
Abstract
Septic acute kidney injury (AKI) is a public health problem with high mortality. Suppression of over-active inflammation is considered as a promising strategy for septic AKI. In this study, we evaluated the prophylactic effect of interleukin (IL)-35, the unique immune-suppressive member of IL-12 cytokine family, on lipopolysaccharide (LPS)-induced AKI in mice, and found that compared with control mice given empty vector, mice pretreated with plasmid encoding IL-35 (pIL-35) significantly improved renal function indicated by reduced blood urea nitrogen (BUN) and serum creatinine (SCr), and obviously alleviated renal pathological changes. To explore the underlying protective mechanisms, we found that pIL-35 treatment could robustly reduce the production of renal pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β), with no significant impact on IL-10, an anti-inflammatory cytokine. Furthermore, our results revealed that IL-35 pretreatment could potentially inhibit the activation of renal NF-κB signaling pathway in LPS-induced AKI mice. Taken together, our study indicated that IL-35 pretreatment could efficiently prevent LPS-induced AKI via inhibiting NF-κB activation and reducing pro-inflammatory cytokine production, and it might represent a novel therapeutic strategy against septic AKI and other inflammatory renal diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Acute kidney injury (AKI), an abrupt deterioration in renal function, is primarily characterized by the disruption in the fluid maintenance, electrolyte homeostasis as well as metabolism, and often diagnosed by a sudden reduce in glomerular filtration rate (GFR), increased serum creatinine (SCr) or oliguria. Generally, AKI is temporary but will have a negative effect on long-term renal function. More importantly, a third of patients who recovered from AKI will develop chronic kidney disease [4]. As revealed by a large-scale meta-analysis, AKI, a common and increasing complication in hospitalized patients with acute illness, occurs in 21.6% adults and 33.7% children [33]. Of note, in recent years, although the mortality of AKI is decreasing, the absolute number of deaths is increasing due to the increased incidence [26]. In sharp contrast, no approved AKI therapy is available so far as a result of limited candidate drugs or biological strategy in clinical trials. Therefore, the development of novel and efficient therapeutic strategies is urgently needed.
There are convincing evidences that sepsis makes the most frequent and important contributor to AKI [1, 23, 31], especially in critically ill patients, who have 50% or more occurrence and high mortality [36]. To date, our understanding of the septic AKI is mainly derived from studies on murine models, among which endotoxin lipopolysaccharide (LPS) injection model was the most widely used [7, 9, 14]. It has been proven that LPS-induced AKI shares many characteristics with human diseases, including augmented pro-inflammatory cytokines, changed hemodynamics as well as direct renal injury [7]. All these alterations result in the accumulation of renal inflammatory cells (neutrophils and macrophages) as well as decreased renal function reflected by reduced glomerular filtration rate (GFR) and increased blood urea nitrogen (BUN) and serum creatinine (SCr) [19, 32]. Therefore, corresponding prophylactic or therapeutic strategies targeting these pathological conditions have been developed [17]. Among them, immune modulation methods have already attracted more and more interests and showed promising potential in murine AKI models [8, 27, 42].
Interleukin (IL)-35, consisting of the p35 subunit of IL-12 (encoded by IL-12 α chain) and Epstein-Barr virus induced 3 (EBi3) chain, is a recently identified member of IL-12 cytokine family. Compared with other members (IL-12, IL-23, and IL-27) which are primarily produced by activated antigen-presenting cells (APC), IL-35 is mainly secreted by regulatory T cells [2, 6], regulatory B cells, [30] as well as plasma cells [30], and it is the unique one strictly functioning as an immune suppressive cytokine in IL-12 family. Therefore, it represents a potential novel target for immune manipulation measures against autoimmune diseases [12, 22] as well as cancers [10, 16, 35]. It has been proven that after binding its receptor, IL-35 could exert its immune suppressive functions by facilitating the differentiation of regulatory T (Treg) [3, 34] and B (Breg) cells [29], inhibiting T cell proliferation [41] and T-Helper 17 (Th17) differentiation [24] as well as manipulating the balance between Treg and Th17 [40], etc. However, a recent study reported that IL-35 inhibited the angiogenesis and inflammation in rheumatoid arthritis by reducing angiopoietin-2 [15], indicating that the exact role of IL-35 in immune modulation is still vastly unclear.
Considering the critical pathological role of over-active inflammation in the initiation and development of septic AKI, it is feasible and practicable to apply immune suppressors or modulators to prevent or alleviate AKI. In the present study, plasmid encoding IL-35 (pIL-35) was injected into mice 7 days before LPS administration, and its prophylactic effects against LPS-induced AKI were evaluated.
MATERIALS AND METHODS
Mice
C57BL/6 (H-2b) male mice, 6–8 weeks old, were obtained from the experimental animal center of Chinese Academy of Science (Shanghai, China) and housed in pathogen-free mouse colonies under room temperature with 12-h light/dark cycles. All animal experiments were carried out in strict according to the guidelines for the Care and Use of Laboratory Animals. The protocol was approved by the Laboratory Animal Ethical Commission of Soochow University.
Plasmid Injection
Plasmid pcDNA3.1-IL-35 (pIL-35) was constructed as previously describe [21]. Mice were given pIL-35, empty vector (pcDNA3.1) or PBS by a hydrodynamic-based gene transfer technique via rapid injection as described by Liu et al. [20]. Briefly, 100 μg plasmid DNA was resolved in 2.0 mL PBS and injected via the tail vein within 5 to 10 s.
Establishment of a Murine Model of LPS-Induced AKI
Seven days post the hydrodynamic delivery of pIL-35, mice were intraperitoneally injected with LPS (Escherichia coli 0111:B4, Sigma-Aldrich, St. Louis, MO) at 10 mg/kg. The dose of LPS was determined by the preliminary experiments in which the minimal dose caused 50% death 24 h post injection was finally chosen. Serum and kidney tissues were collected at 12, 24, or 48 h post LPS injection and subjected to the determination of renal function, histological observation as well as cytokine analysis.
Measurement of Serum Creatinine (SCr) and Blood Urea Nitrogen (BUN)
Serum samples of mice were obtained via retroorbital venous plexus, and levels of SCr and BUN were determined by a BS-800 Chemistry Analyzer (Mindray).
Histological Examination
Kidney tissues were removed at indicated time points post LPS injection followed by fixation, section as well as hematoxylin and eosin staining. Sections were examined by two independent investigators in a blinded manner, and renal tubular injury was assessed using previously described 0–4 scale [9], in which 0, none; 1, < 10%; 2, 10–25%; 3, 25–75%; or 4, > 75%. Each group contained 5–6 animals.
ELISA Assays
Kidney tissues were homogenized in 0.5 ml RIPA buffer. After centrifugation in a cooled micro-centrifuge, the supernatants were collected and the total protein concentration was determined and adjusted to 1 mg/ml, and then subjected to ELISA assays with mouse ELISA Kits for TNF-α, IL-6 as well as IL-1β (eBioscience).
Western Blot
The supernatants of kidney homogenates were collected and subjected to SDS-PAGE. After transferring onto nitrocellulose membranes and blocking with 3% BSA-PBST, the blots were probed with primary antibodies against IKK, phosphorylated IKK (p-IKK), NF-κB p65 and phosphorylated p65 (p-p65) (CST, rabbit anti mouse). After washing with PBST, membranes were further incubated with an appropriate HRP-labeled goat anti-rabbit secondary antibody (Southern Biotech). The blots were developed with enhanced chemiluminescence (Thermo Scientific Pierce) according to the manufacturer’s instructions.
Statistical Analysis
All data are expressed as mean ± SEM. Statistical significance of differences in 2 groups or more than 2 groups was respectively determined by t test or one-way ANOVA followed by Bonferroni test using GraphPad Prism version 5.0 (GraphPad Software Incorporated). P values less than 0.05 were considered statistically significant.
RESULTS
IL-35 Pretreatment Significantly Alleviated LPS-Induced Renal Injury
Mice were injected with LPS 7 days after hydrodynamic tail vein injection of pIL-35, and SCr and BUN levels were evaluated at 12, 24 and 48 h later. As shown in Fig. 1, the levels of SCr and BUN in the control group obviously increased at as early as 12 h post LPS treatment and continued to increase as time went by, while these two indices in the pIL-35 pretreated group were potentially reduced at all indicated time points, indicating that pIL-35 could protect against LPS-induced damage of renal function. In line with these data, improved renal pathological changes were also observed in pIL-35 pre-treated mice. As shown in Fig. 2, after LPS injection, control group showed severe renal pathological lesions including prominent infiltration of inflammatory cell in glomeruli, loss of brush border, and tubule dilatation in both proximal and distal tubules. In contrast, these damages were remarkably attenuated in pIL-35 pre-treated group. All these data indicated that IL-35 could efficiently prevent LPS-induced AKI.

IL-35 pretreatment improved serum BUN and SCr in LPS-induced AKI mice. Serum was collected at indicated time points after LPS injection, and the levels of BUN (a) and SCr (b) were detected. Each group contained 5 or 6 mice. Each experiment was repeated for 3 times and the representative data were shown. * P < 0.05; ** P < 0.01; *** P < 0.001.

IL-35 pretreatment alleviated renal pathological injury in LPS-induced AKI mice. Paraffin sections of renal tissues were prepared and histological alterations was revealed by HE staining. a One representative section was shown for each group, magnification ×200. b Kidney injury score. Each experiment was repeated for 3 times and the representative data were shown. N.D. not detected, * P < 0.05; ** P < 0.01; *** P < 0.001.
IL-35 Pretreatment Significantly Decreased Renal Pro-inflammatory Cytokine Production in LPS-Induced AKI Mice
Over-active inflammation plays an important pathological role in AKI development to investigate whether IL-35 pre-treated could impact the inflammatory responses in LPS-induced AKI mice; renal levels of pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) were determined. As shown in Fig. 3, compared with the control group, pIL-35 pretreatment robustly decreased the renal TNF-α, IL-6, and IL-1β production, suggesting that IL-35 could potently inhibit the excessive local renal pro-inflammatory cytokine production in LPS-induced AKI mice.

IL-35 pretreatment reduced renal pro-inflammatory cytokine production in LPS-induced AKI mice. Renal tissues were collected 12, 24, and 48 h after LPS injection for ELISA determination of TNF-α (a), IL-6 (b), and IL-1β (c). Each group contained 5 or 6 mice. Each experiment was repeated for 3 times and the representative data were shown. * P < 0.05; ** P < 0.01; *** P < 0.001.
IL-35 Pretreatment Had No Significant Impact on the Production of Anti-inflammatory Cytokine IL-10 in Renal Tissues
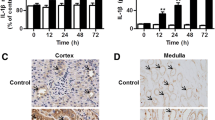
As previous study showed that IL-35 could increase the serum anti-inflammatory cytokine IL-10 which possesses the potent ability to suppress immune responses [24], here, we tried to find out whether the IL-35-caused decreased pro-inflammatory cytokine production could be attributed to the increased IL-10 production. As shown in Fig. 4, consistent with previous study [7], obviously increased renal IL-10 level was evidenced in LPS-induced AKI mice, while comparable IL-10 production was also evidenced in IL-35 pre-treated mice, indicating that IL-35 did not impact the renal IL-10 production, in another words, IL-35 did not exert its anti-inflammation effect via promoting renal IL-10 production.

IL-35 pretreatment had no significant impact on renal anti-inflammatory cytokine IL-10 production. Renal tissues were collected 12, 24, and 48 h after LPS injection for ELISA determination of IL-10. Each group contained 5 or 6 mice. Each experiment was repeated for 3 times and the representative data were shown. n.s. no significance.
IL-35 Pretreatment Obviously Inhibited the Renal NF-κB Pathway Activation
As pro-inflammatory cytokine production was under the control of NF-κB signaling pathway, therefore, we next investigated the influence of IL-35 on the renal NF-kB pathway activation. As shown in Fig. 5, IL-35 pre-treated could obviously inhibited the phosphorylation of IKK and NF-κB p65 components as early as 12 h post LPS injection, and this suppressive effect could be sustained to 48 h, indicating the persistent inhibitive function of IL-35 on the renal NF-κB pathway activation.

IL-35 pretreatment inhibited the activation of renal NF-κB signaling pathway in LPS-induced AKI mice. a Protein levels of renal IKK, phosphorylated IKK (p-IKK), NF-κB p65, and phosphorylated NF-κB p65 (p-p65) were detected by Western blot assays. b Semi-quantification analysis of renal expression of p-IKK and p-p65. Each experiment was repeated for 3 times and the representative data were shown. ** P < 0.01; *** P < 0.001.
DISCUSSION
AKI is a complicated and multi-factor involved disease and characterized by rapid loss of renal function. Causes of AKI are generally categorized into 3 groups: pre-renal (reducing blood supply to the kidneys), renal (damaging kidney tissue, e.g., accident, sepsis) or post-renal (blocking urine leaving the kidney). A previous study reported that about 50% AKI are related to sepsis [39]. Besides, the combination of sepsis could greatly increase the mortality of acute renal failure from 45 to 70% [13]. So far, only activated protein C has been approved by FDA as the therapy to sepsis, and increasing concern has been raised about its efficiency and safety. Meanwhile, to date, no efficient AKI treatment strategies are available. All these dramatized the urgent need to deepen our understanding of septic AKI pathogenesis and to develop novel prophylactic or therapeutic measures.
Excessive inflammation is considered as the critical factor that initiates and aggravates AKI [39]. Moreover, AKI could also lead to vigorous inflammation in both renal and extrarenal organs [11], further exacerbates the renal dysfunction and tissue damages. Based on this, prevention or alleviation of over-active inflammation at AKI initiation phrase represents a promising therapeutic strategy [14] and its feasibility and efficiency has also been evidenced in murine AKI models [39].
IL-35, the recently identified and processing immune suppressive member of IL12 family, has been well-known for its immune-suppressive effects on adaptive immune responses, such as inhibiting T cell proliferation, promoting the induction and expansion of Treg cells [6] and re-balancing Treg and Th17 [40]; therefore, IL-35 has been increasingly applied to the development of novel control measures against autoimmunity and inflammatory diseases [12, 22]. However, the impact of IL-35 on innate immune responses is still not clear.
A recent study revealed that lack of IL-35 enhanced LPS-induced airway eosinophilia in EBI3-deficient mice [18], suggesting that IL-35 also possess the ability to regulate innate immunity. In the present study, we found that IL-35 pre-treated could significantly decrease the renal production of pro-inflammatory cytokines TNF-α, IL-6 as well as IL-1β which play important pathological roles in LPS-induced AKI [5, 25, 37], further confirming the suppressive effect of IL-35 on inflammation responses as reported by Kanai et al. [18]. In addition, our study further extended the innate immunity modulation ability of IL-35 from lung to kidney tissues. In line with the reduced pro-inflammatory cytokines, IL-35 pre-treated potentially inhibited renal NF-κB signaling pathway activation, evidenced by the decreased phosphorylated IKK and NF-κB p65.
Previous studies showed that IL-35 could promote the serum level of anti-inflammatory cytokine IL-10 [24], which could potentially inhibit tissue inflammation via inhibit NF-κB pathway [28]. However, in this study, we did not observe the obvious alteration in renal IL-10 level, indicating that IL-35 was not dependent on IL-10 to exert its anti-inflammation effect. Osamu Yoshie and his colleagues [18] demonstrated that IL-35 possessed the ability to inhibit the production of CCL11 and CCL24 of lung epithelia cells. As CCL11 is under the control of NF-κB signaling pathway [38], in their LPS-induced acute airway inflammation model, it is possible that IL-35 might suppress this chemokine production via inhibiting NF-κB. Besides, since infiltrating inflammatory cells are the major source of pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β), IL-35 might also suppress renal inflammation in our AKI model via inhibiting NF-κB-associated chemokine production, leading to the fever infiltrating inflammatory cells and consequent decreased pro-inflammatory cytokine levels, which further weakened NF-κB-associated renal inflammation, alleviated tissue damage and improved renal function.
Taken together, in this study, we investigated the preventive effect of IL-35 on LPS-induced AKI and found that it could obviously improve renal function and pathological changes. This efficient prophylactic effect could be ascribed to its ability to suppress NF-κB pathway activation as well as associated pro-inflammatory cytokine production, albeit of no significant impact on anti-inflammatory cytokine IL-10 production. Our study indicated that IL-35 may represent a promising and feasible preventive or curative strategy for septic AKI or other renal inflammatory diseases. In addition, it would also help us to gain a better understanding of the modulation effects of IL-35 on innate immune responses.
References
Bagshaw, S.M., K.B. Laupland, C.J. Doig, G. Mortis, G.H. Fick, M. Mucenski, T. Godinez-Luna, L.W. Svenson, and T. Rosenal. 2005. Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: A population-based study. Critical Care 9 (6): R700–R709. doi:10.1186/cc3879.
Bardel, E., F. Larousserie, P. Charlot-Rabiega, A. Coulomb-L'Hermine, and O. Devergne. 2008. Human CD4+ CD25+ Foxp3+ regulatory T cells do not constitutively express IL-35. Journal of Immunology 181 (10): 6898–6905.
Castellani, M.L., A. Anogeianaki, P. Felaco, E. Toniato, M.A. De Lutiis, B. Shaik, M. Fulcheri, et al. 2010. IL-35, an anti-inflammatory cytokine which expands CD4+CD25+ Treg cells. Journal of Biological Regulators and Homeostatic Agents 24 (2): 131–135.
Chawla, L.S., R.L. Amdur, S. Amodeo, P.L. Kimmel, and C.E. Palant. 2011. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney International 79 (12): 1361–1369. doi:10.1038/ki.2011.42.
Chawla, L.S., M.G. Seneff, D.R. Nelson, M. Williams, H. Levy, P.L. Kimmel, and W.L. Macias. 2007. Elevated plasma concentrations of IL-6 and elevated APACHE II score predict acute kidney injury in patients with severe sepsis. Clinical Journal of the American Society of Nephrology 2 (1): 22–30. doi:10.2215/CJN.02510706.
Collison, L.W., C.J. Workman, T.T. Kuo, K. Boyd, Y. Wang, K.M. Vignali, R. Cross, D. Sehy, R.S. Blumberg, and D.A. Vignali. 2007. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 450 (7169): 566–569. doi:10.1038/nature06306.
Doi, K., A. Leelahavanichkul, P.S. Yuen, and R.A. Star. 2009. Animal models of sepsis and sepsis-induced kidney injury. The Journal of Clinical Investigation 119 (10): 2868–2878. doi:10.1172/JCI39421.
Duann, P., E.A. Lianos, J. Ma, and P. H. Lin. 2016. Autophagy, Innate Immunity and Tissue Repair in Acute Kidney Injury. Int J Mol Sci 17 (5). doi:10.3390/ijms17050662.
Fan, H.Y., D. Qi, C. Yu, F. Zhao, T. Liu, Z.K. Zhang, M.Y. Yang, L.M. Zhang, D.Q. Chen, and Y. Du. 2016. Paeonol protects endotoxin-induced acute kidney injury: Potential mechanism of inhibiting TLR4-NF-kappaB signal pathway. Oncotarget 7 (26): 39497–39510. doi:10.18632/oncotarget.8347.
Friedman, A., and K.L. Liao. 2015. The role of the cytokines IL-27 and IL-35 in cancer. Mathematical Biosciences and Engineering 12 (6): 1203–1217. doi:10.3934/mbe.2015.12.1203.
Grigoryev, D.N., M. Liu, H.T. Hassoun, C. Cheadle, K.C. Barnes, and H. Rabb. 2008. The local and systemic inflammatory transcriptome after acute kidney injury. Journal of the American Society of Nephrology 19 (3): 547–558. doi:10.1681/ASN.2007040469.
Guan, S.Y., R.X. Leng, M.I. Khan, H. Qureshi, X.P. Li, D.Q. Ye, and H.F. Pan. 2016. Interleukin-35: a potential therapeutic agent for autoimmune diseases. Inflammation. doi:10.1007/s10753-016-0453-9.
Heresi, G.A. 2004. Acute renal failure and sepsis. The New England Journal of Medicine 351 (22): 2347–2349 author reply 2347-2349.
Hu, J., and J. Liu. 2016. Licochalcone A attenuates lipopolysaccharide-induced acute kidney injury by inhibiting NF-kappaB activation. Inflammation 39 (2): 569–574. doi:10.1007/s10753-015-0281-3.
Jiang, S., Y. Li, T. Lin, L. Yuan, Y. Li, S. Wu, L. Xia, H. Shen, and J. Lu. 2016. IL-35 inhibits angiogenesis through VEGF/Ang2/Tie2 pathway in rheumatoid arthritis. Cellular Physiology and Biochemistry 40 (5): 1105–1116. doi:10.1159/000453165.
Jin, L., X. Xu, B. Ye, M. Pan, Z. Shi, and Y. Hu. 2015. Elevated serum interleukin-35 levels correlate with poor prognosis in patients with clear cell renal cell carcinoma. International Journal of Clinical and Experimental Medicine 8 (10): 18861–18866.
Joannidis, M., and H.P. Kierdorf. 2014. Acute kidney injury and its treatment. Med Klin Intensivmed Notfmed 109 (5): 322–323. doi:10.1007/s00063-013-0337-9.
Kanai, K., A.M. Park, H. Yoshida, I. Tsunoda, and O. Yoshie. 2017. IL-35 suppresses lipopolysaccharide-induced airway eosinophilia in EBI3-deficient mice. Journal of Immunology 198 (1): 119–127. doi:10.4049/jimmunol.1600506.
Leelahavanichkul, A., A.C. Souza, J.M. Street, V. Hsu, T. Tsuji, K. Doi, L. Li, et al. 2014. Comparison of serum creatinine and serum cystatin C as biomarkers to detect sepsis-induced acute kidney injury and to predict mortality in CD-1 mice. American Journal of Physiology. Renal Physiology 307 (8): F939–F948. doi:10.1152/ajprenal.00025.2013.
Liu, F., Y. Song, and D. Liu. 1999. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Therapy 6 (7): 1258–1266. doi:10.1038/sj.gt.3300947.
Liu, Y., Y. Wu, Y. Wang, Y. Cai, B. Hu, G. Bao, H. Fang, et al. 2015. IL-35 mitigates murine acute graft-versus-host disease with retention of graft-versus-leukemia effects. Leukemia 29 (4): 939–946. doi:10.1038/leu.2014.310.
Manzoor, F., M.C. Johnson, C. Li, R.J. Samulski, B. Wang, and R. Tisch. 2016. Beta-cell-specific IL-35 therapy suppresses ongoing autoimmune diabetes in NOD mice. European Journal of Immunology. doi:10.1002/eji.201646493.
Neveu, H., D. Kleinknecht, F. Brivet, P. Loirat, and P. Landais. 1996. Prognostic factors in acute renal failure due to sepsis. Results of a prospective multicentre study. The French study group on acute renal failure. Nephrology, Dialysis, Transplantation 11 (2): 293–299.
Niedbala, W., X.Q. Wei, B. Cai, A.J. Hueber, B.P. Leung, I.B. McInnes, and F.Y. Liew. 2007. IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. European Journal of Immunology 37 (11): 3021–3029. doi:10.1002/eji.200737810.
Paladino, J.D., J.R. Hotchkiss, and H. Rabb. 2009. Acute kidney injury and lung dysfunction: a paradigm for remote organ effects of kidney disease? Microvascular Research 77 (1): 8–12. doi:10.1016/j.mvr.2008.09.001.
Rewa, O., and S.M. Bagshaw. 2014. Acute kidney injury-epidemiology, outcomes and economics. Nature Reviews. Nephrology 10 (4): 193–207. doi:10.1038/nrneph.2013.282.
Schefold, J.C., G. Filippatos, G. Hasenfuss, S.D. Anker, and S. von Haehling. 2016. Heart failure and kidney dysfunction: epidemiology, mechanisms and management. Nature Reviews. Nephrology 12 (10): 610–623. doi:10.1038/nrneph.2016.113.
Schottelius, A.J., M.W. Mayo, R.B. Sartor, and A.S. Baldwin Jr. 1999. Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kappaB DNA binding. The Journal of Biological Chemistry 274 (45): 31868–31874.
Shen, H., C. Wang, E. Fan, Y. Li, W. Zhang, and L. Zhang. 2014. Upregulation of interleukin-35 subunits in regulatory T cells in a murine model of allergic rhinitis. ORL: Journal for Otorhinolaryngology and Its Related Specialties 76 (5): 237–247. doi:10.1159/000369141.
Shen, P., T. Roch, V. Lampropoulou, R.A. O'Connor, U. Stervbo, E. Hilgenberg, S. Ries, et al. 2014. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507 (7492): 366–370. doi:10.1038/nature12979.
Silvester, W., R. Bellomo, and L. Cole. 2001. Epidemiology, management, and outcome of severe acute renal failure of critical illness in Australia. Critical Care Medicine 29 (10): 1910–1915.
Song, S., M. Meyer, T.R. Turk, B. Wilde, T. Feldkamp, R. Assert, K. Wu, A. Kribben, and O. Witzke. 2009. Serum cystatin C in mouse models: a reliable and precise marker for renal function and superior to serum creatinine. Nephrology, Dialysis, Transplantation 24 (4): 1157–1161. doi:10.1093/ndt/gfn626.
Susantitaphong, P., D.N. Cruz, J. Cerda, M. Abulfaraj, F. Alqahtani, I. Koulouridis, B.L. Jaber, and Nephrology Acute Kidney Injury Advisory Group of the American Society of. 2013. World incidence of AKI: a meta-analysis. Clinical Journal of the American Society of Nephrology 8 (9): 1482–1493. doi:10.2215/CJN.00710113.
Tarique, M., C. Saini, R.A. Naqvi, N. Khanna, and D.N. Rao. 2016. Increased IL-35 producing Tregs and CD19+IL-35+ cells are associated with disease progression in leprosy patients. Cytokine 91: 82–88. doi:10.1016/j.cyto.2016.12.011.
Turnis, M.E., D.V. Sawant, A.L. Szymczak-Workman, L.P. Andrews, G.M. Delgoffe, H. Yano, A.J. Beres, P. Vogel, C.J. Workman, and D.A. Vignali. 2016. Interleukin-35 limits anti-tumor immunity. Immunity 44 (2): 316–329. doi:10.1016/j.immuni.2016.01.013.
Uchino, S., J.A. Kellum, R. Bellomo, G.S. Doig, H. Morimatsu, S. Morgera, M. Schetz, et al. 2005. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA 294 (7): 813–818. doi:10.1001/jama.294.7.813.
Xu, C., A. Chang, B.K. Hack, M.T. Eadon, S.L. Alper, and P.N. Cunningham. 2014. TNF-mediated damage to glomerular endothelium is an important determinant of acute kidney injury in sepsis. Kidney International 85 (1): 72–81. doi:10.1038/ki.2013.286.
Yang, J., O.E. Hawkins, W. Barham, P. Gilchuk, M. Boothby, G.D. Ayers, S. Joyce, M. Karin, F.E. Yull, and A. Richmond. 2014. Myeloid IKKbeta promotes antitumor immunity by modulating CCL11 and the innate immune response. Cancer Research 74 (24): 7274–7284. doi:10.1158/0008-5472.CAN-14-1091.
Zarjou, A., and A. Agarwal. 2011. Sepsis and acute kidney injury. Journal of the American Society of Nephrology 22 (6): 999–1006. doi:10.1681/ASN.2010050484.
Zhang, F., M.Y. Li, Y.T. Lan, and C.B. Wang. 2016. Imbalance of Th17/Tregs in rats with smoke inhalation-induced acute lung injury. Scientific Reports 6: 21348. doi:10.1038/srep21348.
Zhao, N., H. Li, Y. Yan, R. Jiang, and X. He. 2016. Mesenchymal stem cells overexpressing IL-35 effectively inhibit CD4+ T cell function. Cellular Immunology. doi:10.1016/j.cellimm.2016.12.001.
Zuk, A., and J.V. Bonventre. 2016. Acute kidney injury. Annual Review of Medicine 67: 293–307. doi:10.1146/annurev-med-050214-013407.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81500572, 81472401), the Science and Technology Key Project on Clinical Medicine of Jiangsu Province (BL2013013), and the Medicine and Hygiene Program of Zhejiang Province (2014KYB231).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Hu, L., Chen, C., Zhang, J. et al. IL-35 Pretreatment Alleviates Lipopolysaccharide-Induced Acute Kidney Injury in Mice by Inhibiting NF-κB Activation. Inflammation 40, 1393–1400 (2017). https://doi.org/10.1007/s10753-017-0582-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-017-0582-9