in an AngII-induced renal injury model. Vitamin D deficiency is recognized to be a poorer-outcome predictor in CKD progression [35]. Accordingly, it is rational to believe that Vitamin D holds promise to achieve better renal cell protection.
References
Stenvinkel, P. 2006. Inflammation in end-stage renal disease: The hidden enemy. Nephrology (Carlton) 11 (1): 36–41. doi:10.1111/j.1440-1797.2006.00541.x.
Holterman, C.E., N.C. Read, and C.R. Kennedy. 2015. Nox and renal disease. Clinical Science (London, England) 128 (8): 465–481. doi:10.1042/CS20140361.
Ruiz-Ortega, M., M. Ruperez, O. Lorenzo, V. Esteban, J. Blanco, S. Mezzano, and J. Egido. 2002. Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney International. Supplement 82: S12–S22. doi:10.1046/j.1523-1755.62.s82.4.x.
Kim, S.M., Y.G. Kim, K.H. Jeong, S.H. Lee, T.W. Lee, C.G. Ihm, and J.Y. Moon. 2012. Angiotensin II-induced mitochondrial Nox4 is a major endogenous source of oxidative stress in kidney tubular cells. PloS One 7 (7): e39739. doi:10.1371/journal.pone.0039739.
Jaattela, M., H. Mouritzen, F. Elling, and L. Bastholm. 1996. A20 zinc finger protein inhibits TNF and IL-1 signaling. Journal of Immunology 156 (3): 1166–1173.
Onizawa, M., S. Oshima, U. Schulze-Topphoff, J.A. Oses-Prieto, T. Lu, R. Tavares, T. Prodhomme, et al. 2015. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nature Immunology 16 (6): 618–627. doi:10.1038/ni.3172.
Verstrepen, L., K. Verhelst, G. van Loo, I. Carpentier, S.C. Ley, and R. Beyaert. 2010. Expression, biological activities and mechanisms of action of A20 (TNFAIP3). Biochemical Pharmacology 80 (12): 2009–2020. doi:10.1016/j.bcp.2010.06.044.
Wertz, I.E., K.M. O'Rourke, H. Zhou, M. Eby, L. Aravind, S. Seshagiri, P. Wu, et al. 2004. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430 (7000): 694–699. doi:10.1038/nature02794.
Wertz, I.E., K. Newton, D. Seshasayee, S. Kusam, C. Lam, J. Zhang, N. Popovych, et al. 2015. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 528 (7582): 370–375. doi:10.1038/nature16165.
Lutz, J., A. Luongle, M. Strobl, M. Deng, H. Huang, M. Anton, M. Zakkar, et al. 2008. The A20 gene protects kidneys from ischaemia/reperfusion injury by suppressing pro-inflammatory activation. J Mol Med (Berl) 86 (12): 1329–1339. doi:10.1007/s00109-008-0405-4.
Lee, E.G., D.L. Boone, S. Chai, S.L. Libby, M. Chien, J.P. Lodolce, and A. Ma. 2000. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 289 (5488): 2350–2354.
Kunter, U., S. Daniel, M.B. Arvelo, J. Choi, T. Shukri, V.I. Patel, C.R. Longo, et al. 2005. Combined expression of A1 and A20 achieves optimal protection of renal proximal tubular epithelial cells. Kidney International 68 (4): 1520–1532. doi:10.1111/j.1523-1755.2005.00564.x.
Yang, Z., Z. Zhong, M. Li, Y. Xiong, Y. Wang, G. Peng, and Q. Ye. 2016. Hypothermic machine perfusion increases A20 expression which protects renal cells against ischemia/reperfusion injury by suppressing inflammation, apoptosis and necroptosis. International Journal of Molecular Medicine 38 (1): 161–171. doi:10.3892/ijmm.2016.2586.
Ketteler, M., G.J. Elder, P. Evenepoel, J.H. Ix, S.A. Jamal, M.H. Lafage-Proust, R. Shroff, et al. 2015. Revisiting KDIGO clinical practice guideline on chronic kidney disease-mineral and bone disorder: A commentary from a kidney disease: Improving global outcomes controversies conference. Kidney International 87 (3): 502–528. doi:10.1038/ki.2014.425.
Zhang, Y., J. Kong, D.K. Deb, A. Chang, and Y.C. Li. 2010. Vitamin D receptor attenuates renal fibrosis by suppressing the renin-angiotensin system. Journal of the American Society of Nephrology 21 (6): 966–973. doi:10.1681/ASN.2009080872.
Tan, X., X. Wen, and Y. Liu. 2008. Paricalcitol inhibits renal inflammation by promoting vitamin D receptor-mediated sequestration of NF-kappaB signaling. Journal of the American Society of Nephrology 19 (9): 1741–1752. doi:10.1681/ASN.2007060666.
Garcia, I.M., L. Altamirano, L. Mazzei, M. Fornes, M.N. Molina, L. Ferder, and W. Manucha. 2012. Role of mitochondria in paricalcitol-mediated cytoprotection during obstructive nephropathy. American Journal of Physiology. Renal Physiology 302 (12): F1595–F1605. doi:10.1152/ajprenal.00617.2011.
Riachy, R., B. Vandewalle, J. Kerr Conte, E. Moerman, P. Sacchetti, B. Lukowiak, V. Gmyr, T. Bouckenooghe, M. Dubois, and F. Pattou. 2002. 1,25-dihydroxyvitamin D3 protects RINm5F and human islet cells against cytokine-induced apoptosis: Implication of the antiapoptotic protein A20. Endocrinology 143 (12): 4809–4819. doi:10.1210/en.2002-220449.
Gregori, S., M. Casorati, S. Amuchastegui, S. Smiroldo, A.M. Davalli, and L. Adorini. 2001. Regulatory T cells induced by 1 alpha,25-dihydroxyvitamin D3 and mycophenolate mofetil treatment mediate transplantation tolerance. Journal of Immunology 167 (4): 1945–1953.
Rice, E.K., G.H. Tesch, Z. Cao, M.E. Cooper, C.N. Metz, R. Bucala, R.C. Atkins, and D.J. Nikolic-Paterson. 2003. Induction of MIF synthesis and secretion by tubular epithelial cells: A novel action of angiotensin II. Kidney International 63 (4): 1265–1275. doi:10.1046/j.1523-1755.2003.00875.x.
Romero, D.G., M. Plonczynski, G.R. Vergara, E.P. Gomez-Sanchez, and C.E. Gomez-Sanchez. 2004. Angiotensin II early regulated genes in H295R human adrenocortical cells. Physiological Genomics 19 (1): 106–116. doi:10.1152/physiolgenomics.00097.2004.
Geilen, C.C., M. Bektas, T. Wieder, V. Kodelja, S. Goerdt, and C.E. Orfanos. 1997. 1alpha,25-dihydroxyvitamin D3 induces sphingomyelin hydrolysis in HaCaT cells via tumor necrosis factor alpha. The Journal of Biological Chemistry 272 (14): 8997–9001.
Zhu, Y., H. Cui, H. Gan, Y. Xia, L. Wang, Y. Wang, and Y. Sun. 2015. Necroptosis mediated by receptor interaction protein kinase 1 and 3 aggravates chronic kidney injury of subtotal nephrectomised rats. Biochemical and Biophysical Research Communications 461 (4): 575–581. doi:10.1016/j.bbrc.2015.03.164.
Wang, Y., D.K. Deb, Z. Zhang, T. Sun, W. Liu, D. Yoon, J. Kong, Y. Chen, A. Chang, and Y.C. Li. 2012. Vitamin D receptor signaling in podocytes protects against diabetic nephropathy. Journal of the American Society of Nephrology 23 (12): 1977–1986. doi:10.1681/ASN.2012040383.
Zhang, Z., W. Yuan, L. Sun, F.L. Szeto, K.E. Wong, X. Li, J. Kong, and Y.C. Li. 2007. 1,25-Dihydroxyvitamin D3 targeting of NF-kappaB suppresses high glucose-induced MCP-1 expression in mesangial cells. Kidney International 72 (2): 193–201. doi:10.1038/sj.ki.5002296.
Chen, Y., J. Zhang, X. Ge, J. Du, D.K. Deb, and Y.C. Li. 2013. Vitamin D receptor inhibits nuclear factor kappaB activation by interacting with IkappaB kinase beta protein. The Journal of Biological Chemistry 288 (27): 19450–19458. doi:10.1074/jbc.M113.467670.
da Silva, C.G., E.R. Maccariello, S.W. Wilson, P. Putheti, S. Daniel, S.M. Damrauer, C.R. Peterson, J.J. Siracuse, E. Kaczmarek, and C. Ferran. 2012. Hepatocyte growth factor preferentially activates the anti-inflammatory arm of NF-kappaB signaling to induce A20 and protect renal proximal tubular epithelial cells from inflammation. Journal of Cellular Physiology 227 (4): 1382–1390. doi:10.1002/jcp.22851.
He, K.L., and A.T. Ting. 2002. A20 inhibits tumor necrosis factor (TNF) alpha-induced apoptosis by disrupting recruitment of TRADD and RIP to the TNF receptor 1 complex in Jurkat T cells. Molecular and Cellular Biology 22 (17): 6034–6045.
Li, M., X. Shi, T. Qian, J. Li, Z. Tian, B. Ni, and F. Hao. 2015. A20 overexpression alleviates pristine-induced lupus nephritis by inhibiting the NF-kappaB and NLRP3 inflammasome activation in macrophages of mice. International Journal of Clinical and Experimental Medicine 8 (10): 17430–17440.
Skaug, B., J. Chen, F. Du, J. He, A. Ma, and Z.J. Chen. 2011. Direct, noncatalytic mechanism of IKK inhibition by A20. Molecular Cell 44 (4): 559–571. doi:10.1016/j.molcel.2011.09.015.
Zilberman-Rudenko, J., L.M. Shawver, A.W. Wessel, Y. Luo, M. Pelletier, W.L. Tsai, Y. Lee, et al. 2016. Recruitment of A20 by the C-terminal domain of NEMO suppresses NF-kappaB activation and autoinflammatory disease. Proceedings of the National Academy of Sciences of the United States of America 113 (6): 1612–1617. doi:10.1073/pnas.1518163113.
Kanamaru, Y., S. Sekine, H. Ichijo, and K. Takeda. 2012. The phosphorylation-dependent regulation of mitochondrial proteins in stress responses. Journal of Signal Transduction 2012: 931215. doi:10.1155/2012/931215.
Zhang, D.W., J. Shao, J. Lin, N. Zhang, B.J. Lu, S.C. Lin, M.Q. Dong, and J. Han. 2009. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325 (5938): 332–336. doi:10.1126/science.1172308.
Arora, D., P.K. Sharma, M.H. Siddiqui, and Y. Shukla. 2017. Necroptosis: Modules and molecular switches with therapeutic implications. Biochimie. doi:10.1016/j.biochi.2017.02.015.
Li, X.H., X.P. Huang, L. Pan, C.Y. Wang, J. Qin, F.W. Nong, Y.Z. Luo, et al. 2016. Vitamin D deficiency may predict a poorer outcome of IgA nephropathy. BMC Nephrology 17 (1): 164. doi:10.1186/s12882-016-0378-4.
Acknowledgments
We thank Lixue Chen, WeixueTang, Jingmei Xie, Yao Xiao, Xiaojuan Deng, and Guangchen Qin at the central laboratory of the First Affiliated Hospital of Chongqing Medical University for their assistance during the experiments.
Author information
Authors and Affiliations
Contributions
Authors’ Contributions
Hongfei Zhao conceived the study and designed the experiments. Hongfei Zhao performed all the experiments, analyzed the data, and wrote the manuscript. Hua Gan and Yunfeng Xia reviewed and revised the paper.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Zhao, H., Xia, Y. & Gan, H. Calcitriol Ameliorates AngiotensinII-Induced Renal Injury Partly via Upregulating A20. Inflammation 40, 1884–1893 (2017). https://doi.org/10.1007/s10753-017-0629-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-017-0629-y