
Abstract
Human β-defensin-2(HBD-2) is one of the two major vertebrate antimicrobial peptide families (α and β), which is highly expressed by proinflammatory induction in the lung and exhibit broad-spectrum antimicrobial activity. We observed that IL-22 receptors high expressed on the membrane of A549 cells; HBD-2 mRNA was expressed in a time- and concentration-dependent manners in A549 cells when treated with IL-22; further studies demonstrated that HBD-2 expression was attenuated by AG490, but to JSH-23, inhibitors of p-STAT3 DNA binding and NF-κB/ p65 subunit nuclear translocation, respectively. These results support that IL-22-mediated signalling pathway of HBD-2 gene expression involved STAT3 but not NF-κB in human alveolar epithelium. These findings provide a new insight into how IL-22 may play an important link between innate and adaptive immunity, thereby anti-infection locally in the alveolar epithelium.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Alveolar epithelium not only serves as a physical barrier against infection, but also secretes a variety of antimicrobial substances that can inhibit or eliminate invading pathogens [1–3]. Small cationic antimicrobial peptides (AMPs) are one of the substances that protect their hosts against a wide range of microorganisms. These peptides are produced by bacteria, insects, plants and vertebrates, and have been recognised as ancient evolutionary conserved molecules that are effectively preserved in mammals [4].
Defensins are a type of important AMPs. They share six highly conserved cystein residues, which form disulfide bridges. On the basis of the different arrangements of cysteine motifs, human defensins are classified as α-defensins, β-defensins and θ-defensins [5]. Six β-defensins (HBD-1to HBD-6) that are produced by epithelial cells have been found in humans [6, 7]. All the β-defensins exhibit a wide range of antimicrobial activity at micromolar concentrations.
HBD-2 expressed by epithelial cells following contact with bacteria, viruses, fungi, or cytokines, such as IL-1 and TNF-α [8], has shown notable antimicrobial activity. A previous study indicated that amongst IL-1α, 1β, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 15, 16, 17, and 18, IFN-γ, GM-CSF, and TNF-α, IL-17 is the most potent inducer of HBD-2 expression in primary human tracheobronchial epithelial (TBE) cells [9]. Although the nature of this regulation has not been adequately elucidated, both the MAPK signalling pathway and the NF-κB transcription factor have been confirmed involved in the regulation [10].
IL-22 belongs to the IL-10 family of cytokines that was recently determined to be produced by activated Th17 cells, predominantly of the prototypic CD45+RO+CD4+ subtype [11]. Emerging evidence indicates that IL-22 targets skin cells, as well as digestive and respiratory organ systems; it also plays an important role in mucosal immunity [12]. In previous research, IL-22 could regulate multiple airway epithelial cytokine secretion in the context of local bacterial infections, as well as increase lung epithelial cell proliferation and transepithelial resistance to injury [13]. However, the physiological functions between IL-22 and alveolar epithelium remain unknown.
HBD-2 has been found elevated in airway epithelium by IL-17, thereby stimulating NF-κB activation [14]. IL-22, as well as IL-17, is also an effect cytokine produced by the Th-17 lineage; both cytokines are crucial for maintaining local control of Gram-negative pulmonary pathogens [13]. IL-22 could induce STAT3 phospholipids in intestinal epithelial cells [15]. We hypothesise that IL-22 can induce HBD-2 expression in alveolar epithelial type II cells involving the NF-κB or STAT3 signalling pathway.
In this study, we initially identify a marked activation of HBD-2 expression in human alveolar epithelial type II cells by IL-22. Further experiments suggest that this stimulation occurs at the transcriptional level through STAT3-dependent and NF-κB-independent mechanisms.
MATERIALS AND METHODS
Reagents and Cell Lines
Recombinant human IL-22 (rhIL-22), monoclonal anti-human IL-22R-allophycocyanin and mouse IgG1 isotype control-APC were purchased from R&D Systems (Minneapolis, USA). STAT3 antibody, phospho-STAT3 antibody, NF-κB p65 antibody and the F(ab’)2 fragment of anti-rabbit IgG (H+L) (Alexa Fluor 488 Conjugate) were obtained from Cell Signaling Technology (Massachusetts, USA). NF-κB activation inhibitor II (JSH-23) and STAT3 DNA binding inhibitor (AG490) were purchased from Calbiochem-Novabiochem (Darmstadt, GER). Alveolar epithelial type II cell lines A549 were provided by the Department of Oncology of the First Affiliated Hospital of Nanjing Medical University.
IL-22R Detection on A549 Cells by Flow Cytometry
Adherent cell lines A549 were pre-treated with 0.25 % Trypsin-EDTA to facilitate removal from their substrates. These cell lines were then incubated in medium for 6 to 8 h on a platform to enable the regeneration of receptors. Flow cytometry was conducted as described above; that is, the A549 cells were treated with monoclonal anti-human IL-22R-APC (R&D Systems Minneapolis, USA) and incubated for 40 min at 4 °C. The cells were centrifuged (1000 rpm, 4 min), and the supernatant was discarded to remove unbound fluorescent antibody. The A549 cells were analysed using a FACScan cytometer, and IL-22R+ cells accounted for a percentage of the total number of cells, as estimated using FACSDiva software.
rhIL-22 Stimulation and RNA Isolation
rhIL-22 was dissolved in PBS with 1 % BSA and added directly to the overnight serum-starved A549 cell cultures (20 ng/ml, following literature). The control treatments had the same amount of added PBS-1 % BSA. Six groups were established: 0 (control), 2, 4, 8, 16 and 24 h groups. Another five groups were set for different concentrations of rhIL-22 (i.e. 0, 4, 20, 100 and 500 ng/ml), with 24 h as the optimal time for rhIL-22 stimulation. RNA was extracted from cultures using RNA TRIzol reagent (Invitrogen), according to the manufacturer’s protocol.
RT-PCR Reaction
Real-time quantitative PCR (RT-PCR) was performed using the ABI 7900HT Fast Real-time PCR System (Applied Biosys-tems, Foster City, CA, USA). Total RNA was isolated from individual cell samples for analysis using TRIzol reagent (Invitro-gen, Carlsbad, CA, USA), according to the manufacturer’s instructions. The isolated total RNA was then quantified by spectrophotometric determination (260 nm). One microgram of total RNA was reverse transcribed to complementary DNA (cDNA) using an RT-PCR kit (Takara Bio Inc., Otsu, Japan), according to the manufacturer’s instructions. The cDNA was immediately analysed or stored at −20 °C, and subsequently amplified using the SYBR Green Master mix (Takara), according to the manufacturer’s protocols. Briefly, the 20 μl reaction system contained 10 μl SYBR Green PCR Master mix, 0.5 μmol/L primers, 2 μl cDNA (diluted 1:5) and diethyl pyrocarbonate water. The sequences of primer sets for RT-PCR are shown: HBD-2 forward: 5′-CCAGCCATCAGCCATGAGGGT-3′; HBD-2 reverse 5′-GGAGCCCTTTCTGAATCCGCA-3′(58 °C, 35 cycles). hGAPDH forward: 5′-CGGAGTCAACGGATTTGGTCGTAT-3′; hGAPDH reverse: 5′-AGCCTTCTCCATGGTG GTGAAGAC-3′(58 °C, 35 cycles). Relative mRNA levels were calculated using the relative standard curve method, in which the amounts of targets normalised to an endogenous reference (GAPDH) were compared. Briefly, the mean ± SD values of replicate samples were calculated. Results are expressed as the relative amount of mRNA in the experimental test samples and normal control samples (all normalised to β-actin). In preliminary experiments, the products were analysed by gel electrophoresis, and a single product was obtained with each primer set. Dissociation curves yielded single peaks.
Inhibitor Treatment, RNA Isolation and RT-PCR Analysis
STAT3 inhibitor AG490 and NF-κB activation inhibitor II JSH-23 were dissolved in DMSO, and added to the cultures 30 min before IL-22 treatment. The control culture media contained similar levels of PBS/1 % BSA. The optimal dose grades for each of the selected inhibitors were determined in reference to current literature and the results of our previous tests. No obvious cytotoxicity (as indicated by trypan blue dye exclusion) was observed in these cultures after the inhibitor treatments. After IL-22 stimulation for the optimal time, RNA was extracted from the cultures and used for RT-PCR expression analysis.
Measurement of HBD-2 Secretion by ELISA
For the quantification of HBD-2 released before and after the addition of inhibitors, BD OptEIA Human HBD-2 Elisa Kit II (BD Biosciences) was used according to the manufacturer’s instructions. For quantification, the reference curves obtained using increasing concentrations of recombinant rat cytokines, were determined in parallel. The protein concentrations in the cell samples were determined using the Bradford dye binding procedure. HBD-2 concentration was calculated as pg mg – 1 of total protein for each sample.
Western Blot Detection of STAT3 Phosphorylation
A549 cells (106) were stimulated with an optimal dose of rhIL-22 for up to 60 min. The cells were then lysed in RIPA Lysis Buffer. Obtained aliquots were boiled for 5 min with loading buffer, resolved on a 10 % SDS-PAGE gel and electrophoretically transferred to Immobilon-P membranes (Millipore). The membranes were blocked for 90 min in TBS (150 mmol/L NaCl and 20 mmol/L Tris, pH 7.5) containing 5 % nonfat dry milk and 0.1 % Tween 20. The membranes were then incubated with rabbit monoclonal anti-p-STAT3 at 4 °C overnight. After incubation with horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibodies (ZSGB-BIO), the proteins were visualised with SuperSignal West Pico (Thermo Scientific).
Immunofluorescence Analysis of p-STAT3 and NF-κB Translocation
For p-STAT3 and NF-κB/p65 staining, the A549 cells were cultured on sterile Lab-Tek II chamber slide systems (Nalge Nunc International) incubated in previously established experimental conditions (to the optimal intervention the concentration of IL-22 to interfere with the p-STAT3 cells 10 min, 30 min, intervention NF-κB cells 15 min, 30 min). After rhIL-22 treatment, the cells were fixed for 15 min at room temperature with 4 % paraformaldehyde in PBS. The Cell Signaling Technology manufacturer’s protocols were followed. The staining was visualised under a Zeiss AxioSkop fluorescence microscope (×40 objective).
Statistical Analysis
Data are expressed as mean ± SE. The number of repeats for each experiment is described under the RESULTS section. Group differences were calculated by t test or one-way ANOVA. A value of p < 0.05 was considered significant.
RESULTS
High Expression of IL-22R on Alveolar Epithelial Type II Cell Lines A549
IL-22 receptors were highly expressed on the membrane of human alveolar epithelial type II cell lines A549. The A549 cells were analysed using a FACScan cytometer, and IL-22R+ cells accounted for a percentage of the total number of cells, as estimated using FACSDiva software. The positive rate was (99.90 ± 0.13)%, (p < 0.05) (Fig. 1).

Quantity of IL-22R+ alveolar epithelial type II cell lines A549 measured by flow cytometry. The positive rate was (99.90 ± 0.13)%; p < 0.05 when compared with isotype IgG1 expression.
Kinetics Studies of IL-22 Effects on HBD-2 mRNA
IL-22 exerted time- and concentration-dependent effects on HBD-2 mRNA expression (Figs. 2 and 3). A time gradient existed between IL-22 stimulation and HBD-2 mRNA expression. A significant expression of HBD-2 mRNA by IL-22 (20 ng/ml) stimulation was observed within 2 h, and the maximum expression was observed at 24 h. A concentration of IL-22 4 ng/ml induced HBD-2 mRNA expression in the A549 cells after 24 h of treatment. Peak expression occurred at 100 ng/ml, with no further increase at 500 ng/ml.

RT-PCR analysis of the level of HBD-2 mRNA expression in A549 cells after rhIL-22 (20 ng/ml) stimulation at different times. PCR products were visualised in ethidium bromide-2 % agarose gels.*p < 0.05 when compared with group 0 h.

RT-PCR was performed for HBD-2 and GAPDH on total RNA isolated from A549 cells after rhIL-22 stimulation for 24 h at various concentrations. PCR products were analysed in ethidium bromide-2 % agarose gels. Values of untreated and IL-22-treated samples were compared.*p < 0.05.
Inhibition of IL-22-Dependent HBD-2 Expression by STAT3 but Not by NF-κB Inhibitors
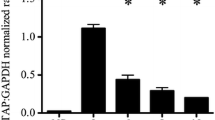
To elucidate possible IL-22-mediated signalling on the regulation of HBD-2 expression, the inhibitors of two signalling pathways were used (Fig. 4). AG490, an inhibitor of the DNA binding activity of p-STAT3, was the most potent inhibitor that reversed the enhancement of IL-22-induced HBD-2 expression. Figure 4 shows that the reversal of AG490 enhancement was concentration dependent, but NF-κB activation inhibitor II (JSH-23) had no suppressive effect on IL-22-induced HBD-2 expression, even at higher doses.

RT-PCR analysis of the effect of two inhibitors on IL-22-induced HBD-2 expression. A549 cells were cultured as described in the text. Cultures were pre-treated with 5 μM (C), 10 μM (D), and 20 μM (E) activation of STAT3 DNA binding inhibitor, AG490, and 2.5 μM (F), 5 μM (G), and 10 μM (H) NF-κB activation inhibitor II, JSH-23 30 min before IL-22 (100 ng/ml) treatment (C–H). Cells were harvested 24 h later. Group A was the control culture (no treatment). Group B was treated only with IL-22. *p < 0.05 when compared with control (A). **p < 0.05 and ***p > 0.05 when compared with IL-22 treatment alone (B).
ELISA Measurement of HBD-2 Protein with Two Inhibitors
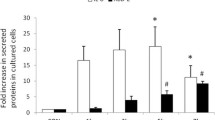
A competitive ELISA demonstrated a significant increase in HBD-2 peptide secretion levels compared with control after 24-h treatment with IL-22 (100 ng/ml) in human alveolar epithelial type II cell lines A549. And after various concentrations of rhIL-22 were added to the inhibitor of AG490 with various concentrations, as indicated, the secretion of HBD-2 significantly decreased in the epithelial cells exposed to IL-22 compared with the epithelial cells subjected to IL-22 treatment alone (Fig. 5). This result was not observed after JSH-23 addition, even at higher doses. As show in Fig. 5, the tendency for IL-22-induced protein release after the two-inhibitor treatment was associated with HBD-2 gene induction.

Competitive ELISA for the quantification of HBD-2 secretion by cultured A549 cells after IL-22 and two-inhibitor treatment. Serial dilutions of synthetic mature HBD-2 peptide were used to generate a standard curve, and the HBD-2 peptide concentration of each experimental group was considered. Two inhibitors were also added to the cultures 30 min before IL-22 treatment. After incubation (24 h), culture fluids, including the wash, were collected and centrifuged to pellet cell debris, and subjected to competitive HBD-2 ELISA. HBD-2 peptide secretion significantly decreased in the AG490 groups at a dose-dependent fashion; this decrease was not observed in JSH-23 exposed to IL-22 compared with JSH-23 subjected to IL-22 treatment alone. *p < 0.05 when compared with control. **p < 0.05 and ***p > 0.05 when compared with IL-22 treatment alone.
IL-22 Induces STAT3 Phosphorylation in A549 Cells
Alveolar epithelial type II cell lines A549 highly expressed IL-22R, making this cell type likely responsive to IL-22. As shown by Western blotting, IL-22 induced STAT3 phosphorylation in the A549 cells. Cell extracts were measured with anti-STAT3 and anti-p-STAT3 antibodies (Fig. 6). STAT3 phosphorylation was detected with the anti-p-STAT3 antibody 5 min after IL-22 treatment and peak stimulation at 30 min.

Western blot analysis of STAT3 tyrosine phosphorylation in alveolar epithelial type II cell lines A549. A549 cells were stimulated with 100 ng/ml rhIL-22 for up to 60 min. After cell lysis, total cell protein was electrophoresed with 10 % SDS-PAGE gel and electrophoretically transferred to Immobilon-P membranes. The membranes were incubated with rabbit monoclonal anti-p-STAT3. After incubation with HRP-conjugated secondary antibodies, the proteins were visualised with ECL.*p < 0.05 when compared with control.
Induction of p-STAT3 but Not NF-κB/p65 Nuclear Translocation by IL-22
To confirm STAT3 activation, induced p-STAT3 nuclear translocation was conducted. Figure 7a shows almost no green staining in the nucleus of the cultured A549 cells before IL-22 treatment. Ten minutes after IL-22 stimulation, the nuclear translocation of p-STAT3 was clearly observed in many of the A549 cells. As a comparison, no visible nuclear translocation of NF-κB/p65 was detected (Fig. 7b). These results are consistent with the STAT3 inhibitor experiment, further suggesting the significant role of IL-22-induced STAT3 activation in the stimulation of HBD-2 gene expression.

a Nuclear translocation of p-STAT3 in alveolar epithelial type II cell lines A549 after IL-22 treatment. A549 cells were plated on a Lab-Tek II chamber slide and treated with IL-22 as described in the MATERIALS AND METHODS section. After 10- and 30-min treatments, these slides were fixed with ice-cold 4 % paraformaldehyde solution overnight, followed by staining with anti-p-STAT3 mAb and Alexa Fluor 488-conjugated rabbit anti-mouse IgG Ab (green fluorescence), as described. These slides were examined under a Zeiss fluorescent microscope with ×40 lens. Control culture without cytokine treatment is shown. b Nonnuclear translocation of the p65 subunit of NF-κB transcriptional factors in A549 cells after IL-22 treatment, as depicted in Fig. 7a.
DISCUSSION
IL-22, induced in mouse T lymphoma cells by IL-9 and having 22 % amino acid identity with IL-10 [16], is an IL-10 family cytokine member that was initially known as an IL-10-related T-cell-derived inducible factor (IL-TIF) when it was first characterised by Dumoutier et al. [12]. Emerging studies have indicated that IL-22 targets the epithelial or parenchymal cells of the lung, gut, skin and kidney [17]. The IL-22 receptor consists of two subunits: IL-22R1 and IL-10R2 [17, 18]. IL-22R is widely expressed on a variety of tissues—lung, skin, liver, colon, kidney, small intestine and pancreas [18]—thereby allowing the cytokine to mediate epithelial innate immunity in response to microbial infections. Although current studies have revealed the significance of IL-22 in host defence against Gram-negative bacteria (in lung and gut) [18–20], evidence shows that IL-22 also plays an important role in autoimmune diseases, such as psoriasis [21, 22]. Therefore, IL-22 exerts complex anti-inflammatory, pro-inflammatory and autoimmune effects—an issue that requires further investigation.
IL-22, mainly secreted by activated Th17 cells, is markedly enhanced in lung after microbial infections and plays a pro-inflammatory role in regulating the production of multiple cytokines. IL-22R knockout mice have impaired abilities of elimination invading pathogens [13]. Furthermore, IL-22 increases lung epithelial cell proliferation and transepithelial resistance to injury [13], which further strengthens the mucosal barrier against microorganisms. These data support the idea that IL-22 mediates the lung inflammatory response from inherent immunity processes to adaptive response mechanisms.
HBD-2 is inducibly expressed in a variety of human mucosa-associated tissues by IL-1α, IL-1β, Gram (+), Gram (−), tumour necrosis factor (TNF), lipopolysaccharides (LPS) and lipoarabinomannan (LAM) [23, 24], especially in mucosal epithelial cells in the skin, lung, gut and genitourinary tract. The known biological effect of HBD-2 in innate immunity is recognised as related to its antimicrobial activity and its chemotactic activity in CCR6-positive immature dendritic and memory T-cells [25]. Other functions of HBD-2 have recently been described: neutralisation of endotoxins, immunomodulation properties and induction of both angiogenesis and wound repair [26].
The exact mechanism by which HBD-2 exhibits antimicrobial activity remains unknown, but a generally accepted finding is that through electrostatic forces, cationic HBD-2 interacts with negatively charged phospholipid head groups on bacterial membrane and causes disruption. The most widely accepted mechanisms are the “barrel stave” model and the “carpet model” [26]. Other suggested mechanisms also include the formation of ionic channels and the activation or blockage of intracellular targets after bacterial membrane permeabilisation [27, 28].
In this study, we observed that IL-22R was highly expressed on A549 cells, and found an IL-22 time- and dose-dependent stimulation of HBD-2 expression in human alveolar epithelial type II cell lines A549. The appearance of this cell line is epithelioid. It behaves similarly to primary alveolar epithelial cells in its response to IL-22 for inducing HBD-2 expression. An IL-22 concentration as lows as 4 ng/ml elicited a significant stimulation of HBD-2 gene expression. Although the up-regulation of HBD-2 by IL-22 has been reported in airway epithelial cells [13], the influence of IL-22 on HBD-2 in alveolar epithelial cells has not been discussed. We further suggest that IL-22 is an important immune mediator and that it acts on lung tissue to regulate host defence responses.
A previous study indicated that IL-17, as well as IL-22 mainly secreted by Th17 cells, induced the up-regulation of HBD-2 expression through NF-κB by airway epithelial cells [8]. In our study, however, IL-22-mediated HBD-2 expression was uninhibited by JSH-23. Our results suggest that AG490 dramatically suppresses IL-22-stimulated HBD-2 expression in a dose-dependent fashion. On the basis of AG490 (>50 μmol/L) inducing apoptosis in the A549 cells (as determined by assay), we reregulated the concentration gradient (5 to 20 μmol/L). Up to a concentration of 20 μmol/L, no effects on total RNA levels or cell viability were found for AG490, indicating that the inhibitory effect observed was not due to cell toxicity (data not shown).
Additional Western blotting and immunofluorescence experiments on the A549 cells were conducted to elucidate the molecular mechanism of this regulation. After 5 min of IL-22 treatment, STAT3 phosphorylation occurred in a time-dependent fashion and peaked at 30 min. Consistent with this idea, the immunofluorescence experiment showed that IL-22 induced STAT3 phosphorylation nuclear translocation but not the NF-κB/p65 subunit at the corresponding. This result also demonstrated that IL-22 enhanced HBD-2 expression by inducing STAT3 phosphorylation and enhancing its nuclear translocation from the cytoplasm.
On the basis of these findings, we propose an IL-22R-dependent JAK2 and STAT3 signalling pathway for the transcription of HBD-2 gene by IL-22 in alveolar epithelial cells. Because IL-22 elevation in lung has been related to local microbial infection [13], such a signalling mechanism may play an important role in regulating the innate and adaptive immune responses in lung. We postulate that a low-level induction of HBD-2 is directly or indirectly stimulated during the early stages of bacterial infection in lung by microbial products, such as LPS, binding to nonspecific receptors, including TLR-4 on epithelial cells. This innate nature of response provides limited protection from bacterial invasion through the antimicrobial activities of HBD-2 [29–31]. When adaptive immune responses develop, however, activated Th17 cells are recruited to the site of infection and locally release IL-22. Such IL-22 can presumably enhance local antimicrobial activity by binding to the IL-22R through a JAK2/STAT3 signalling pathway-dependent mechanism for the high gene expression of HBD-2 and/or other cytokines.
In summary, this paper demonstrated a newly identified role of IL-22 in directly mediating HBD-2 gene expression in alveolar epithelial cells. We also confirmed that this effect on HBD-2 occurs mostly through STAT3 signalling events. Subsequent studies should elucidate whether the other pathways are also involved in IL-22-induced HBD-2 gene expression. Our results widen the spectrum of known interleukins that can stimulate lung defensin expression and serve as valid evidence of a novel IL-22-related mechanism in coordinating inherent and acquired immune responses.
LIMITATIONS
We did not use human alveolar epithelial type II cells to do this work, because they are too difficult to be acquired and cultivated. Although the molecular characteristics of cell lines A549 are similar to alveolar epithelial type II cells, the hypothesis posed by our authors remains to be validated in primary cell in our future research.
Abbreviations
- IL:
-
Interleukin
- HBD:
-
Human β-defensin
- TBE:
-
Tracheobronchialepithelial
- AMPs:
-
Antimicrobial peptides
- IFN-γ:
-
Interferon-γ
- GM-CSF:
-
Granulocyte-macrophage colony-stimulatingfactor
- TNF-α:
-
Tumor necrosis factor α
- PBS:
-
Phosphate-buffered saline
- LPS:
-
Lipopolysaccharides
- LAM:
-
Lipoarabinomannan
References
Bartlett, J.A., A.J. Fischer, and P.B. McCray. 2008. Innate immune functions of the airway epithelium. Contributions to Microbiology 15: 147–163.
Hiemstra, P.S. 2007. The role of epithelial beta-defensins and cathelicidins in host defense of the lung. Experimental Lung Research 33: 537–542.
Laube, D.M., S. Yim, L.K. Ryan, K.O. Kisich, and G. Diamond. 2006. Antimicrobial peptides in the airway. Current Topics in Microbiology and Immunology 306: 153–182.
Lai, Y., and R.L. Gallo. 2009. MPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends in Immunology 30: 131–141.
Yang, D., Z.H. Liu, P. Tewary, Q. Chen, G. de la Rosa, and J.J. Oppenheim. 2007. Defensin participation in innate and adaptive immunity. Current Pharmaceutical Design 13: 3131–3139.
McCormick, T.S., and A. Weinberg. 2010. Epithelial cell-derived antimicrobial peptides are multifunctional agents that bridge innate and adaptive immunity. Periodontology 2000 54: 195–206.
Hollox, E.J., and J.A. Armour. 2008. Directional and balancing selection in human betadefensins. BMC Evolutionary Biology 8: 113.
Wehkamp, K., L. Schwichtenberg, J.M. Schröder, and J. Harder. 2006. Pseudomonas aeruginosa- and IL-1β-mediated induction of human beta-defensin-2 in keratinocytes is controlled by NF-kappaB and AP-1. Journal of Investigative Dermatology 126: 121–127.
Kao, C.Y., Y. Chen, P. Thai, S. Wachi, F. Huang, C. Kim, et al. 2004. IL-17 markedly up-regulates beta-defensin-2 expression in human airway epithelium via JAK and NF-kappaB signaling pathways. Journal of Immunology 173: 3482–3491.
Yoon, Y.M., J.Y. Lee, D. Yoo, Y.S. Sim, Y.J. Kim, Y.K. Oh, et al. 2010. Bacteroides fragilis enterotoxin induces human beta-defensin-2 expression in intestinal epithelial cells via a mitogen-activated protein kinase/I kappaB kinase/NF-kappaB-dependent pathway. Infection and Immunity 78: 2024–2033.
Korn, T., E. Bettelli, M. Oukka, and V.K. Kuchroo. 2009. IL-17 and Th17 cells. Annual Review of Immunology 27: 485–517.
Aujla, S.J., and J.K. Kolls. 2009. IL-22: a critical mediator in mucosal host defense. Journal of Molecular Medicine 87: 451–454.
Aujla, S.J., Y.R. Chan, M. Zheng, M. Fei, D.J. Askew, D.A. Pociask, et al. 2008. IL-22 mediates mucosal host defense against gram-negative bacterial pneumonia. Nature Medicine 14: 275–281.
Kao, C.Y., C. Kim, F. Huang, and R. Wu. 2008. Requirements for two proximal NF-kappaB binding sites and IkappaB-zeta in IL-17A-induced human beta-defensin 2 expression by conducting airway epithelium. Journal of Biological Chemistry 283: 15309–15318.
Brand, S., F. Beigel, T. Olszak, K. Zitzmann, S.T. Eichhorst, J.M. Otte, et al. 2006. IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. American Journal of Physiology - Gastrointestinal and Liver Physiology 290: G827–G838.
Dumoutier, L., J. Louahed, and J.C. Renauld. 2000. Cloning and characterization of IL-10-related T cell derived inducible factor (IL-TIF), a novel cytokine structurally related to IL-10 and inducible by IL-9. Journal of Immunology 164: 1814–1819.
Wolk, K., and R. Sabat. 2006. Interleukin-22: a novel T and NK cellderived cytokine that regulates the biology of tissue cells. Cytokine & Growth Factor Reviews 17: 367–380.
Wolk, K., S. Kunz, E. Witte, M. Friedrich, K. Asadullah, and R. Sabat. 2004. IL-22 increases the innate immunity of tissues. Immunity 21: 241–254.
Sugimoto, K., A. Ogawa, E. Mizoguchi, Y. Shimomura, A. Andoh, A.K. Bhan, et al. 2008. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. Journal of Clinical Investigation 118: 534–544.
Zheng, Y., P.A. Valdez, D.M. Danilenko, Y. Hu, S.M. Sa, Q. Gong, et al. 2008. Interleukin 22 mediates early host defense against attaching and effacing bacterial pathogens. Nature Medicine 14: 282–289.
Wolk, K., E. Witte, K. Warszawska, G. Schulze-Tanzil, K. Witte, S. Philipp, et al. 2009. The Th17 cytokine IL-22 induces IL-20 production in keratinocytes: a novel immunological cascade with potential relevance in psoriasis. European Journal of Immunology 39: 3570–3581.
Ma, H.L., S. Liang, J. Li, L. Napierata, T. Brown, S. Benoit, et al. 2008. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. Journal of Clinical Investigation 118: 597–607.
Harder, J., U. Meyer-Hoffert, L.M. Teran, L. Schwichtenberg, J. Bartels, S. Maune, Mucoid, et al. 2000. Pseudomonas aeruginosa, TNF-alpha, and IL-1beta, but not IL-6, induce human beta-defensin-2 in respiratory epithelia. American Journal of Respiratory Cell and Molecular Biology 22: 714–721.
Mendez-Samperio, P., L. Alba, and A. Trejo. 2007. Mycobacterium bovismediated induction of human beta-defensin-2 in epithelial cells is controlled by intracellular calcium and p38MAPK. Journal of Infection 54: 469–474.
Rohrl, J., D. Yang, J.J. Oppenheim, and T. Hehlgans. 2010. Specific binding and chemotactic activity of mBD4 and its functional orthologue HBD2 to CCR6-expressing cells. Journal of Biological Chemistry 285: 7028–7034.
Zaiou, M. 2007. Multifunctional antimicrobial peptides: therapeutic targets in several human diseases. Journal of Molecular Medicine 85: 317–329.
Brogden, K.A. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nature Reviews Microbiology 3: 238–250.
Cudic, M., and L. Otvos Jr. 2002. Intracellular targets of antibacterial peptides. Current Drug Targets 3: 101–106.
MacRedmond, R., C. Greene, C.C. Taggart, N. McElvaney, and S. O’Neill. 2005. Respiratory epithelial cells require Toll-like receptor 4 for induction of human beta-defensin 2 by lipopolysaccharide. Respiratory Research 6: 116.
Romano Carratelli, C., N. Mazzola, R. Paolillo, S. Sorrentino, and A. Rizzo. 2009. Toll-like receptor-4 (TLR4) mediates human beta-defensin-2 (HBD-2) induction in response to Chlamydia pneumoniae in mononuclear cells. FEMS Immunology and Medical Microbiology 57: 116–124.
Donnarumma, G., I. Paoletti, E. Buommino, M.R. Iovene, L. Tudisco, V. Cozza, et al. 2007. Anti-inflammatory effects of moxifloxacin and human beta-defensin 2 association in human lung epithelial cell line (A549) stimulated withlipopolysaccharide. Peptides 28: 2286–2292.
Acknowledgments
We thank the Department of Oncology of the First Affiliated Hospital of Nanjing Medical University for providing alveolar epithelial type II cell lines A549 and Cui Ping Liu for expert technical help.
Conflict of Interest
None of the authors of this study has any financial or commercial conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, A., Gan, Y., Wang, R. et al. IL-22 Up-Regulates β-Defensin-2 Expression in Human Alveolar Epithelium via STAT3 but Not NF-κB Signaling Pathway. Inflammation 38, 1191–1200 (2015). https://doi.org/10.1007/s10753-014-0083-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-014-0083-z