
Abstract
Species proliferate through evolutionary mechanisms but coexist through ecological dynamics. As such, it might be expected that mechanisms of speciation and species maintenance jointly influence the settlement of ecological communities, a process called community assembly. Disentangling the relative contribution of evolutionary and ecological dynamics might be a difficult task, particularly so for the tropical biotas due to their extreme diversity and large knowledge gaps. Here, we explore genetic diversity and distribution of 23 freshwater shrimp species of the genera Caridina and Macrobrachium in Sundaland to examine patterns of species co-occurrence based on 1583 observations across 19 sites in Java and Bali islands. DNA-based species delimitation methods applied to 204 cytochrome oxidase I sequences detected 30 operational taxonomic units and a few cases of deep intraspecific divergence. Species co-occurrence and phylogenetic community structure show no departure from expectations under a random distribution of species in landscapes and support a lottery model of community assembly. Species age estimates expand beyond the geological settlement of Sundaland, suggesting that species proliferation and community assembly are driven by mechanisms acting at distinct spatial and temporal scales.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Species proliferate through evolutionary dynamics and further persist in landscapes through a variety of ecological dynamics (Ricklefs, 1987; Ricklefs & Schluter, 1993; McPeek, 2008). The mechanisms of species aggregation in ecological communities, often referred to as community assembly, result from the integration of eco-evolutionary dynamics of species proliferation and persistence through time (Kraft et al., 2007; Emerson & Gillespie, 2008; Kembel, 2009; Hubert et al., 2015a). By acknowledging the overlap of speciation and ecological dynamics through space and time, recent studies of community assembly have increasingly incorporated species phylogenetic relationships to address the nature of community assembly mechanisms (Emerson & Gillespie, 2008; Cavender-Bares et al., 2009; Vamosi et al., 2009). Phylogenetic community structure analyses have been successfully applied to the study of community assembly for terrestrial biotas, particularly for plants (Cavender-Bares et al., 2006; Webb et al., 2006; Kraft et al., 2008). In turn, community assembly of freshwater biotas has attracted much less attention in the literature; however, historical contingencies, niche diversity and temporal heterogeneity have been identified as potential drivers (Vamosi & Vamosi, 2006; Vamosi et al., 2009; Hoverman et al., 2011; Brown & Milner, 2012; Hauffe et al., 2016).
Most of the previous studies on community assembly of freshwater biotas have been focused on temperate biomes; meanwhile, dynamics in tropical biotas have been largely overlooked. This is due, in part, to the fact that studying community assembly dynamics is particularly challenging in tropical biotas where taxonomic knowledge gaps and historical contingencies may have confounding effects on the nature of the ecological dynamics in ecological communities (Hebert et al., 2004; Smith et al., 2007, 2008). This situation is well exemplified by the freshwater biotas of Southeast Asia (SEA) that are extremely rich (De Grave et al., 2008; Hubert et al., 2015b), and result from a complex geological and volcanic history that prompted the settlement of some of the world’s largest insular biodiversity hotspots (Myers et al., 2000; Hoffman et al., 2010). This is particularly true in Sundaland, which includes the islands of Java, Sumatra, Borneo and Bali, and has been repeatedly connected to the continent during the sea-level low stand associated with glacial times (Voris, 2000; Woodruff, 2010). In such a mosaic of contiguous continental and insular aquatic systems, riverine habitats have been colonized by a large diversity of strictly freshwater organisms, through temporary connections to the continent, and freshwater organisms having a marine stage in their life cycle (Kottelat et al., 1993; De Grave et al., 2008; Keith et al., 2015). This diversity of life history traits and dispersal abilities is well exemplified by the shrimps genera Caridina (Atyidae) and Macrobrachium (Palaemonidae). South East Asia (SEA) hosts more than 50% of the diversity of Caridina and Macrobrachium, and both genera include species displaying a variety of reproductive strategies, scattered along a r/K continuum (MacArthur & Wilson, 1967; Pianka, 1970) and broadly classified into Abbreviated Larval Development (ALD) or Elongated Larval Development (ELD) strategies (Cai, 2003; De Grave et al., 2008; Wowor et al., 2009).
In the present study, we sampled Caridina and Macrobrachium species in the Islands of Java and Bali to examine diversity patterns and community assembly in a mosaic of watersheds that have been varyingly connected to the continent during glacial times and, as such, potentially hosting a variety of reproductive strategies. The community composition of Caridina and Macrobrachium assemblages was examined across 19 sites in Java and Bali through DNA barcoding (i.e., use of the mitochondrial cytochrome oxidase I gene for species identification [Hebert et al., 2003]) with the following objectives: (1) explore diversity and species co-occurrence patterns to detect potential departures from random species aggregation that may result from biotic interactions such as competitive interactions (Hutchinson, 1959; MacArthur & Levins, 1967) or life history trade-offs (MacArthur & Wilson, 1967), (2) explore phylogenetic community structure in order to detect a potential departure of the distribution of cladogenetic events from expected at random that may result from the overlap of speciation dynamics and species aggregation in ecological communities (Gillespie, 2004; Emerson & Gillespie, 2008) and (3) explore the potential consequences of reproductive strategies on the accumulation of genetic divergence among populations. Considering the lack of prior studies on evolutionary community ecology of Macrobrachium and Caridina assemblages in this area, this study, based on newly generated DNA barcodes and occurrence data, aims at identifying trends in diversity patterns to be further explored at wider spatial scale in SEA freshwater ecosystems.
Materials and methods
Sampling and collection management
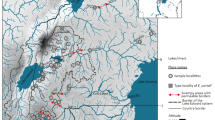
Specimens from the genera Caridina and Macrobrachium were sampled using tray net (mesh size 0.2 mm) and electroshocking at 19 sites in Java and Bali during a field expedition conducted by the authors in April, 2014 (Fig. 1A). A total of 1583 specimens were collected and preserved in a 95% ethanol solution (Table S1). Sampling information, including geographic coordinates, was recorded for each of the sites visited. Tissues were taken from abdominal muscle samples for further genetic analyses. Both tissues and voucher specimens were deposited in the Museum Zoologicum Bogoriense (MZB), Research Centre for Biology (RCB), Indonesian Institute of Sciences (LIPI). Specimens were identified to the species level according to the identification keys (Cai, 2003; Wowor, 2004), and their valid names updated according to De Grave and Fransen (De Grave & Frasen, 2011). Once species were delimitated and identified using morphological characters, a total of 285 specimens were selected for DNA barcoding. For each species, several specimens were selected for each site in order to cover the intraspecific genetic diversity at the study sites and further validate or reject the initial set of identification hypotheses based on morphological characters. The comprehensiveness of the sampling was further examined through accumulation curve analysis as implemented in the Barcode of Life Data System (Ratnasingham & Hebert, 2007).

Sampling scheme. A location of the 19 collection sites visited during the present study, B accumulation curves for species (green) and OTUs (blue)
Sequencing and international repositories
Genomic DNA was extracted using a Qiagen DNeasy 96 tissue extraction kit following the manufacturer’s specifications. A 654-bp segment from the 5’ region of the cytochrome oxidase I gene (COI) was amplified using primers cocktails C_FishF1t1/C_FishR1t1 including M13 tails (Ivanova et al., 2007). PCR amplifications were done on a Veriti 96-well Fast (ABI-Applied Biosystems) thermocycler with a final volume of 10.0 μl containing 5.0 μl buffer 2X, 3.3 μl ultrapure water, 1.0 μl each primer (10 μM), 0.2 μl enzyme Phire® Hot Start II DNA polymerase (5U) and 0.5 μl of DNA template (~ 50 ng). Amplifications were conducted as follows: initial denaturation at 98°C for 5 min followed by 30 cycles denaturation at 98°C for 5 s, annealing at 56°C for 20 s and extension at 72°C for 30 s, followed by a final extension step at 72°C for 5 min. The PCR products were purified with ExoSap-IT® (USB Corporation, Cleveland, OH, USA) and sequenced in both directions. Sequencing reactions were performed using the “BigDye® Terminator v3.1 Cycle Sequencing Ready Reaction,” and sequencing was performed on an ABI 3130 DNA Analyzer (Applied Biosystems). The sequences and collateral information have been deposited in BOLD (Ratnasingham & Hebert, 2007) in the project BICA “Barcoding Indonesian Crustaceans” (Table S2) and DNA sequences were submitted to GenBank (Accession numbers MN526034 to MN526237).
DNA-based species delimitation and genetic diversity
The DNA barcode reference library was first analyzed to check for a potential mismatch between species delimitation hypotheses based on morphological characters and DNA barcodes. DNA sequences divergence was calculated using the Kimura 2-parameter (K2P) model (Kimura, 1980) and further used to construct a midpoint-rooted neighbor-joining (NJ) tree to provide a graphic representation of the species divergence as implemented in the Sequence Analysis module of BOLD (Ratnasingham & Hebert, 2007). Sequence divergence was considered below and above species boundaries by calculating the maximum intraspecific distance and the minimum interspecific distance. The distribution of both distances was examined through bins of 1% to visualize a potential overlap between intraspecific and interspecific genetic distances. The presence of a barcoding gap that is the absence of overlap between the distributions of the maximum intraspecific and the minimum interspecific distance (Meyer & Paulay, 2005) was further checked by plotting both distances for each species. For each species, haplotype diversity (h) and nucleotide diversity (π) were calculated using the R package pegas 0.1 (Paradis, 2010).
Several alternate methods have been proposed for delimitating molecular lineages that all have in common to detect departure from mutation–drift equilibrium in branching events (Pons et al., 2006; Puillandre et al., 2012; Ratnasingham & Hebert, 2013; Kekkonen & Hebert, 2014). Each of these methods is prone to pitfalls, and combining different approaches is increasingly used to circumvent potential pitfalls arising from, for instance, uneven sampling among species (Kekkonen & Hebert, 2014; Kekkonen et al., 2015; Blair & Bryson, 2017). We used four sequence-based methods of species delimitation to establish a final delimitation scheme based on a 50% consensus among methods. For the sake of clarity, species identified based on morphological characters are referred to as morphological species, while species delimitated by DNA sequences are referred to as operational taxonomic units (OTU), defined as diagnosable molecular lineages (Avise, 1989; Moritz, 1994; Vogler & DeSalle, 1994). OTUs were delimitated using the following algorithms: (1) Refined Single Linkage (RESL) as implemented in BOLD and used to produce Barcode Index Numbers (BIN) (Ratnasingham & Hebert, 2013), (2) Automatic Barcode Gap Discovery (ABGD) (Puillandre et al., 2012) using the initial DNA sequence alignment, (3) Poisson Tree Process (PTP) in its multiple rates version (mPTP) as implemented in the stand-alone software mptp_0.2.3 (Zhang et al., 2013; Kapli et al., 2017) and (4) General Mixed Yule-Coalescent (GMYC) in its multiple rate version (mGMYC) as implemented in the R package Splits 1.0-19 (Fujisawa & Barraclough, 2013).
The mPTP algorithm uses a phylogenetic tree as an input file; thus, a maximum likelihood (ML) tree was first reconstructed using RAxML (Stamatakis, 2014) based on a GTR + I substitution model. An ultrametric and fully resolved tree was also reconstructed using the Bayesian approach implemented in BEAST 2.4.8 (Bouckaert et al., 2014) to be further used for OTUs delineation using the mGMYC algorithm. The best-fit substitution model for the Bayesian analysis was selected using JMODELTEST 2.1.7 (Darriba et al., 2012). Two Markov chains of 10 million each were run independently using a Yule pure birth model tree prior with the monophyly of each genus enforced. Previous substitution rate estimates for shrimps mitochondrial genes ranged from 1.1 to 1.4%/Myr (Knowlton et al., 1992; Knowlton & Weigt, 1998). These estimates are close to the 1.2%/Myr substitution rate that has been previously observed at COI for vertebrates (Bermingham et al., 1997). BEAST reconstructions were done using a relaxed clock exponential model with a canonical 1.2%/Myr substitution rate. Trees were sampled every 5000 states after an initial pre-burning period of 1 million, and both runs were combined using LogCombiner 2.4.8 (Bouckaert et al., 2014) after a burn in period of 10% of the sampled trees. The maximum credibility tree was constructed using TreeAnnotator 2.4.7 (Bouckaert et al., 2014). Duplicated sequences were removed prior to the Bayesian analysis using RAxML.
Once OTUs were delineated, the potential impact of cryptic diversity, introgressive hybridization or overlapping morphological characters was estimated by examining the distribution of genetic distances for the morphology-based and DNA-based delimitation schemes. We further quantified the match among methods and their relative power using the match ratio and the Relative Taxonomic Index of Congruence index (Rtax) following Blair and Bryson (Blair and Bryson, 2017). The match ratio is a measure of concordance among methods and is defined as twice the number of matches divided by the sum of the number of delimitated OTUs and the number of morphological species (Arhens et al., 2016). The Rtax index quantifies the relative power of a method to infer all estimated speciation events and is defined as the number of speciation events identified by a method divided by the total number of speciation events identified by the different methods (Miralles & Vences, 2013).
Species co-occurrence and phylogenetic relatedness within communities
Morphological species and OTU occurrences were recorded across the 19 sampling sites (Fig. 1A; Tables S1 & S2). A hierarchical cluster of sites was established based on dissimilarities in species occurrence using the R package Vegan (Oksanen et al., 2007). The significance of the correlation between dissimilarity and geographic distance among sites was tested with a Mantel test (Mantel, 1967). The matrix of geographic distance was derived from the geographic coordinates using the R package Geosphere (Hijmans et al., 2016). Patterns of species aggregation were further explored using the probabilistic model of (Veech, 2013) as implemented in the R package Cooccur (Griffith et al., 2016). This approach provides the expected probabilities of species co-occurrence under a null model of random association that can be used to identify departures related to repulsion or association dynamics among species.
The signature of non-random dynamics of species co-occurrence on species phylogenetic relatedness within communities was examined by calculating the Mean Phylogenetic Distance (i.e., MPD, the average phylogenetic distance among species within sites) and the Mean Nearest Taxon Distance (i.e., MNTD, the average phylogenetic distance among the most closely related species within sites) as implemented in the R package Picante (Kembel et al., 2010). The departure of phylogenetic distances from those expected under random patterns of species co-occurrence was calculated based on 1000 random samplings of species from the regional pool. A hierarchical cluster based on the matrix of average phylogenetic distance among sites was further established using the R Stats ver. 3.1.2 package (R_Core_Team, 2014). The correlation between average phylogenetic distance and geographic distance among sites was tested through a Mantel test (Mantel, 1967).
Results
DNA barcoding, genetic divergence and species delimitation
A total of 1583 specimens were collected across the 19 sampling sites visited. Identifications based on the taxonomic keys established by Cai (2003) and Wowor (2004) resulted in the delimitation of 23 morphological species out of which a single ALD species was observed, Macrobrachium pilimanus (De Man, 1879) in Java (Fig. 1A, sites 1, 2, 3 and 5). The species accumulation curve indicates that a ceiling has been reached for morphological species, hence suggesting that a comprehensive sample of the species diversity has been obtained (Fig. 1B). A set of 204 sequences was successfully obtained. All the sequences were above 500 bp of length and no codon stops were detected, suggesting that the sequences collected represent functional coding regions. Large levels of genetic divergence are observed within morphological species as the mean genetic distance peaks at 4.77% between conspecifics and reaches 27.68% when considering the maximum distance (Table 1, Fig. 2A, D). These values overlap those observed among morphological species within genera as the mean genetic distance reaches 24.8% among congeneric morphological species. The inspection of the NJ tree confirmed this general trend and indicates that the high genetic distances within morphological species are due to deep divergences in 5 taxa (Fig. S1). Haplotype sharing is also observed as 0 minimum genetic distances are detected among congeneric morphological species (Table 1). The NJ tree highlights three mismatches between individual assignments based on morphology and sequences clustering, including Caridina typus H. Milne Edwards, 1837 with an individual nested within C. brachydactyla De Man, 1908, C. appendiculata Jalihal & Shenoy, 1998 with an individual nested within C. brachydactyla and C. papuana Nobili, 1905 with an individual nested within C. serratirostris De Man, 1892 (Fig. S1).

Summary distributions of K2P genetic distances including the maximum intraspecific and minimum interspecific distances. A–C distribution of maximum intraspecific and minimum interspecific distances. D–F, plotting of maximum intraspecific and minimum interspecific. A, D based on morphological species. B, E based on operational taxonomic units (OTUs). C, F based on OTUs and excluding the three cases of conflicting assignment between morphology and DNA barcodes clustering
The number of delimitated OTUs ranged from 30 with ABGD to 33 with mGMYC (Fig. 3, Table 2), with 18 morphological species displaying fully convergent delimitation scheme across all methods, two morphological species with one method yielding conflicting results (Caridina gracilipes De Man, 1892 and C. peninsularis Kemp, 1918) and three morphological species with conflicting scheme among methods (C. gracilirostris De Man, 1892, Macrobrachium esculentum (Thallwitz, 1891) and M. pilimanus). The consensus delimitation scheme identified 30 OTUs within the 23 morphological species recognized here, with 19 morphological species displaying 1 OTU, two morphological species displaying two OTUs (C. brachydactyla, C. gracilipes), one morphological species displaying three OTUs (C. papuana) and one morphological species displaying four OTUs (M. pilimanus). The match ratio was similar among methods ranging from 0.63 for RESL to 0.679 in mGMYC with the highest value observed for the consensus delimitation scheme and the highest resolution power observed for mGMYC with a Rtax of 0.943 (Table 2). Similarly to that of species, OTUs accumulation curves indicate that a ceiling has been reached (Fig. 1B).

Bayesian chronogram constructed using 654 bp of the mitochondrial cytochrome oxidase I gene including 95% Highest Posterior Density intervals for median node ages, Barcode Index Numbers (BINs), species delimitation schemes based on RESL (BIN), ABGD, mPTP, mGMYC and the consensus scheme (Final), species and OTUs distribution across the 19 sites. Larval development categories refer to the Abbreviated Larval Development (ALD) in brown and Elongated Larval Development (ELD) in green. Elevation categories correspond to the maximum elevation observed per OTUs and separated into classes of 100 m ranging from 0 to 100 m in yellow, 201–300 m in red and above 301 m in brown. Distribution of each OTU is given across the 19 sites sampled with presence represented by black square and absence represented by white square. Sites in yellow correspond to Java Island, sites in green to Bali Island. Reconstruction based on the 140 unique sequences selected using RAxML. 1including specimen BICA146-14 of C. appendiculata and specimen BICA114-14 of C. typus. 2including specimen BICA051-14 of C. papuana
The distributions of the maximum intraspecific and minimum interspecific genetic distances confirmed the relative influence of cryptic molecular lineages and haplotype sharing in shaping genetic divergence patterns (Fig. 2). A first examination of the genetic distances based on morphological species confirms that the distributions of maximum intraspecific and minimum interspecific distances largely overlap (Fig. 2A). This trend was much less pronounced, however, if plotting both distances on an individual basis (Fig. 2D). Accounting for cryptic diversity by considering OTUs reduces the overlap between the distributions of the maximum intraspecific and minimum interspecific distances (Fig. 2B, E). Accounting for both cryptic diversity and the three cases of haplotype sharing further reduced the overlap (Fig. 2C); however, a barcoding gap is observed only if maximum intraspecific and minimum interspecific distances are plotted individually (Fig. 2F). Genetic diversity largely varies among morphological species with haplotype diversity ranging from 0 (C. brevicarpalis De Man, 1892, M. australe (Guérin & Méneville, 1838), M. bariense (De Man, 1892) and M. gracilirostre (Miers, 1875)) to 1 (C. celebensis De Man, 1892, C. gracilirostris, C. weberi De Man, 1892 and M. pilimanus) and nucleotide diversity ranging from 0 (C. brevicarpalis, M. australe (Guérin-Méneville, 1838), M. bariense and M. gracilirostre) to 0.081 (M. pilimanus) (Table 2).
Species richness and co-occurrence
The species richness varies among sites, ranging from one morphological species for 6 of the visited sites to eight morphological species for one site (Fig. 4A). Species commonness also largely varies with nine morphological species distributed in a single site and one morphological species distributed in 8 sites (Fig. 4C). The distribution of OTUs richness per sites is similar to that of morphological species (Fig. 4B); however, OTUs distribution is skewed toward rare OTUs present at a single site (Fig. 4D). The hierarchical cluster recovered using sites dissimilarity based on OTUs occurrence clearly separates sites 1, 2, 4 and 5 (Fig. 5A) that correspond to the sites hosting M. pilimanus OTUs with an ALD strategy (Fig. 3). Clustering patterns among sites hosting OTUs with ELD strategies show no clear trends; however, a significant correlation between sites dissimilarity and geographic distance was detected (Mantel test; P value = 0.003). The analysis of species co-occurrence using the probabilistic model of Veech (2013) revealed that, out of the 435 pairs of OTUs analyzed, only 6 were significantly associated (Fig. 5B). This co-occurrence analysis was further performed on the two larval development categories (ELD, ALD) and resulted in a significant departure of the observed co-occurrence from expected at random (observed probability = 0, expected probability = 0.166, P value < 0.001). The OTUs show no clear relationship with their distribution between islands and across the 19 sites (Fig. 3). Examining the distribution of species phylogenetic relatedness within sites returned nonsignificant departures from expected by chance of the Mean Phylogenetic Distance (MPD) and Mean Nearest Taxon Distance (MNTD) (Table 3). The hierarchical cluster constructed using the average phylogenetic distances among sites, and derived from the Bayesian tree, clearly separates sites 1, 2, 4 and 5 that host M. pilimanus with an ALD strategy (Fig. 5C). The correlation between the average phylogenetic and geographic distances among sites was significant (Mantel test, P value = 0.009).

Distribution of species and OTUs richness per site, A, B, respectively, and distribution of species and OTUs across the 19 sites, C, D, respectively

Analysis of beta-diversity among sites and co-occurrence patterns including the hierarchical cluster based on dissimilarity matrix among sites (A), Veech (2013) matrix of co-occurrence (B) and average phylogenetic distance among sites (C). Only OTUs involved in pairwise comparisons with co-occurrence patterns departing from expected at random at included (C)
Discussion
Species co-occurrence and phylogenetic community structure are consistent with a lottery model of recruitment and community assembly
The lack of significant departure of the species phylogenetic relatedness within communities is consistent with a lottery model of recruitment as previously described for communities where stochastic dispersal is sustained by early stages of the life cycle such as in coral reef fishes (Sale, 1977; Sale & Dybdal, 1978; Sale & Williams, 1982; Hubert et al., 2011) or tropical trees (Webb, 2000; Hubbell, 2001). This pattern was expected considering that 95% of the visited sites host communities composed exclusively of ELD species dispersing through their early life stages. In fact, ELD species invest their reproductive effort into producing many offspring at the cost of low survival rates, usually resulting in stochastic fluctuations of recruitment success and population size through time (Gotelli, 1991; Hanski, 1991). In such biological models, dispersal occurs at random and species turnover is a function of geographic distance among connected sites (Hubbell, 2001; Alonso et al., 2006; Chisholm & Lichstein, 2009). The detection of a significant correlation between sites dissimilarity or average phylogenetic distance among sites and geographic distance seems to confirm that dispersal is limited to a random diffusion into neighboring watersheds. This pattern is surprising, however, considering that most ELD species sampled here have been reported outside of the study area, expanding into the neighboring Wallacea and Sahul provinces. The detection of high levels of cryptic diversity at the scale of the present study, however, questions the taxonomic status of the ELD species with large distribution ranges as recently highlighted (de Mazancourt et al., 2019). This pattern of isolation by distance may have several origins depending on the relative contribution of historical contingency during community assembly and colonization that usually produces stable patterns through time, and the stochasticity of a lottery model eventually leading to a dynamic equilibrium between dispersal and extinction (Emerson & Gillespie, 2008; Vamosi et al., 2009; Hubert et al., 2011). Considering the species age inferred here, the persistence of small-size populations through millions of years is unlikely, particularly so in changing landscapes such as those of Java and Bali islands that have experienced major rearrangements during their history through volcanic activities and associated geological changes (Lohman et al., 2011). The present pattern seems consistent with a dynamic equilibrium between dispersal and extinction, and the isolation by distance pattern is likely to result from limited random diffusion of larvae into neighboring watersheds.
No evidence that species diversification and current coexistence are driven by the same mechanisms
The settlement of Sundaland in its modern configuration happened during the last 10 Myr and was initiated with the isolation of Borneo from the SEA continent through tectonic plate drift (Lohman et al., 2011). The final establishment of Sundaland, particularly the southern part including Java and the lesser Sunda island (e.g., Bali), is much more recent and happened during the last 5 Myr. Recent phylogeographic studies on primary freshwater organisms in South Sundaland suggest that the settlement of the populations of Java and Bali started at 3 Myr and most populations were fragmented during the last glacial cycle (Hutama et al., 2017; Hubert et al., 2019). In addition, several cases of in situ diversification were previously reported for primary freshwater fishes in Java (Hutama et al., 2017; Hubert et al., 2019). The present study shows a markedly distinct pattern that is likely to be due to the peculiar dispersal strategies of Caridina and Macrobrachium that disperse through the diffusion of their early life stages in the marine environment. In fact, the present study shows no evidence of interactions between the mechanisms of species diversification and species coexistence. The ages of most of the morphological species referred to here expand beyond the estimated age of settlement of Sundaland and Lesser Sunda Islands, which started around 10 Myr (Lohman et al., 2011). Except for the two OTUs within C. peninsularis and C. gracilipes, the species pair including M. bariense and M. esculentum and three OTUs in M. pilimanus, most species are older than 10 Myr and actually predate the settlement of Sundaland Islands. Furthermore, previous cases of in situ diversification among freshwater species described in Java are consistent with a marked influence of landscape fragmentation through the rise of volcanic arches, resulting in allopatric distribution between sister species (Hutama et al., 2017; Hubert et al., 2019). The dominance of geographic speciation through allopatric divergence was also reported for other freshwater species at wider spatial scales in Sundaland (De Bruyn et al., 2005, 2013). Such mechanisms of species proliferation might be expected to impact species co-occurrence patterns and phylogenetic community structure, resulting in phylogenetic over-dispersal (Hubert et al., 2011) and negative species associations. The present pattern of lottery assembly and species age inferred here suggests that mechanisms of species proliferation and community assembly do not overlap in space and time for the Caridina and Macrobrachium communities in Java and Bali.
Potential evolutionary consequences of life history traits on the accumulation of genetic divergence and species occurrence
The present study evidences several cases of deep genetic divergence within morphological species, a trend that has been previously described for several shrimp families including Atyidae (Baker et al., 2004; Page & Hughes, 2007; von Rintelen et al., 2007; von Rintelen et al., 2010) and Palaemonidae (De Bruyn et al., 2005; Liu et al., 2007; Wowor et al., 2009; Castelin et al., 2017). Several cases of morphologically similar, yet genetically divergent, lineages are identified, particularly in Macrobrachium pillimanus, the only species of the present study characterized by an ALD strategy. Larvae of species with ALD strategy are bottom dwellers with only a few zoeal stages that show little tolerance to salinity fluctuations and as such, have limited dispersal abilities (Wowor et al., 2009). This larval development may be expected to increase population differentiation by limiting gene flow among populations and to induce faster rates of genetic drift (Kimura, 1983; Tajima, 1983). The detection of highly divergent OTUs in M. pillimanus, all restricted to a single watershed, and the high haplotype and nucleotide diversities seem to confirm that the species is accumulating genetic diversity at a faster rate than other species with ELD strategies.
The co-occurrence and distribution patterns of M. pillimanus also depart from those observed for ELD species that are usually restricted to a single site and largely coexist with other ELD species. Macrobrachium pillimanus is one of the few species observed at multiple sites and is the only species that was not sampled together with other Caridina and Macrobrachium species. This pattern suggests that ALD and ELD strategies do not co-occur within sites despite their contiguous distributions within the same watersheds. In fact, species with ALD strategy exhibit faster growth rates during their early life stages as a consequence of both higher energetic stocks due to the large size of the eggs and less energy invested into zoeal metamorphoses (Cai, 2003; Wowor et al., 2004). From an ecological perspective, their limited dispersal abilities due to their abbreviated larval stages might be balanced by higher competitive skills, a trade-off that may translate into a competition/colonization trade-off (Calcagno et al., 2006; Logue et al., 2011).
Potential limits and perspectives
Several limitations of our study, which may have confounding effects on the interpretation of present results, should be acknowledged. Species age estimates are particularly old considering the age of the geological context of Sundaland and the globally limited range distribution observed here for both Caridina and Macrobrachium species. It is likely that our species age estimates are inflated due to the limited spatial scope of the study and the lack of actual sister species; however, this potential bias involves speciation events that happened at a wider spatial scale than examined here (de Mazancourt et al., 2019). Along the same line, deep divergence and old age of cladogenetic events have been previously reported for the families Atyidae and Palaemonidae, a trend that is consistent with the present phylogenetic inferences (Page & Hughes, 2007; von Rintelen et al., 2007; de Mazancourt et al., 2019). Expanding the spatial scope might be expected to add more closely related species and lead to more accurate species age estimates. It is unlikely, however, that the results on phylogenetic community structure will be affected at the scale under scrutiny as the metrics were used to detect potential departure from random species aggregation through randomization procedures, and our inferences were not based on the absolute values of these metrics. As such, our results are more prone to bias due to diversity gaps during sampling than to phylogenetic inferences per se. The accumulation curves for both species and OTUs show, however, that a ceiling was reached, and significant bias due to a substantial amount of unsampled species is unlikely. Along the same line, it should be acknowledged that Sundaland ecosystems have been significantly disrupted during the last decades (Schipper et al., 2008; Hoffman et al., 2010). In Java for instance, numerous dams have been constructed for the cultivation of rice in terraces. These dams are usually built for irrigation purposes and consist of structures a few meters high with a moderate slope and a constant flow of freshwater throughout the year. Many of these dams have been visited during the present survey, and ELD species have been collected upstream of many of those structures. In addition, species with ELD strategies are able to climb large waterfalls (Keith et al., 2010, 2013; de Mazancourt et al. 2017) and as such, anthropogenic perturbations per se are unlikely to account for the present patterns of species co-occurrence.
The main limit of the present study is the scarcity of ALD species and replicates of sites including them. Three of the four OTUs detected in M. pillimanus correspond to a single sequence (i.e., singleton) that exhibits high genetic divergence ranging from 1.3 to 5.5% (BOLD:ACS9506, BOLD:ACS9365, BOLD:ACS9366). Highly divergent singletons are varyingly delineated as independent lineages by the available algorithms of species delimitation (Fujisawa & Barraclough, 2013; Kekkonen & Hebert, 2014; Kekkonen et al. 2015), and the conflicting delimitation schemes recovered by the four methods confirm the difficulties in accurately delimitating OTUs represented by a singleton. As such, the validity of the OTUs consisting in singletons calls for a specific assessment based on more comprehensive sampling schemes. Along the same line, the generality of a potential competition/colonization trade-off between ALD and ELD species calls for a broader assessment at larger spatial scales with ALD species replicates.
Conclusion
The present study highlights the complexity of the interactions between spatial and temporal scales and their consequences on species co-occurrence and diversity patterns in Java and Bali communities of Macrobrachium and Caridina. The lack of ALD replicates in the present study limits the exploration of the eco-evolutionary consequences of reproductive strategies on species co-occurrence; however, the spatial segregation of M. pilimanus from other ELD species within the same watersheds suggests that the coexistence of species with varying reproductive strategies might be driven by a competition/colonization trade-off. For ELD species, however, a pattern of random co-occurrence is observed that matches a lottery model of recruitment as expected for r strategists. From a molecular evolutionary perspective, the higher genetic distances and nucleotide diversity in M. pilimanus suggest that ALD species accumulates genetic diversity at a faster rate, potentially as a consequence of their lower dispersal abilities, hence higher fragmentation of the populations, and potentially smaller population sizes. In the absence of information about local scale community ecology dynamics among freshwater shrimps of insular SEA, this study provides crucial clues about community dynamics that point to several aspects that will deserve more attention in the near future, such as the geographic scale of speciation and its potential influence on community assembly.
References
Alonso, D., R. S. Etienne & A. J. McKane, 2006. The merits of neutral theory. Trends in Ecology & Evolution 21: 451–457.
Arhens, D., T. Fujisawa, H. J. Krammer, J. Eberle, S. Fabrizi & A. P. Vogler, 2016. Rarity and incomplete sampling in DNA-based species delimitation. Systematic Biology 65: 478–494.
Avise, J. C., 1989. Molecular Markers, Natural History and Evolution. Chapman & Hall, New York.
Baker, A. M., D. A. Hurwood, M. Krogh & J. M. Hughes, 2004. Mitochondrial DNA signatures of restricted gene flow within divergent lineages of an atyid shrimp (Paratya australiensis). Heredity (Edinburgh) 93: 196–207.
Bermingham, E., S. McCafferty & A. P. Martin, 1997. Fish biogeography and molecular clocks: perspectives from the Panamanian isthmus. In Kocher, T. D. & C. A. Stepien (eds), Molecular Systematics of Fishes. CA Academic Press, San Diego: 113–128.
Blair, C. & J. R. W. Bryson, 2017. Cryptic diversity and discordance in single-locus species delimitation methods within horned lizards (Phrynosomatidae: Phrynosoma). Molecular Ecology Resources 17: 1168–1182.
Bouckaert, R. R., J. Heled, D. Kühnert, T. Vaughan, C.-H. Wu, D. Xie, M. A. Suchard, A. Rambaut & A. J. Drummond, 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLOS Computational Biology 10: e1003537.
Brown, L. E. & A. M. Milner, 2012. Rapid loss of glacial ice reveals stream community assembly processes. Global Change Biology 18: 2195–2204.
Cai, Y., 2003. Systematic of Freshwater Shrimps of the Family Atyidae De Haan, 1849 (Crustacea: Decapoda: Caridea: Palaemonidae) of East and Southeast Asia. National University of Singapore, Singapore.
Calcagno, V., N. Mouquet, P. Jarne & P. David, 2006. Coexistence in a metacommunity: the competition-colonization trade-off is not dead. Ecology Letters 9: 897–907.
Castelin, M., G. Marquet, G. Zimmerman, V. de Mazancourt & P. Keith, 2017. Genetic and morphological evidence for cryptic diversity in Macrobrachium australe and resurrection of M. ustulatus (Crustacea, Palaemonidae). European Journal of Taxonomy 289: 1–27.
Cavender-Bares, J., A. Keen & B. Miles, 2006. Phylogenetic structure of Floridian plant communities depends on taxonomic and spatial scale. Ecology 87: S109–S122.
Cavender-Bares, J., K. H. Kozak, P. V. A. Fine & S. W. Kembel, 2009. The merging of community ecology and phylogenetic biology. Ecology Letters 12: 693–715.
Chisholm, R. & J. Lichstein, 2009. Linking dispersal, immigration and scale in the neutral theory of biodiversity. Ecology Letters 12: 1385–1393.
Darriba, D., G. L. Taboada, R. Doallo & D. Posada, 2012. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods 9: 772.
De Bruyn, M., E. Nugroho, M. M. Hossain, J. B. Wilson & P. Mather, 2005. Phylogeographic evidence for the existence of an ancient biogeographic barrier: the isthmus of Kra seaway. Heredity (Edinburgh) 94: 370–378.
De Bruyn, M., L. Rüber, S. Nylinder, B. Stelbrink, N. R. Lovejoy, S. Lavoué, T. Heok Hui, E. Nugroho, D. Wowor, P. K. L. Ng, M. N. Siti Azizah, T. Von Rintelen, R. Hall & G. R. Carvalho, 2013. Paleo-drainage basin connectivity predicts evolutionary relationships across three Southeast Asian biodiversity hotspots. Systematic Biology 62: 398–410.
De Grave, S. & C. H. J. M. Frasen, 2011. Carideorum catalogus: the recent species of the Dendrobranchiate, Stenopodidean, Procarididean and Caridean shrimps (Crustacea: Decapoda). Zoologische Mededeelingen 85: 195–589.
De Grave, S., Y. Cai & A. Anker, 2008. Global diversity of shrimps (Crustacea: Decapoda: Caridea) in freshwater. Hydrobiologia 595: 287–293.
de Mazancourt, V., W. Klotz, G. Marquet, B. Mos, D. C. Rogers & P. Keith, 2019. The complex study of complexes: the first well-supported phylogeny of two species complexes within genus Caridina (Decapoda: Caridea: Atyidae) sheds light on evolution, biogeography, and habitat. Molecular Phylogenetics and Evolution 131: 164–180.
de Mazancourt, V., G. Marquet & P. Keith, 2017. The “Pinocchio shrimp effect”: first evidence of rostrum length variation in the environment in Caridina (Crustacea: Decapoda: Atyidae). Journal of Crustacean Biology 37: 243–248.
Emerson, B. C. & R. G. Gillespie, 2008. Phylogenetic analysis of community assembly and structure over space and time. Trends in Ecology & Evolution 23: 519–530.
Fujisawa, T. & T. G. Barraclough, 2013. Delimiting species using single-locus data and the generalized mixed Yule coalescent approach: a revised method and evaluation on simulated data sets. Systematic Biology 62: 707–724.
Gillespie, R., 2004. Community assembly through adaptive radiation in Hawaiian spiders. Science 303: 356–359.
Gotelli, N., 1991. Metapopulation models: the rescue effect, the propagule rain, and the core-satellite hypothesis. American Naturalist 138: 768–776.
Griffith, D. M., J. A. Veech & C. J. Marsh, 2016. Cooccur: probabilistic species co-occurrence analysis in R. Journal of Statistical Software 69: 1–7.
Hanski, I., 1991. Single-species metapopulation dynamics: concepts, models and observations. Biological Journal of the Linnean Society 42: 17–38.
Hauffe, T., C. Albrecht & T. Wilke, 2016. Assembly processes of gastropod community change with horizontal and vertical zonation in ancient Lake Ohrid: a metacommunity speciation perspective. Biogeosciences 13: 2901–2911.
Hebert, P. D. N., A. Cywinska, S. L. Ball & J. R. de Waard, 2003. Biological identifications through DNA barcodes. Proceedings of the Royal Society of London Series B 270: 313–321.
Hebert, P. D. N., E. H. Penton, J. M. Burns, D. H. Janzen & W. Hallwachs, 2004. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proceedings of the National Academy of Sciences of the United States of America 101: 14812–14817.
Hijmans, R. J., E. Williams, & C. Vennes. 2016. Package ‘geosphere’.
Hoffman, M., C. Hilton-Taylor, A. Angulo, M. Böhm, T. M. Brooks, S. H. M. Butchart, K. E. Carpenter, J. Chanson, B. Collen, N. A. Cox, et al., 2010. The impact of conservation on the status of the world’s vertebrates. Science 330: 1503–1509.
Hoverman, J. T., C. J. Davis, E. E. Werner, D. K. Skelly, R. A. Relyea & K. L. Yurewicz, 2011. Environmental gradients and the structure of freshwater snail communities. Ecography 34: 1049–1105.
Hubbell, S. P., 2001. The Unified Neutral Theory of Biodiversity and Biogeography. Princeton University Press, Princeton.
Hubert, N., V. Calcagno, R. S. Etienne & N. Mouquet, 2015a. Metacommunity speciation models and their implication for diversification theory. Ecology Letters 18: 864–881.
Hubert, N., A. Kadarusman, F. Wibowo, D. Busson, S. Caruso, N. Sulandari, L. Nafiqoh, L. Rüber, J. C. Pouyaud, F. Avarre, R. Herder, P. Keith Hanner & R. K. Hadiaty, 2015b. DNA barcoding Indonesian freshwater fishes: challenges and prospects. DNA Barcodes 3: 144–169.
Hubert, N., D. Lumbantobing, A. Sholihah, H. Dahruddin, F. Busson, S. Sauri, R. K. Hadiaty & P. Keith, 2019. Revisiting species boundaries and distribution ranges of Nemacheilus spp. (Cypriniformes: Nemacheilidae) and Rasbora spp. (Cypriniformes: Cyprinidae) in Java, Bali and Lombok through DNA barcodes: implications for conservation in a biodiversity hotspot. Conservation Genetics 20: 517–529.
Hubert, N., E. Paradis, H. Bruggemann & S. Planes, 2011. Community assembly and diversification in Indo-Pacific coral reef fishes. Ecology and Evolution 1(3): 229–277.
Hutama, A., H. Dahruddin, F. Busson, S. Sauri, P. Keith, R. K. Hadiaty, R. Hanner, B. Suryobroto & N. Hubert, 2017. Identifying spatially concordant evolutionary significant units across multiple species through DNA barcodes: Application to the conservation genetics of the freshwater fishes of Java and Bali. Global Ecology and Conservation 12: 170–187.
Hutchinson, G. E., 1959. Homage to Santa Rosalia, or why there so many kinds of animals. American Naturalist 93: 145–159.
Ivanova, N. V., T. S. Zemlak, R. H. Hanner & P. D. N. Hébert, 2007. Universal primers cocktails for fish DNA barcoding. Molecular Ecology Resources 7: 544–548.
Kapli, P., S. Lutteropp, J. Zhang, K. Kobert, P. Pavlidis, A. Stamatakis & T. Flouri, 2017. Multi-rate poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 33: 1630–1638.
Keith, P., G. Marquet, C. Lord, D. Kalfatak & E. Vigneux, 2010. Poissons et Crustacés d’eau Douce du Vanuatu. Société Française d’Ichtyologie, Paris.
Keith, P., G. Marquet, P. Gerbeaux, E. Vigneux & C. Lord, 2013. Poissons et Crustacés d’eau Douce de Polynésie. Société Française d’Ichthyologie, Paris.
Keith, P., C. Lord & K. Maeda, 2015. Indo-Pacific Sicydiinae Gobies: Biodiversity, Life Traits and Conservation. Société Française d’Ichtyologie, Paris.
Kekkonen, M. & P. D. N. Hebert, 2014. DNA barcode-based delineation of putative species: efficient start for taxonomic workflows. Molecular Ecology Resources 14: 706–715.
Kekkonen, M., M. Mutanen, L. Kaila, M. Nieminen & P. D. N. Hebert, 2015. Delineating species with DNA barcodes: a case of taxon dependent method performance in moths. PLoS ONE 10: e0122481.
Kembel, S. W., 2009. Disentangling niche and neutral influences on community assembly: assessing the performance of community phylogenetic structure tests. Ecology Letters 12: 949–960.
Kembel, S. W., P. D. Cowan, M. R. Helmus, W. K. Cornwell, H. Morlon, D. D. Ackerly, S. P. Blomberg & C. O. Webb, 2010. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26: 1463–1464.
Kimura, M., 1980. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution 15: 111–120.
Kimura, M., 1983. The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge.
Knowlton, N., L. A. Weight, L. A. Solorzano, D. K. Mills & E. Bermingham, 1992. Divergence of proteins, mitochondrial DNA and reproductive compatibility across the Isthmus of Panama. Science 260: 1629–1632.
Knowlton, N. & L. A. Weigt, 1998. New dates and new rates for divergence across the Isthmus of Panama. Proceedings of the Royal Society London B 265: 2257–2263.
Kottelat, M., A. J. Whitten, S. R. Kartikasari & S. Wirjoatmodjo, 1993. Freshwater Fishes of Western Indonesia and Sulawesi. Periplus, Singapore.
Kraft, N. J., W. K. Cornwell, C. O. Webb & D. D. Ackerly, 2007. Trait evolution, community assembly, and the phylogenetic structure of ecological communities. The American Naturalist 170: 271–283.
Kraft, N. J., R. Valencia & D. D. Ackerly, 2008. Functional traits and niche-based tree community assembly in an Amazonian forest. Science 322: 580–582.
Liu, M.-Y., Y.-X. Cai & C.-S. Tzeng, 2007. Molecular systematics of the freshwater prawn genus Macrobrachium Bate, 1868 (Crustacea: Decapoda: Palaemonidae) inferred from mtDNA sequences, with emphasis on East Asian species. Zoological Studies 46: 272.
Logue, J. B., N. Mouquet, P. Hannes, H. Hillebrand & T. metacommunity working group, 2011. Empirical approaches to metacommunities: a review and comparison with theory. Trends in Ecology & Evolution 26: 482–491.
Lohman, K., M. De Bruyn, T. Page, K. Von Rintelen, R. Hall, P. K. L. Ng, H.-T. Shih, G. R. Carvalho & T. Von Rintelen, 2011. Biogeography of the Indo-Australian archipelago. Annual Review of Ecology, Evolution, and Systematics 42: 205–226.
MacArthur, R. H. & R. Levins, 1967. The limiting similarity, convergence, and divergence of coexisting species. The American Naturalist 101: 377–387.
MacArthur, R. H. & E. O. Wilson, 1967. The Theory of Island Biogeography. Princeton University Press, Princeton, NJ.
Mantel, N., 1967. The detection of disease clustering and a generalized regression approach. Cancer Research 27: 209–220.
McPeek, M. A., 2008. The ecological dynamics of clade diversification and community assembly. The American Naturalist 172: e270–e284.
Meyer, C. & G. Paulay, 2005. DNA barcoding: error rates based on comprehensive sampling. PLoS Biology 3: 2229–2238.
Miralles, A. & M. Vences, 2013. New metrics for comparison of taxonomies eveal striking discrepancies among species delimitation methods in Madascincus lizards. PLoS ONE 8: e68242.
Moritz, C., 1994. Defining “Evolutionary Significant Units” for conservation. Trends in Ecology & Evolution 9: 373–375.
Myers, N., R. A. Mittermeier, C. G. Mittermeier, G. A. B. da Fonseca & J. Kent, 2000. Biodiversity hotspots for conservation priorities. Nature 403: 853–858.
Oksanen, J., R. Kindt, P. Legendre, B. O’Hara & M. H. H. Stevens, 2007. The vegan package. Community Ecology Package 10: 631–637.
Page, T. J. & J. M. Hughes, 2007. Radically different scales of phylogeographic structuring within cryptic species of freshwater shrimp (Atyidae: Caridina). Limnology and Oceanography 52: 1055–1066.
Paradis, E., 2010. Pegas: an R package for population genetics with an integrated modular approach. Bioinformatics 26: 419–420.
Pianka, E. R., 1970. On r and K selection. The American Naturalist 104: 592–597.
Pons, J., T. G. Barraclough, J. Gomez-Zurita, A. Cardoso, D. P. Duran, S. Hazell, S. Kamoun, W. D. Sumlin & A. P. Vogler, 2006. Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Systematic Biology 55: 595–606.
Puillandre, N., A. Lambert, S. Brouillet & G. Achaz, 2012. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Molecular Ecology 21: 1864–1877.
R_Core_Team, 2014. R: A Language and Environment for Statistical Computing. R Core Team, Vienna.
Ratnasingham, S. & P. D. N. Hebert, 2007. BOLD: the barcode of life data system (www.barcodinglife.org). Molecular Ecology Resources 7: 355–364.
Ratnasingham, S. & P. D. N. Hebert, 2013. A DNA-based registry for all animal species: the barcode index number (BIN) system. PLoS ONE 8: e66213.
Ricklefs, R. E., 1987. Community diversity: relative roles of local and regional processes. Science 235: 167–171.
Ricklefs, R. E. & D. Schluter, 1993. Species Diversity in Ecological Communities: Historical and Geographical Perspectives. University of Chicago Press, Chicago.
Sale, P. F., 1977. Maintenance of high diversity in coral reef fish communities. The American Naturalist 111: 337–359.
Sale, P. F. & R. Dybdal, 1978. Determinants of community structure for coral reef fishes in isolated coral heads at lagoonal and reef slope sites. Oecologia 34: 57–74.
Sale, P. F. & D. M. Williams, 1982. Community structure of coral reef fishes: are the patterns more than those expected by chance? The American Naturalist 120: 121–127.
Schipper, J., J. S. Chanson, F. Chiozza, N. A. Cox, M. Hoffmann, V. Katariya, J. Lamoreux, A. S. L. Rodrigues, S. N. Stuart & H. J. Temple, 2008. The status of the world’s land and marine mammals: diversity, threat, and knowledge. Science 322: 225–230.
Smith, A. M., J. J. Rodriguez, J. B. Whitfield, A. R. Deans, D. H. Janzen, W. Hallwachs & P. D. N. Hebert, 2008. Extreme diversity of tropical parasitoid wasps exposed by iterative integration of natural history, DNA barcoding, morphology, and collections. Proceedings of the National Academy of Sciences of the United States of America 105: 12359–12364.
Smith, M. A., D. M. Wood, D. H. Janzen, W. Hallwachs & P. D. N. Hebert, 2007. DNA barcodes affirm that 16 species of apparently generalist tropical parasitoid flies (Diptera, Tachinidae) are not all generalists. Proceedings of the National Academy of Sciences of the United States of America 104: 4967–4972.
Stamatakis, A., 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313.
Tajima, F., 1983. Evolutionary relationships of DNA sequences in finite populations. Genetics 105: 437–460.
Vamosi, J. C. & S. M. Vamosi, 2006. Body size, rarity, and phylogenetic community structure: insights from diving beetle assemblages of Alberta. Diversity and Distributions 13: 1–10.
Vamosi, S. M., B. Heard, J. C. Vamosi & C. O. Webb, 2009. Emerging patterns in the comparative analysis of phylogenetic community structure. Molecular Ecology 18: 572–592.
Veech, J. A., 2013. A probabilistic model for analysing species co-occurrence. Glob. Ecol. Biogeogr. 22: 252–260.
Vogler, A. P. & R. DeSalle, 1994. Diagnosing units of conservation management. Conservation Biology 6: 170–178.
Von Rintelen, K., T. Von Rintelen & M. Glaubrecht, 2007. Molecular phylogeny and diversification of freshwater shrimps (Decapoda, Atyidae, Caridina) from ancient Lake Poso (Sulawesi, Indonesia): the importance of being colourful. Molecular Phylogenetics and Evolution 45: 1033–1041.
von Rintelen, K., M. Glaubrecht, C. D. Schubart, A. Wessel & T. von Rintelen, 2010. Adaptive radiation and ecological diversification of Sulawesi’S ancient lake shrimps. Evolution 64: 3287–3299.
Voris, H. K., 2000. Maps of Pleistocene sea levels in Southeast Asia: shorelines, river systems and time durations. Journal of Biogeography 27: 1153–1167.
Webb, C. O., 2000. Exploring the phylogenetic structure of ecological communities: an example for rain forest trees. The American Naturalist 156: 146–155.
Webb, C. O., G. S. Gilbert & M. J. Donoghue, 2006. Phylodiversity-dependent seedling mortality, size structure, and disease in a Bornean rain forest. Ecology 87: S123–S131.
Woodruff, D. S., 2010. Biogeography and conservation in Southeast Asia: how 2.7 million years of repeated environmental fluctuations affect today’s patterns and the future of the remaining refugium-phase biodiversity. Biodiversity and Conservation 19: 919–941.
Wowor, D., 2004. A Systematic Revision of the Freshwater Prawns of the Genus Macrobrachium (Crustacea: Decapoda: Caridea: Palaemonidae) of Sundaland. National University of Singapore, Singapore.
Wowor, D., Y. Cai & P. K. L. Ng, 2004. Crustacea: Decapoda, Caridea. In Yule, C. M. & Y. H. Sen (eds), Freshwater Invertebrates of the Malaysian region. Academy of Sciences Malaysia, Kuala Lumpur.
Wowor, D., V. Muthu, R. Meier, M. Balke, Y. Cai & P. K. L. Ng, 2009. Evolution of life history traits in Asian freshwater prawns of the genus Macrobrachium (Crustacea: Decapoda: Palaemonidae) based on multilocus molecular phylogenetic analysis. Molecular Phylogenetics and Evolution 52: 340–350.
Zhang, J., P. Kapli, P. Pavlidis & A. Stamatakis, 2013. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29: 2869–2876.
Acknowledgements
The authors wish to thank Dr. Siti Nuramaliati Prijono, Dr. Bambang Sunarko, Dr. Witjaksono, Mohammad Irham Msc., Dr. Marlina Adriyani, Ruliyana Susanti, Dr. Rosichon Ubaidillah, Dr. Hari Sutrisno and Muhamad Syamsul Arifin Zein Msc, at Research Centre for Biology (RCB-LIPI); Jean-Paul Toutain, Robert Arfi, Valérie Verdier and Jean-François Agnèse from the “Institut de Recherche pour le Développement”; Joel Le Bail and Nicolas Gascoin at the French embassy in Jakarta for their continuous support. We are thankful to late Renny Kurnia Hadiaty and Sopian Sauri at RCB-LIPI, Sumanta at IRD Jakarta for their help during the field sampling in East Java and Bali. Part of the present study was funded by the Institut de Recherche pour le Développement (UMR 226 ISEM and IRD through incentive funds), the MNHN (UMR BOREA), the RCB-LIPI, the French Ichthyological Society (SFI), the Foundation de France and the French embassy in Jakarta. The Indonesian Ministry of Research and Technology approved this study, and field sampling was conducted according to the research permits 097/SIP/FRP/SM/IV/2014 for Philippe Keith, 60/EXT/SIP/FRP/SM/XI/2014 for Frédéric Busson and 41/EXT/SIP/FRP/SM/VIII/2014 for Nicolas Hubert. Sequence analysis was aided by funding from the government of Canada through Genome Canada and the Ontario Genomics Institute in support of the International Barcode of Life project. We thank Paul Hébert and Evgeny Zakharov as well as BOLD and CCDB staffs at the University of Guelph for their valuable support. We are also very grateful to our editor, Diego Fontaneto, as well as the two anonymous reviewers for their insightful feedbacks on the earlier version of the manuscript. This publication has ISEM Number 2019-189 SUD.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Diego Fontaneto
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10750_2019_4127_MOESM2_ESM.xlsx
Occurrence data for the 23 species included in the present study and based on 1583 observations across 19 sites. Supplementary material 2 (XLSX 11 kb)
10750_2019_4127_MOESM5_ESM.xlsx
Occurrence data for the 30 OTUs detected in the present study across the 19 sites. Supplementary material 5 (XLSX 12 kb)
Rights and permissions
About this article

Cite this article
Hernawati, R., Nurhaman, U., Busson, F. et al. Exploring community assembly among Javanese and Balinese freshwater shrimps (Atyidae, Palaemonidae) through DNA barcodes. Hydrobiologia 847, 647–663 (2020). https://doi.org/10.1007/s10750-019-04127-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-019-04127-7