
Abstract
Glucocorticosteroids (GCs) are basic drugs in therapy of a number of diseases, including chronic diseases of the respiratory system. They are the most important anti-inflammatory drugs in the treatment of asthma. GCs after binding to the glucocorticoid receptor (GR) form the complex (transcription factor), which acts on promoter and regulatory parts of genes enhancing the expression of anti-inflammatory proteins and decreasing the proinflammatory protein synthesis, including numerous cytokines mediating inflammation in the course of asthma. Non-sensitivity or resistance to GCs favours an increase in the TGF-β expression. This cytokine plays a central role in asthma inducing fibroblast differentiation and extracellular matrix synthesis. TGF-β isoforms, 1, 2 and 3, are located on chromosome 19q13, 1q41 and 14q24, respectively. GCs reduce TGF-β 1 and TGF-β 2 production and significantly decrease the expression of upregulated TGF-β 1 and TGF-β 2 mRNA induced by exogenous TGF-β. In asthma, TGF-β may play a role in the development of the peribronchiolar and subepithelial fibrosis, which contributes to a significant clinical exacerbation of asthma. Therefore, it is possible that NR3C1 glucocorticoid receptor gene polymorphisms could exert varied effects on the TGF-β mRNA expression and fibrotic process in lungs of asthmatic patients. The aim of the study was to evaluate the impact of polymorphic forms (Tth111I, BclI, ER22/23EK, N363S) of the NR3C1 gene on the level of the TGF-β 1 mRNA expression. A total of 173 patients with asthma and 163 healthy volunteers participated in the study. Genotyping of Tth111I, BclI, ER22/23EK, and N363S polymorphisms of the NR3C1 gene was performed by using PCR-HRM and PCR-RFLP techniques. TGF-β mRNA was assessed by real time RT-PCR. Tth111I SNP significantly (p = 0.0115) correlated with the TGF-β 1 mRNA expression level. The significance of AA and GG genotypes of Tth111I SNP in increasing and decreasing the level of the TGF-β 1 mRNA expression was demonstrated. Both BclI SNP and ER22/23EK SNP did not affect the expression level of the cytokine analysed. The N363S SNP AA genotype of NR3C1 gene statistically significantly influenced the increase in the level of the TGF-β 1 mRNA expression. Thus, SNPs of NR3C1 gene play an important regulatory function in the bronchi of patients suffering from asthma. In the case of the occurrence of Tth111I and N363S polymorphic forms of the gene studied, a reduced ability of GCs to inhibit the TGF-β 1 expression can be observed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Glucocorticosteroids (GCs) are the basic and most important anti-inflammatory drugs [1–3]. They inhibit phagocytosis and lysosomal breakdown; reduce the number of lymphocytes, eosinophils, and monocytes; and block IgE-dependent histamine and leukotriene release [2–4]. They also brake the synthesis and release of such cytokines as interferon-gamma, interleukins (IL-1, IL-2, IL-3, IL-6), TNF-α (tumor necrosis factor-α), and GM-CSF (granulocyte-macrophage colony-stimulating factor). By inhibiting phospholipase A2 activity through lipocortin, glucocorticosteroids do not allow to release arachidonic acid and in consequence to synthesize inflammatory mediators (leukotrienes and prostaglandins) [2–5].
A normal response to GC treatment in asthmatic patients is a key element of complex therapy [5]. Unfortunately, the effects of biological action of glucocorticosteroids are determined individually. This individual predisposition causes that side effects can be quickly observed in some patients while using even low doses of these drugs. However, there are a number of patients who do not demonstrate any side effects despite taking high doses of glucocorticosteroids. This phenomenon proves that there is a wide range of drug doses, which can be used in order to achieve total asthma control. In a healthy population, approximately 2.3 % of people exhibit resistance to GCs; hypersensitivity to this group of drugs has been found in 6.6 % of people [5–9]. Resistance to the treatment with glucocorticosteroids results from several complex mechanisms which differ in particular patients. A decreased response to GCs in asthmatic patients has been explained by the occurrence of mutations and polymorphisms (single nucleotide polymorphism, SNP) in the glucocorticoid receptor NR3C1 gene, by the reduced glucocorticoid receptor (GR) expression, their weakened ability to bind DNA and ligand, as well as by overproduction of transcription factors [6–8, 10–14].
The occurrence of SNP(s) in NR3C1 gene can modify the response of asthmatic patients to GC treatment. A few polymorphisms may be involved in the pathogenesis of this phenomenon [1, 3–6].
The Tth111I polymorphism is localized in the region of the NR3C1 gene promoter and converts C > T in the promoter region. It is the restriction fragment length polymorphism (RFLP) coupled with ER22/23EK SNP [15–19]. The Tt111II SNP alone is likely to be non-functional. Its effects are observed only when coupled with ER22/23EK. It is believed that the resistance to glucocorticosteroids and a normal metabolic profile in Tth111I carriers are a result of the presence of the ER22/23EK polymorphism [16, 19, 20].
The BclI polymorphism is localized in intron 2 of the NR3C1 gene. Intron 2 (B) is located between exon 2 and 3 of the NR3C1 gene. It causes the conversion of C > G in the promoter region and is RFLP. The BclI polymorphism is coupled with two other SNPs, Intron B 33389 (rs33389) and Intron B 33388 (rs33388) [15, 19, 21, 22]. It significantly affects the process of alternative splicing of the NR3C1 gene and increases sensitivity to GCs in this mechanism. It concerns both BclI CG and BclI GG. SNP enhances the GC-dependent genomic mechanisms which in complexes with GR influence glucocorticoid response element (GRE) sequences in promoter and regulatory parts of gene coding protein synthesized in response of cells to glucocorticosteroid action [19, 20, 23].
The ER22/23EK polymorphism is located in exon 2 of the NR3C1 gene. It consists of two connected with each other nucleotide transitions in codons 22 and 23 [19, 20, 24, 25]. Silent transition in codon 22 does not convert amino acid (G > A; position 198; codon GAG and GAA code glutamic acid), while the transition in codon 23 leads to conversion of arginine to lysine (G > A; AGG > AAG; position 200), which can result in the third-order change in the domain structure of the glucocorticoid receptor, responsible for activation of transcription [13, 19, 26]. The transition in codon 23 can cause the second-order change in the mRNA structure of the glucocorticoid receptor and in consequence the initiation of translation from 1 or 27 methionine [27, 28]. Moreover, the described change in codon 23 may have an impact on mRNA stability for the glucocorticoid receptor [27, 28]. The ER22/23EK polymorphism induces structural changes in the receptor region A/B of the glucocorticosteroids as well as functional changes within the activation function 1 (AF1) domain, which affects the GR activity and allows to interact with several transcription factors. It exhibits a strong coupling with the functional polymorphism in exon 9β of the NR3C1 gene, which results in bilateral interactions and the mutual regulation of functional effects. ER22/23EK can be investigated with the use of restriction enzyme by PCR-RFLP method (RFLP) [15, 16, 18, 19, 24, 25, 29].
The N363S polymorphism is located in exon 2 of the NR3C1 gene. The occurrence of SNP causes changes in the nucleotide sequence AAT > AGT, which results in the conversion of asparagine to serine in codon 363 of GR. It induces structural changes in the A/B region of the GR as well as functional changes within the activation functional domain AF1. The N363S polymorphism modulates various protein regulatory systems. It inhibits activity of NF-ƘB (nuclear factor kappa-light-chain-enhancer of activated B cells) and stimulates the production of I-ƘB inhibitors, thus, interfering with the IL-2 suppression. N363S affects the degree of phosphorylation of the GC receptor. N363S SNP is characterized by an increased ability to transactivate gene encoding proteins synthesized in response of cells to GC action. This is the RFL polymorphism [13, 19, 24, 25, 30–35].
Due to the changed NR3C1 gene expression by SNP, the production of many factors involved in the pathogenesis of inflammation in asthma such as TGF-beta (β) (transforming growth factor beta), GM-CSF, VEGF (vascular endothelial growth factor), fibroblast growth factor, cysteinyl leukotrienes, and interleukins (IL-1, IL-2, IL-3, IL-4, IL-5, IL-12, IL-13, IL-16, IL-17) has not been decreased [1, 3–6]. In this context, the role of TGF-beta in the regulation of chronic bronchial inflammation in asthmatic patients is very important. This cytokine is responsible for the growth, differentiation, cell migration, formation, and degradation of extracellular matrix elements, chemotaxis processes, remodelling, and cell apoptosis in the bronchi [36–42]. TGF-beta occurs in 5 isoforms. Three of them: TGF-beta 1, TGF-beta 2, and TGF-beta 3 are coded by different genes. In vitro isoforms 1–3 bind together and activate the same membranous receptors of TGF-beta (serine-threonine kinases). Smad proteins are responsible for transmitting the signal to the cellular nucleus and affect various transcription factors. TGF-β through signaling Smad2 and Smad3 protein pathway exerts an impact on the expression of approximately 500 target genes, and through Smad1, Smad5 and Smad8 proteins affects subsequent 500 target genes [36–38, 43–45]. In bronchoalveolar lavage (BAL), levels of TGF-β1 are increased in asthmatic patients [46]. TGF-β has been shown to be essential for the transformation of fibroblasts into myofibroblasts, which are major producers of collagen [46]. Some experimental studies indicated that adenoviral-mediated gene transfer of TGF-β1 in the rodent lung caused severe lung fibrosis with the deposition of extracellular matrix [46]. The phosphorylation of TGF-β type I (activin receptor-like kinase-5; ALK-5) and type II receptors is characteristic of the activation of the TGF-β signal transduction pathways [46]. The cascade and subsequent phosphorylation and translocation of Smad 2 and Smad 3 to the cell nucleus affect the regulation of gene transcription [46]. Therefore, it is possible that glucocorticosteroids could exert varied effects on the fibrotic process [47]. Due to the fact that GCs may block TGF-beta production by changing mRNA levels and c-Jun [47], it is important to recognize the SNP effect in the NR3C1 gene on the level of the TGF-beta mRNA expression. GCs significantly inhibit TGF-beta1 and TGF-beta2 production [47]. They may also decrease expression of the upregulated TGF-beta1 and TGF-beta2 mRNA induced by exogenous TGF-beta 1, TGF-beta 2, or TGF-beta 3 but do not affect Smads [47].
The aim of the study was to evaluate the impact of the occurrence of polymorphic forms (Tth111I, BclI, ER22/23EK, N363S) of the NR3C1 gene on the level of the TGF-beta 1 mRNA expression in the group of asthmatic patients in the Polish population as compared to a control group (healthy subjects).
MATERIALS AND METHODS
Approval of the Research Review Board
The study was approved by the local ethics committee (Consent of the Research Review Board at the Medical University of Lodz, Poland, No. RNN/133/09/KE and No. RNN/31/14/KE). At the commencement of the study, the participants were invited to attend it voluntarily. Before enrolment, written informed consent was obtained from every patient.
Patients and control subjects
Asthma patients were recruited from among the patients treated in the Department of Internal Medicine, Asthma and Allergy, the Department of Pneumology and Allergology, and the Specialist Outpatient Department of Pulmonary Diseases and Allergology at N. Barlicki Memorial Teaching Hospital No. 1 of the Medical University of Lodz, Poland.
Healthy volunteers were included in the study from the general population. They were selected on random basis.
A total of 336 subjects including 173 patients (51.49 %) suffering from asthma participated in the study. The remaining subjects constituted a control group. The proportion of gender in both groups was similar (63.58 % of women in the group with asthma vs 62.58 % in the control group, p = 0.8415). The mean age (±SD) in the group of patients was 50.03 ± 15.76 years, and in the control group, it was 45.60 ± 16.55 years. The mean FEV1(%) (forced expiratory volume in 1 s) value among asthmatic patients was 72.96 ± 20.33 and 95.07 ± 12.64 in the controls.
The inclusion criteria for participating in the study group were as follows: the patient’s informed consent and appropriate spirometry results enabling the correct interpretation and diagnosis of asthma according to GINA (The Global Initiative For Asthma) Report [19, 24, 30, 48, 49]. The patients were excluded from the study on the basis of the following criteria: treatment with rifampicin, phenobarbital, phenytoin and ephedrine, as well as exacerbations of the disease due to infections. The subjects qualified to the control group met all of the following criteria: no data from subjective and objective examinations confirming the presence of asthma, other pulmonary diseases, allergy, atopy and hypersensitivity to non-steroid anti-inflammatory drugs (NSAIDs), no spirometry results confirming airway obstruction, negative results of skin prick tests with common allergens, no first-degree relatives with asthma, allergy or atopy, no treatment with rifampicin, phenobarbital, phenytoin, and ephedrine [19, 24, 30].
Spirometry
Functional tests of the respiratory system were carried out according to ERS (European Respiratory Society)/ATS (American Thoracic Society) standards [19, 24, 30, 50].
Skin Prick Tests
Skin prick tests were performed according to the guidelines of EAACI (European Academy of Allergy and Clinical Immunology) [51].
Asthma Control Test
The level of asthma control was assessed with Asthma Control Test (ACT™) recommended by the GINA Report. Asthma control level was calculated on the basis of the following patients’ results obtained in ACT: 00–19 points—no asthma control, 20–24 points—partially controlled asthma and 25 points—well-controlled asthma [5, 19, 48, 49, 52, 53].
BMI
Obesity was evaluated by means of the body mass index (BMI) [54, 55].
Isolation of DNA
Venous blood samples were collected from the participants onto EDTAK3 (ethylenediaminetetraacetate) vacuum blood collection tubes (SARSTEDT AG & Co.; Nümbrecht, Germany). DNA was obtained from the peripheral blood leukocyte fraction. The genetic material was isolated using QIAamp DNA Blood Mini Kit (QIAGEN Inc., USA) according to the guidelines provided by the manufacturer.
NR3C1 Gene Tth111I Polymorphism Genotyping with PCR-HRM Method
The Tth111I polymorphism of the NR3C1 gene was genotyped using the PCR-HRM method with application of LightScanner® 32 System, that is the fastest Real-Time PCR combined with the most accurate Hi-Res Melting® (Idaho Technology Inc., Utah, USA). Exponential amplification of DNA segments for Tth111I polymorphism was carried out using a forward primer (5′-GGA TGA ATC CCT ATC TGA GTG-3′) and a reverse primer (5′-GGC CAC AAC AAT AAC CCA GTA-3′) according to the standard PCR protocol. Starter binding to complementary DNA matrix sites was conducted at 58 °C [19].
The first stage of HRM analysis involved amplification of the investigated DNA fragment containing the analyzed Tth111I SNP on the 1:50 matrix using a forward primer (5′-GCA GAG GTG GAA ATG AAG GTG-3′) and a reverse primer (5′-GGA GTG GGA CAT AAA GCT ATG ACA-3′), then denaturation and slow renaturation to form a heteroduplex. At the last stage, the mixture was subjected to precise denaturation in the presence of intercalating stain, and identification of DNA fragments (Tth111I SNP) was based on the analysis of melting curves [19]. The LightScanner® High Sensitivity Master Mix (Idaho Technology Inc., Utah, USA) was used for the reaction. It is a specialized master mix containing LCGreen Plus® dye and internal temperature calibrators. The obtained product was subjected to internal control using a molecular probe C3 labeled carbon at the 3ʹ-terminal portion (labeled, 3′ blocked oligonucleotide; 5′-ATG TAT TCA GAC TCA GTC AAG GCA AGG ACC T[SpcC3]-3′) [38–42]. The selected SNP samples were verified once again by sequencing.
NR3C1 Gene BclI Polymorphism Genotyping with the PCR-RFLP Method
Amplification of the DNA fragment containing BclI polymorphism of the NR3C1 gene was conducted using starters with the following sequences: a forward primer (5′-GAG AAA TTC ACC CCT ACC AAC-3′) and a reverse primer (5′-AGA GCC CTA TTC TTC AAA CTG-3′) according to the standard PCR protocol [19, 22, 25, 56]. Starter binding to complementary DNA matrix sites was conducted at 56 °C. BclI restriction enzyme (Fermentas International Inc., Burlington, Canada) was used for digestion of the amplification product containing the Bc1I polymorphism [19, 22, 25, 56]. Hydrolysis of the PCR product with the restriction enzyme was conducted for 24 h at 55 °C. DNA fragments containing 263 and 151 base pairs identified as a set of representative, typical (wild type) alleles were obtained, as well as segments with 418, 263, and 155 base pairs. The RFLP product of 418-bp length was identified as a set of polymorphic alleles. RFLP products were separated by electrophoresis on 8 % polyacrylamide gel, stained with etidium bromide and observed in UV light (Image Master Pharmacia Biotech Video Documentation System (VDS), Pharmacia Biotech, Sweden) [19, 22, 25]. Representative, typical homozygotes, as well as heterozygotes were sequenced and used as internal control.
NR3C1 Gene ER22/23EK Polymorphism Genotyping with the PCR-RFLP Method
Amplification of the DNA fragment containing ER22/23EK polymorphism of the NR3C1 gene was conducted using starters with the following sequences: a forward primer (5′-TGC ATT CGG AGT TAA CTA AAA-3′) and a reverse primer (5′-ATC CCA GGT CAT TTC CCA TCA-3′) [19, 56]. Starter binding to complementary DNA matrix sites was conducted at 56 °C. MnlI restriction enzyme (Fermentas International Inc., Burlington, Canada) was used for digestion of the amplification product containing the ER22/23EK polymorphism [19, 56]. Hydrolysis of the PCR product with the restriction enzyme was conducted for 24 h at 37 °C. DNA fragments with 149 and 163 bp (and shorter fragments containing 50, 49, and 35 base pairs) were obtained as a set of representative, typical (wild type) alleles, whereas segments of 163 and 184 bp (and shorter fragments containing 50 and 49 base pairs) constituted a set of polymorphic alleles. RFLP products were separated by electrophoresis on 8 % polyacrylamide gel, stained with ethidium bromide and observed in UV light (Image Master Pharmacia Biotech Video Documentation System (VDS), Pharmacia Biotech, Sweden) [19, 22, 24, 25, 30]. Representative, typical homozygotes, as well as heterozygotes were sequenced and used as internal control.
NR3C1 Gene N363S Polymorphism Genotyping with the PCR-RFLP Method
Exponential amplification of DNA segments for the N363S polymorphism was carried out using a forward primer (5′-CCA GTA ATG TAA CAC TGC CCC-3′) and a reverse primer (5′-TTC GAC CAG GGG AAG TTC AGA-3′) according to the standard PCR protocol. Starter binding to complementary DNA matrix sites was conducted at 56 °C. Amplified DNA sequences of 357 bp length were obtained. The material was incubated with FastDigest® Tsp509I (TasI) restriction enzyme (Fermentas International INC., Burlington, Canada) at 65 °C for 1 h. DNA fragments of 135, 73, 70, 60 and 19 bp were obtained as a set of representative, typical (wild type) alleles, whereas segments of 135, 92 (73 + 19 bp), 70, and 60 bp constituted the set of polymorphic alleles [24, 25, 30, 56]. RFLP products were separated by electrophoresis on 8 % polyacrylamide gel 1:20, stained with ethidium bromide, and observed in UV light (Image Master Pharmacia Biotech Video Documentation System (VDS), Pharmacia Biotech, Sweden) [19, 22, 24, 25, 30]. Representative, typical homozygotes, as well as heterozygotes were sequenced and used as internal control.
TGF-beta (β) mRNA Expression
An amount of 10 μg total RNA was extracted from the peripheral blood lymphocytes using TRI Reagent® Solution (Ambion, NY, USA) according to the standard acid-guanidinium-phenol-chloroform method [57]. The extracted RNA was analyzed by agarose gel electrophoresis and only cases with preserved 28S, 18S, and 5S ribosomal RNA bands, indicating good RNA quality, were used in the study. Total RNA was digested with DNase (GIBCO, Life Technologies, NY, USA) at room temperature for 15 min. The amount of purified RNA was determined using spectrophotometry at 260 nm in a Nanodrop analyzer (ND-100, Nanodrop Technologies, Wilmington, DE, USA) [36]. The purity was verified according to the 260/280 nm ratio, with values between 1.8 and 2.1 indicating that RNA quality was optimal and suitable for quantitative real-time PCR (qRT-PCR) [36, 57].
Reverse transcription of 1 μg RNA was performed using an AccuScript PfuUltraII RT-PCR kit (Agilent Technologies, CA, USA). The complementary DNA (cDNA) was subjected to real-time quantitative PCR using gene-specific primers (5′-GGT ACC TGA ACC CGT GTT GCT-3′ and 5′-TGT TGC TGT ATT TCT GGT ACA GCT C-3′ Sigma-Aldrich, Germany) for TGFβ-1 and (5′-AGC CAC ATC GCT CAG ACA-3′ and 5′-GCC CAA TAC GAC CAA ATC C-3′ IBB PAN, Polska) for GAPDH using a Brilliant II SYBR Green QRT-PCR Master Mix Kit (Stratagene, CA, USA). Amplification was performed using the normal two steps and a standard thermal profile. Primer annealing temperature was 61 °C, and primer annealing time was 20 s. An Agilent Technologies Stratagene Mx3000P was used for the PCR reaction. For each sample, the C T (threshold cycle) values were calculated with the help of Mx-Pro software. The RT-PCR amplification of the TGFβ-1 gene was compared to that of GAPDH (glyceraldehyde 3-phosphate dehydrogenase), a house-keeping reference gene, and ΔC T values were determined (ΔCT = CT,GENE - CT,GAPDH). Real-time PCR data was automatically calculated with the data analysis module. The results were analyzed according to the 2−ΔΔCT method [36, 58, 59]. Validation of PCR efficiency was performed with a standard curve [36].
Statistical Analysis
Nominal variables were given as numbers with appropriate percentage whereas continuous variables as means with standard deviations. The chi-square test with Yate’s correction and the Fisher’s exact test with Freeman-Halton extension were used to test the associations. Continuous variables were analyzed using the Mann-Whitney’s U test or analysis of variance (ANOVA) depending on the number of compared groups. Correlation coefficients were calculated with the Spearman’s rank test. The Hardy-Weinberg equilibrium (HWE) test of SNP was performed using Michael H. Court’s (2005–2008) online calculator (http://www.tufts.edu/~mcourt01/Documents/Court%20lab%20-%20HW%20calculator.xls). P levels lower than 0.05 were considered as statistically significant. Statistica 10.0 package (Statsoft, Tulsa, OK, USA) was used for statistical analysis.
The genotyping was performed by two investigators who were unaware of the phenotypes.
RESULTS
Frequencies of the NR3C1 gene polymorphic forms in the population studied (controls and cases) were evaluated. Frequencies of particular SNP genotypes and single polymorphic alleles of the glucocorticoid receptor gene were calculated. Table 1 presents frequencies of genotypes and particular alleles of SNP(s) studied.
TGF-β 1 mRNA expression values were evaluated separately for controls and cases as well as combined together. The data is presented in Table 2.
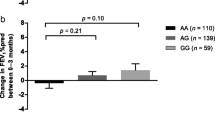
The impact of the NR3C1 gene polymorphic forms on the level of the TGF-β 1 expression in the populations studied was analyzed. Significant correlations between the Tth111I SNP genotypes and the level of the TGF-β 1 mRNA expression were found. There were statistically significant correlations between particular genotypes (the GG and AA homozygotes) of the SNP analyzed (p = 0.0103). It was stated that Tth111I SNP significantly (p = 0.0115) correlated with the level of the TGF-β 1 mRNA expression [R 2 = 0.10; beta coefficient (AA) = 0.39 (95 % confidence interval (CI) −0.70 to −0.08); beta coefficient (AG) = 0.10 (95 % CI −0.21 to 0.41)]. The AA genotype of Tth111I correlated with the highest level of the TGF-β 1 mRNA expression, while the GG genotype correlated with its lowest level. Figure 1 presents the detailed correlation between the TGF-β 1 mRNA expression levels and NR3C1 gene polymorphic forms.

The impact of the NR3C1 gene Tth111I SNP genotypes on the TGF-β 1 mRNA expression level. A detailed description can be found in the text. The authors’ own drawing-up.
No statistically significant correlation was observed between the BclI SNP genotypes and the level of the TGF-β 1 mRNA expression as well as between particular genotypes of SNP studied. BclI SNP did not correlate (p = 0.9753) with the level of the TGF-β 1 mRNA expression [R 2 = 0.01; beta coefficient (GC) = 0.08 (95 % CI −0.23 to 0.39); beta coefficient (CC) = −0.10 (95 % CI −0.42 to 0.21)]. The detailed relationships of the TGF-β 1 mRNA expression level to SNP of the NR3C1 gene are presented in Fig. 2.

The effect of BclI SNP genotypes of the NR3C1 gene on the level of the TGF-β 1 mRNA expression. The detailed description in the text. The authors’ own drawing-up.
There was no statistically significant correlation observed between the ER22/23EK SNP genotypes and the level of the TGF-β 1 mRNA expression as well as between particular genotypes of this SNP. ER22/23EK SNP was found not to correlate (p = 0.4898) with the TGF-β 1 mRNA expression level [R 2 = 0.02; beta coefficient (GA) = −0.15 (95 % CI −0.36 to 0.07)]. The detailed relationships of the level of the TGF-β 1 mRNA expression to the NR3C1 gene SNP is shown in Fig. 3.

The effect of ER22/23EK SNP genotypes of the NR3C1 gene on the level of the TGF-β 1 mRNA expression. The detailed description in the text. The authors’ own drawing-up.
Statistically significant correlations between N363S SNP genotypes and the level of the TGF-β 1 mRNA expression, as well as between particular genotypes (AA homozygote and AG heterozygote) of the SNP studied (p = 0.0103) were noted. N363S SNP significantly (p = 0.0119) correlated with the level of the TGF-β 1 mRNA expression [R 2 = 0.10; beta coefficient (AA) = −0.32 (95 % CI −0.55 to −0.08); beta coefficient (AG) = −0.01 (95 % CI −0.24 to 0.22)]. The AA genotype of N363S SNP was found to correlate with the highest TGF-β 1 mRNA expression level, while the AG genotype correlated with the lowest level. The detailed correlations of the TGF-β 1 mRNA expression level for SNP of the NR3 C1 gene are shown in Fig. 4.

The effect of the N363S SNP genotypes of the NR3C1 gene on the level of the TGF-β 1 mRNA expression. The detailed description in the text. The authors’ own drawing-up.
Moreover, it was observed that the level of clinical control of asthma symptoms evaluated by ACTTM was affected by other variables. A significant effect of BMI value (p = 0.0075) on the loss of control of the disease symptoms was detected. Figure 5 presents the correlation between the level of asthma control (ACTTM) and the BMI values.

The diagram showing the correlation between ACTTM and BMI values. The detailed description in the text. The authors’ own drawing-up.
Furthermore, it was found that cigarette smoking significantly (p = 0.0081) affected (β = −0.20) loss of control expressed by scores obtained in the ACTTM test. The association between allergy and asthma prevalence was also assessed. Allergy was found to worsen asthma control and the control level depended on the number of allergens (p = 0.0028). Obviously, the greater the number of allergens the patient is sensitized to, the less the ACTTM scores are (R = −0.22).
Figure 6 presents the correlation between the ACTTM score values and the number of allergens.

The diagram showing the correlation between ACTTM scores and the number of allergens. The detailed description in the text. The authors’ own drawing-up.
DISCUSSION
Asthma is a complex disease involving numerous cells and cell-secreted substances. It is a chronic, inflammatory disease of the respiratory system, determined multifactorially. The complex etiopathogenesis, leading to the development of bronchial hyperresponsiveness, recurrent episodes of wheezing, cough, and dyspnea, is determined by interactions of hereditary and environmental factors. The clinical picture results from the complicated gene-gene and gene-environment interactions [5, 19, 24, 30].
From the clinical point of view, determination of the level of patient’s hypersensitivity to GCs is very important. In part of patients resistant to GCs, familial occurrence of hypersensitivity to this group of drugs can be noticed. The disease is characterized by the presence of high concentration of cortisol in blood, without symptoms of Cushing’s syndrome. High 24-h excretion of free cortisol can be observed. Resistance to GCs is familially determined by the occurrence of mutations in the NR3C1 gene, which change the amino acid content in structural receptor regions containing functional domains determining its biological action. A normal value of cortisol concentration is noted in patients with asthma and lowered response to GCs, as opposed to patients with familial resistance to GCs (in whom changes in the cortisol concentration in blood are recorded). They do not demonstrate characteristic features of Addison disease either. [5, 8, 11, 13, 20, 31, 60].
Inefficient treatment with GCs in some patients can result from increased density of complement receptors on monocytes. In these patients, a lack of change in the expression of these receptors after GC administration has been seen, which is a characteristic reaction in steroid-sensitive patients [5, 61].
In patients resistant to treatment, chronic inflammatory process, through cytokines IL-2 and IL-4, activates p38MAP kinases (p38 mitogen-activated protein kinase), which phosphorylate GR decreasing its affinity to GCs [62, 63]. Other kinases such as MAPK (mitogen-activated protein kinase) affect GR affinity to GCs. Extracellular kinase, MAPK, controlled by a signal resulting from T lymphocyte excitation by coreceptors or superantigens affects phosphorylation of the GC receptor and regulates its functions. This may lead to enhancement of the GRβ expression. After stimulation of T lymphocytes by IL-2, JAK3/STAT5 (Janus kinase 3/signal transducer and activator of transcription 5) is an element exerting effect on GR activity. This reaction is associated with the improper SOCS (suppressor of cytokine signaling) expression. These patients, after receiving GCs do not demonstrate an elevated level of phosphotyrosine associated with the growth factor, which results in intensified inflammation [8, 62]. Patients with lowered sensitivity to GCs are characterized by the increased cellular activity of JNK (c-Jun N-terminal kinase). In asthmatic patients, a decreased expression of double MAPK (MKP-1) phosphatase reduces the ability to counteract p38MAPK and JNK activation. This leads to impairment of biological balance of p38MAPK/MKP-1 and lack of sensitivity to glucocorticosteroids. Phosphorylation pathways dependent on this group of kinases may contribute to inhibiting GC activity [8, 62].
Bearing in mind the above complex model of inducing lack of sensitivity to GCs, it is worth noticing that the significant impact of the NR3C1 gene polymorphic forms on the TGF-β 1 mRNA expression level has been indicated in the present study. Two of four analysed SNPs significantly clinically correlated with the TGF-β 1 expression. Both Tth111I SNP and N363S SNP affected the increase in the level of the TGF-β 1 mRNA expression, thus leading to intensification of inflammation in the course of asthma. The Tth111I AA genotype and the N363S AA genotype most strongly induced the increase in TGF-β 1 values. Due to the fact that NR3C1 gene SNPs intensify non-sensitivity to GCs, it should be emphasized that Tth111I and N363S significantly modulate the level of TGF-β 1 in patients with asthma, who are treated with GCs. The product of the expression of two A alleles of each polymorphism studied, Tth111I and N363S, appeared to be a determinant most strongly correlated with the increase in the TGF-β 1 level and the loss of asthma control. Hence, the Tth111I SNP AA genotype and the N363S SNP AA genotype are significant molecular risk factors for the development of severe and resistant to treatment asthma. Tth111I SNP is associated with the increased cortisol level in the blood observed during the day and the reduced cortisol secretion after administration of 1 mg dexamethasone (DEX) [19, 20, 23]. It is believed that resistance to GCs and a normal metabolic profile in carriers of Tth111I SNP is a result of the occurrence of the ER22/23EK polymorphism in these patients [16, 19, 20]. Numerous studies confirm the role of N363S SNP in increasing the ability of the GCs-GR complex to transactivate target genes [19, 24, 30, 35, 64]. Patients with an uncontrolled inflammatory phenotype and moderate and severe asthma are characterized by a higher frequency of the AA variant of the NR3C1 gene N363S polymorphism as compared to the control group, which is associated with the reduced sensitivity of cells to GCs [19, 24, 30, 64]. In the population of patients with uncontrolled asthma, a lower frequency of AG and GG polymorphic variants of N363S SNP was noted, which was related to a weakened response to anti-inflammatory treatment with GCs. The measurable effect of the lack of occurrence of N363S polymorphic variants of the NR3C1 gene was the loss of control of asthma symptoms, confirmed by ACT™ results. The occurrence of codon 363 AG or GG variants correlates with the increase in anti-inflammatory effects of treatment with GCs and the lower risk of uncontrolled asthma (severe and moderate) [19, 24, 30, 64]. Therefore, GR non-sensitivity to GCs caused by Tth111I and N363S polymorphic forms of the NR3C1 gene results in a significant clinical lack of inhibition of the TGF-β 1 mRNA expression in asthma, which was confirmed in the present study by the control group (healthy volunteers). An increase in the TGF-β 1 expression is a clinically and prognostically unfavorable phenomenon, which leads to progression of the disease and contributes to maintaining bronchial obstruction. It is worth emphasizing that TGF-β apart from Smad protein-dependent transduction pathway can also affect MAPK [37]. Moreover, TGF-β blocks the expression of cytokines D2, E, CDK4 and c-myc in T lymphocytes, influencing cell division processes. TGF-β also inhibits the T1 and T2 lymphocyte differentiation through the inhibition of the expression of transcription factors T-bet and STAT-4 (signal transducer and activator of transcription 4) in T1 lymphocytes and GATA-3 (trans-acting T-cell-specific transcription factor GATA-3) as well as STAT-6 in T2 lymphocytes [37]. It should be added that TGF-β 1 plays an important role in the epithelial-mesenchymal transition (EMT) mechanism, which is responsible for the accumulation of subepithelial mesenchymal cells, thereby contributing to increased contractile cell mass and airway hyperresponsiveness [65]. In this process, epithelial cells lose their typical cell-cell junctions and cell polarity and acquire a more mesenchymal phenotype [65]. Various cytokines, such as interleukins, enhance EMT in respiratory epithelial cells in a TGF-β1-dependent manner [65]. This mechanism promotes airway remodelling in asthma. EMT process increases airway smooth muscle mass and TGF-β1 signaling in the lungs [65], which has been confirmed in many experimental studies on models of animal chronic house dust mite-driven allergic airway inflammation [65]. Remodelling of the bronchi in the EMT mechanism is characterized by specific changes in the bronchi in a form of loss of epithelial markers including cytokeratins, tight junction proteins, and E-cadherin. In this process, the presence of mesenchymal markers such as vimentin and N-cadherin, and increased expression of the Snail, Twist, and Zeb transcription factors have been observed [65]. Our clinical observations require further experimental studies to objectify these phenomena.
In asthmatic patients, the occurrence of non-sensitivity to glucocorticosteroids poses a difficult therapeutic problem. Not all patients from this group reveal a complete resistance to GCs. Drug action is manifested after administration of high doses, higher than standardly recommended. There is a certain group of patients with asthma in whom even optimal treatment does not lead to total control of asthma [8, 19, 24, 30, 66].
Bearing in mind clinical implications concerning the occurrence of NR3C1 gene polymorphic forms as well as the reduced response to GCs and increase in the level of TGF-β 1 expression, it should be stated that the treatment of asthmatic patients with different levels of sensitivity to GCs poses a complex diagnostic and therapeutic problem. In this group of patients, researchers recommend verification of the diagnosis, evaluation of patients’ compliance with doctor’s orders, and elimination of additional environmental factors (e.g., reduction in body mass and efficient treatment of allergy—immunotherapy) and concomitant diseases [6, 66, 67]. Confirmation of asthma diagnosis and monitoring of the control level of asthma symptoms are recommended.
SUMMARY
Current knowledge and recognition of molecular mechanisms inducing different levels of resistance to glucocorticosteroids provides a chance for more efficient therapy and its adjustment to patients’ needs. Non-sensitivity to GCs is determined multifactorially. Both genetic (e.g., NR3C1 gene polymorphic forms, expression level of transcription factors, proinflammatory proteins) as well as environmental factors (allergy, atopy, smoking, obesity, inflammation) determine a proper response to this group of drugs. Therefore, therapy should be individualized and based on holistic evaluation of the clinical state of the patient.
The present study showed a significant impact of polymorphic forms (Tth111I and N363S) of the NR3C1 gene on intensification of asthma inflammation measured by the level of the TGF-β 1 expression. AA genotypes of Tth111I and N363S SNPs are significant risk factors for an increase in the TGF-β 1 mRNA expression level. Thereby, they can contribute to the epithelial-mesenchymal transition, resulting in unfavorable bronchial remodelling and promotion of hyperesponsiveness. The increase in the level of TGF-β 1 was observed in the group of studied patients with asthma, which could enhance the bronchial remodelling, lower the values of spirometric parameters, worsen the disease control, lead to maintenance of bronchial obstruction, and worsen quality of life. Moreover, attention was paid to such individual factors as obesity (BMI) or allergy, which correlated with the loss of control of asthma symptoms (reduced scores in the ACTTM test).
The future development of new molecular diagnostic tests, new anti-inflammatory drugs, and the use of efficient forms of targeted therapy give hope for better quality of health and life of patients with asthma [6, 8, 68–70].
References
Gajewski, P., and A. Szczeklik. 2013. Interna Szczeklika. Kraków: Medycyna Praktyczna Press.
Empendium, Leki. 2014. Medycyna Praktyczna na ekranie. Kraków: Medycyna Praktyczna Press.
Fal, A. 2010. Alergia, choroby alergiczne, astma. Kraków: Wydawnictwo Medycyna Praktyczna Press.
Antczak, A., M. Myśliwiec, and P. Pruszczyk. 2009. Wielka InternaPulmonologia. Warszawa: Wydawnictwo Medical Tribune Polska Press.
Droszcz, W. 2007. Astma. Warszawa: Wydawnictwo Lekarskie PZWL Press.
Ito, K., K. Chung, and I. Adcock. 2006. Update on glucocorticoid action and resistance. Journal of Allergy and Clinical Immunology 117: 522–543.
van der Velden, V. 1998. Glucocorticoids: mechanisms of action and anti-inflammatory potential in asthma. Mediators of Inflammation 7: 229–237.
Ian, M., I. Adcock, P. Ford, P. Bhavsar, T. Ahmad, and K. Chung. 2008. Steroid resistance in asthma: mechanisms and treatment options. Current Allergy and Asthma 8: 171–178.
Lamberts, S., A. Huizenga, P. de Lange, F. de Jong, and J. Koper. 1996. Clinical aspects of glucocorticoid sensitivity. Steroids 61: 157–160.
Leung, D., and J. Bloom. 2003. Update on glucocorticoid action and resistance. Journal of Allergy and Clinical Immunology 111: 3–22.
Kino, T., M. De Martino, E. Charmandari, M. Mirani, and G. Chrousos. 2003. Tissue glucocorticoid resistance/hypersensitivity syndromes. Journal of Steroid Biochemistry and Molecular Biology 85: 457–467.
Grzanka, A., and B. Rogala. 2000. Molecular mechanism of glucocorticoids and difficult asthma. Allergy, Asthma, and Immunology 5: 247–252.
DeRijk, R., M. Schaaf, and E. de Kloet. 2002. Glucocorticoid receptor variants: clinical implications. Journal of Steroid Biochemistry and Molecular Biology 81: 103–122.
Luisi, B., W. Xu, Z. Otwinowski, L. Freedman, K. Yamamoto, et al. 1991. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 352: 497–505.
Derijk, R., and E. de Kloet. 2008. Corticosteroid receptor polymorphisms: determinants of vulnerability and resilience. European Journal of Pharmacology 583: 303–311.
Nicolaides, N., Z. Galata, T. Kino, G. Chrousos, and E. Charmandari. 2010. The human glucocorticoid receptor: molecular basis of biologic function. Steroids 75: 1–12.
Detera-Wadleigh, S., I. Encio, D. Rollins, D. Coffman, and D. Wiesch. 1991. A TthIII1 polymorphism on the 5′ flanking region of the glucocorticoid receptor gene (GRL). Nucleic Acids Research 19: 1960.
Rosmond, R., Y. Chagnon, M. Chagnon, L. Pérusse, C. Bouchard, et al. 2000. A polymorphism of the 5-flanking region of the glucocorticoid receptor gene locus is associated with basal cortisol secretion in men. Metabolism 49: 1197–1199.
Panek, M., T. Pietras, A. Fabijan, M. Miłanowski, L. Wieteska, et al. 2013. Effect of glucocorticoid receptor gene polymorphisms on asthma phenotypes. Experimental and Therapeutic Medicine 5: 572–580.
van Rossum, E., and S. Lamberts. 2004. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Progress in Hormone Research 59: 333–357.
Stevens, A., D. Ray, E. Zeggini, S. John, H. Richards, et al. 2004. Glucocorticoid sensitivity is determined by a specific glucocorticoid receptor haplotype. Journal of Clinical Endocrinology and Metabolism 89: 892–897.
Pietras, T., M. Panek, D. Tworek, K. Oszajca, R. Wujcik, et al. 2011. The Bcl I single nucleotide polymorphism of the human glucocorticoid receptor gene h-GR/NR3C1 promoter in patients with bronchial asthma: pilot study. Molecular Biology Reports 38: 3953–3958.
De Iudicibus, S., R. Franca, S. Martelossi, A. Ventura, and G. Decorti. 2011. Molecular mechanism of glucocorticoid resistance in inflammatory bowel disease. World Journal of Gastroenterology 17: 1095–1108.
Panek, M., T. Pietras, A. Antczak, P. Górski, P. Kuna, et al. 2012. The role of functional single nucleotide polymorphisms of the human glucocorticoid receptor gene NR3C1 in Polish patients with bronchial asthma. Molecular Biology Reports 39: 4749–4757.
Pietras, T., M. Panek, I. Kuprys-Lipinska, K. Oszajca, R. Wujcik, et al. 2010. Frequencies of Bcl I, E22E, and N363S of h-GR/NR3C1 restriction fragment length polymorphisms of glucocorticoid receptor gene in Polish adult population. Medical Science Monitor 16: 475–479.
de Lange, P., J. Koper, N. Huizenga, A. Brinkmann, F. de Jong, et al. 1997. Differential hormone- dependent transcriptional activation and repression by naturally occurring human glucocorticoid receptor variants. Molecular Endocrinology 11: 1156–1164.
van Rossum, E., P. Voorhoeve, S. te Velde, J. Koper, H. Delemarre-van de Waal, et al. 2004. The ER22/23EK polymorphism in the glucocorticoid receptor gene is associated with a benefi cial body composition and muscle strength in young adults. Journal of Clinical Endocrinology and Metabolism 89: 4004–4009.
Bulas, M., J. Trelińska, M. Stolarska, M. Zubowska, F. Pierlejewski, et al. 2008. Association of steroid receptor gene (NR3C1) polymorphism with clinical course of lymphoproliferative disorders in children—preliminary results. Onkologia Polska 2: 77–81.
Syed, A., J. Irving, C. Redfern, A. Hall, N. Unwin, et al. 2006. Association of glucocorticoid receptor polymorphism A3669G in exon 9beta with reduced central adiposity in women. Obesity 14: 759–764.
Panek, M., T. Pietras, A. Antczak, A. Fabijan, M. Przemęcka, et al. 2012. The N363S and I559N single nucleotide polymorphisms of the h-GR/NR3C1 gene in patients with bronchial asthma. International Journal of Molecular Medicine 30: 142–150.
Bray, P., and R. Cotton. 2003. Variations of the human glucocorticoid receptor gene (NR3C1): pathological and in vitro mutations and polymorphisms. Human Mutation 21: 557–568.
Feng, J., J. Zheng, W. Bennett, L. Heston, I. Jones, et al. 2000. Five missense variants in the amino-terminal domain of the glucocorticoid receptor: no association with puerperal psychosis or schizophrenia. American Journal of Medical Genetics 96: 412–417.
Russcher, H., P. Smit, E. van den Akker, E. van Rossum, A. Brinkman, et al. 2005. Two polymorphisms in the glucocorticoid receptor gene directly affect glucocorticoid-regulated gene expression. Journal of Clinical Endocrinology and Metabolism 90: 5804–5810.
Karl, M., S. Lamberts, S. Detera-Wadleigh, I. Encio, C. Stratakis, et al. 1993. Familial glucocorticoid resistance caused by a splice site deletion in the human glucocorticoid receptor gene. Journal of Clinical Endocrinology and Metabolism 76: 683–689.
van den Akker, E., H. Russcher, E. van Rossum, A. Brinkmann, F. de Jong, et al. 2006. Glucocorticoid receptor polymorphism affects transrepression but not transactivation. Journal of Clinical Endocrinology and Metabolism 91: 2800–2803.
Panek, M., T. Pietras, J. Szemraj, A. Fabijan, and P. Kuna. 2013. Identification and association of TGFβ-1 expression in patients with asthma in a Polish population—Lodz metropolitan area study. International Journal of Biochemistry and Molecular Biology 31: 67–74.
Stępień-Wyrobiec, O., A. Hrycek, and G. Wyrobiec. 2008. Transforming growth factor beta (TGF-beta): its structure, function, and role in the pathogenesis of systemic lupus erythematosus. Postepy Higieny I Medycyny Doswiadczalnej 62: 688–693.
Bierie, B., and H.L. Moses. 2006. TGF β: the molecular Jekyll and Hyde of cancer. Nature Reviews Cancer 6: 506–520.
Flanders, K.C., and J.K. Burmester. 2003. Medical applications of TGF-β. Clinical Medicine & Research 1: 13–20.
Horowitz, J.C., D.Y. Lee, M. Waghray, V.G. Keshamouni, P.E. Thomas, et al. 2004. Activation of the prosurvival phosphatidylinositol 3-kinase/AKT pathway by TGF-β1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. Journal of Biological Chemistry 279: 1359–1367.
Kim, S.G., H.S. Jong, T.Y. Kim, J.W. Lee, N.K. Kim, et al. 2004. TGF-β1 induces apoptosis through Fas ligand -independent activation of the Fas death pathway in human gastric SNU-620 carcinoma cells. Molecular Biology of the Cell 15: 420–434.
Lee, K.Y., and S.C. Bae. 2002. TGF-β-dependent cell growth arrest and apoptosis. Journal of Biochemistry and Molecular Biology 35: 47–53.
Fleisch, M.C., C.A. Maxwell, and M.H. Barcellos-Hoff. 2006. The pleiotropic roles of transforming growth factor beta in homeostasis and carcinogenesis of endocrine organs. Endocrine-Related Cancer 13: 379–400.
Lu, L.Y., H.H. Cheng, P.K. Sung, J.J. Yeh, Y.L. Shiue, et al. 2004. Single-nucleotide polymorphisms of transforming growth factor-β1 gene in Taiwanese patients with systemic lupus erythematosus. Journal of Microbiology, Immunology and Infection 37: 145–152.
Rider, C.C. 2006. Heparin/heparan sulphate binding in the TGF-β cytokine superfamily. Biochemical Society Transactions 34: 458–460.
McMillan, S.J., G. Xanthou, and C.M. Lloyd. 2005. Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect on the Smad signaling pathway. Journal of Immunology 174: 5774–80.
Wen, F.Q., T. Kohyama, C.M. Sköld, Y.K. Zhu, X. Liu, et al. 2002. Glucocorticoids modulate TGF-beta production. Inflammation 26: 279–290.
Global Initiative for Asthma 2007. 2008. GINA executive summery. European Respiratory Journal 31: 143–178.
Guidelines for the Diagnosis and Management of Asthma. National Asthma Education and Prevention Program (NAEPP) Expert Panel Report 3. Summary Report 2007. NIH Publication Number 08–5846 (2007) Bethesda: National Institutes of Health, National Heart, Lung and Blood Institute Press.
Miller, M., J. Hankinson, V. Brusasco, F. Crapo, R. Burgos, et al. 2005. ATS/ERS task force: standardization of lung function testing. European Respiratory Journal 26: 319–338.
Dreborg, S., A. Backman, and A. Bosomba. 1989. Skin tests used for type I allergy testing. Position paper, EAACI subcommittee. Allergy 44: 1–59.
Panek, M., T. Pietras, and P. Kuna. 2012. Contemporary management of COPD in the view of the new GOLD guidelines. Terapia 271: 71–77.
Nathan, R.A., C.A. Sorkness, M. Kosinski, M. Schatz, J.T. Li, et al. 2004. Development of the test: a survey for assessing asthma control. Journal of Allergy and Clinical Immunology 113: 59–65.
BMI Classification. Global Database on Body Mass Index (2012) World Health Organization Press.
Eknoyan, G. 2008. Adolphe Quetelet (1796–1874)—the average man and indices of obesity. Nephrology, Dialysis, Transplantation 23: 47–51.
Szabó, V., G. Borgulya, T. Filkorn, J. Majnik, I. Bányász, et al. 2007. The variant N363S of glucocorticoid receptor in steroid-induced ocular hypertension in Hungarian patients treated with photorefractive keratectomy. Molecular Vision 13: 659–666.
Chomczynski, P., and N. Sacchi. 1987. Single - step method of RNA isolation by acid guanidinium thiocyanate- phenol—chloroform extraction. Analytical Biochemistry 162: 156–159.
Livak, K.J., and T.D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2 − ΔΔCT method. Methods 25: 402–408.
Winer, J., C.K. Jung, I. Shackel, and P.M. Williams. 1999. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Analytical Biochemistry 270: 41–49.
Lipsett, M., G. Chrousos, M. Tomita, D. Brandon, and D. Loriaux. 1985. The defective glucocorticoid receptor in man and nonhuman primates. Recent Progress in Hormone Research 41: 199–247.
Carmichael, J., I. Paterson, P. Diaz, G. Crompton, A. Kay, et al. 1981. Corticosteroid resistance in chronic asthma. British Medical Journal 282: 1419–1422.
Barnes, P. 2006. Corticosteroid effects on cell signalling. European Respiratory Journal 27: 413–426.
Corrigan, C., P. Brown, N. Barnes, S. Szefler, J. Tsai, et al. 1991. Glucocorticoid resistance in chronic asthma. Glucocorticoid pharmacokinetics, glucocorticoid receptor characteristics, and inhibition of peripheral blood T cell proliferation by glucocorticoids in vitro. American Review of Respiratory Disease 144: 1016–1025.
Huizenga, N., J. Koper, P. De Lange, H. Pols, R. Stolk, et al. 1998. A polymorphism in the glucocorticoid receptor gene may be associated with and increased sensitivity to glucocorticoids in vivo. Journal of Clinical Endocrinology and Metabolism 83: 144–151.
Johnson, J.R., M. Nishioka, J. Chakir, P.A. Risse, I. Almaghlouth, et al. 2013. IL-22 contributes to TGF-β1-mediated epithelial-mesenchymal transition in asthmatic bronchial epithelial cells. Respiratory Research 14: 118.
Komisja Chorób Układu Oddechowego Komitetu Patofizjologii Klinicznej Polskiej Akademii Nauk. Przewodnicząca Komisji: prof. Ryszarda Bożena Chazan. Stanowisko Komisji Chorób Układu Oddechowego Komitetu Patofizjologii Klinicznej Polskiej Akademii Nauk. Astma ciężka. (2009) Warszawa: Polska Akademia Nauk Press.
Bel, E., and A. ten Brinke. 2005. A rational approach to the management of severe refractory asthma. Treatments in Respiratory Medicine 4: 365–379.
Chanez, P., S. Wenzel, G. Anderson, J. Anto, E. Bel, et al. 2007. Severe asthma in adults: what are the important questions? Journal of Allergy and Clinical Immunology 119: 1337–1348.
Barnes, P. 2004. New drugs for asthma. Nature Reviews Drug Discovery 3: 831–844.
Adcock, I., K. Chung, G. Caramori, and K. Ito. 2006. Kinase inhibitors and airway inflammation. European Journal of Pharmacology 533: 118–132.
Acknowledgments
The authors of the paper wish to thank Grzegorz Uraziński, M.Sc. Eng. in Architecture, for preparation of the figures and his kind assistance in graphic rendering of research materials. The authors of the study thank Joanna Molinska MA for helping in administrative work. Lastly, we offer our acknowledgements to all of those who supported us in any respect during the completion of the project
Author information
Authors and Affiliations
Corresponding author
Additional information
Artur Fabijan and Jan Zioło contributed equally to this work.
Rights and permissions
About this article

Cite this article
Panek, M., Pietras, T., Fabijan, A. et al. The NR3C1 Glucocorticoid Receptor Gene Polymorphisms May Modulate the TGF-beta mRNA Expression in Asthma Patients. Inflammation 38, 1479–1492 (2015). https://doi.org/10.1007/s10753-015-0123-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-015-0123-3