
Abstract
Spartina alterniflora Losiel. is a highly invasive species found on the Chinese coast. To characterize the evolutionary mechanisms underlying the success of S. alterniflora in China, we examined the distribution and structure of genetic variation among three native populations at their source locations and five non-native populations in China, at both the chloroplast and nuclear microsatellite loci. Both cpDNA and microsatellite data revealed comparable genetic diversity and population differentiation between the native and introduced regions, which is consistent with the deliberate historical planting of heterogeneous founding groups in China. Bayesian clustering analysis showed that the best two clusters in the introduced region correspond to populations from the Atlantic coast and Gulf coast, respectively, instead of the three ecotypes proposed by a previous study. Investigating genetic composition of individuals suggested that most of the non-native plants might be of Atlantic origin, while some individuals in the Hong Kong population could be genetic admixtures of Atlantic and Gulf origin. This study indicated varied genetic components among populations in China, which imply different sources for the present Chinese populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The rapid spread of exotic weeds has received considerable attention within the international community, and has mobilized substantial ecological and genetic research (Lee, 2002). Identification of the source of invasive populations and reconstruction of invasion histories can facilitate the search for appropriate biological control agents (Roderick & Navajas, 2003) and identify post-introduction evolutionary changes (Amsellem et al., 2000; McCauley et al., 2003). Historical and observational data on the spread of invasive populations are often sparse, but even when there is good documentary evidence, molecular genetic data can offer unique insights into the sources, routes, and mechanisms of spread (Hoos et al., 2010; Lombaert et al., 2010). Knowledge of the relative levels of genetic diversity and structure in native and exotic ranges of invasive plants can further our understanding of the processes underlying early demographic establishment and evolutionary adaptation following invasion (Verhoeven et al., 2010). Theory predicts that introduced species will show lower levels of intrapopulation diversity and higher levels of population differentiation than their native counterparts (Brown and Marshall, 1981). However, on a case by case basis, empirical data show that the magnitude of these changes varies greatly (Amsellem et al., 2000; McCauley et al., 2003; Provan et al., 2005). A review of invasive population genetic studies found that exotic populations with multiple introduction sources are more common than those with a single origin (Bossdorf et al., 2005). The comparison of populations from both the introduced and native ranges will be of great value when an invasion involves hybridization, increased genetic diversity, or admixture (intraspecific hybridization) following secondary contact (Blum et al., 2007; Dlugosch et al., 2008).
Spartina alterniflora Lois. (Poaceae, Chloridoideae) is a smooth cordgrass native to the Atlantic and Gulf coasts of North America, and has been used in coastal restoration programs in many countries (Daehler & Strong, 1996). For such a purpose, hundreds of individuals and 0.5 kg of seeds of S. alterniflora were collected from Florida (FL), Georgia (GA) and North Carolina (NC) in the United States, and introduced into China in late 1979 (Xu & Zhuo, 1985). Because of its strong reproductive capacity, dispersal range and competitive ability (Xu & Zhuo, 1985; Deng et al., 2006; An et al., 2007), S. alterniflora expanded rapidly in China and has caused native species loss, habitat destruction, and millions of dollars of economic loss each year. Thus, the species has been listed in the top nine most notorious invasive plants in China (Zhi et al., 2007). Although local government and scientists have made intensive efforts to control and/or eradicate this species using physical, chemical, biological, and integrated methods, the population is still rapidly expanding (Li et al., 2006; Deng et al., 2007). Three ecotypes, namely FL, GA, and NC ecotypes (abbreviated as one letter descriptor: F, G, and N in Xu & Zhuo, 1985), were identified in Chinese S. alterniflora based on their source populations, supported by physiological (Qin et al., 1985; Chen & Chung, 1990) and morphological characteristics (Xu & Zhuo, 1985). However, it was reported that the three forms of S. alerniflora do not differ genetically, and thus are not ecotypes, but simply reflect differences in the environment to which they are exposed and are thus ecophenes (Jiang et al., 1985).
The genetic characteristics of populations can profoundly impact their range expansion capacity (Lee, 2002). Several attempts have been made to characterize genetic variation within and among S. alterniflora populations in native and introduced regions (Stiller & Denton, 1995; Ayres et al., 1999; O’Brien & Freshwater, 1999; Anttila et al., 2000; Travis et al., 2002, 2004; Travis & Hester, 2005; Deng et al., 2007). However, there are still only a few detailed studies comparing the genetic diversity between native and non-native populations of S. alterniflora, especially in Chinese populations. This knowledge gap largely reflects the logistical difficulties of collecting material over large spatial scales, which such work usually entails. Nevertheless, Blum et al. (2007) demonstrated that such studies are necessary if we are to gain an increased understanding of the population dynamics of invasive S. alterniflora. In Blum’s study, genetic comparisons between native (Atlantic and Gulf coast) and non-native (Pacific coast) populations of S. alterniflora showed reciprocal interspecific hybridization with a native congener in San Francisco Bay (California, USA), and admixture following secondary contact among previously allopatric native populations in Willapa Bay (Washington, USA). Considering that the initial population (c. 1,000 m2) of the Chinese smooth cordgrass was a mixture of three source populations originally introduced from the United States, An et al. (2007) assumed that hybrids with high environmental tolerance and high growth rate were produced from these allopatric native populations and that they have spread widely in Chinese coastal areas. However, a comprehensive analysis of putative source populations from the United States and non-native populations in China has yet to be conducted.
In this study, we sampled S. alterniflora from three native populations, located at or close to the three well-documented source locations, and five populations along the coast of China. We examined the genetic diversity and population structure of plants from native and introduced regions using both chloroplast and nuclear molecular markers. Further, we investigated the genetic relatedness between the samples. The major objectives were to: (1) estimate the amount, distribution, and structure of genetic variation in native and non-native populations; (2) investigate underlying genetic differentiation between three ecotypes proposed by a previous study in China, which correspond to the three source populations in the United States; (3) test if intraspecific admixture among allopatric native populations has occurred, based on molecular evidence.
Materials and methods
Sample collection and DNA extraction
Plant material was collected from five non-native populations in five provinces of China: Jiangsu (JS), Shanghai (SH), Fujian (FJ), Hong Kong (HK), and Taiwan (TW) and three native populations in three states of United States: NC, GA, and FL (Table 1; Figs. 1, 2). The sampling sites in the United States were located at or close to the well-documented source locations for S. alterniflora in China, e.g., Morehead City (NC), Altamaha Estuary (GA), and Tampa Bay (FL) (Xu & Zhuo, 1985). At each site, individuals were sampled close and perpendicular to shorelines from areas accessible on foot and separated by a minimum of 10 m apart to minimize the possibility of resampling large clones. For each individual, a leaf sample 10 cm in length was collected with a scissor and stored in plastic bags with silica gels for DNA extraction (Doyle & Doyle, 1987). A piece of leaf tissue weighing approximately 0.3 g was used to extract genomic DNA using the CTAB method of Doyle & Doyle (1987). The quantity and quality of genomic DNA were estimated spectrophotometrically using NanoDrop ND-100 (NanoDrop Technologies Inc., Rockland, DE, USA), as well as visually by ethidium bromide staining on agarose gels.
Chloroplast DNA sequencing and analysis
The trnT-trnF chloroplast intergenic spacer region (partial) was amplified and sequenced in this study considering this spacer was also used in the study of Blum et al. (2007) which provided a detailed cpDNA haplotypes (chlorotypes) information across 30 populations in the United States. We randomly selected 8–14 individuals from each population to conduct PCR amplification and sequencing using the universal primers listed in Taberlet et al. (1991). Amplification reactions contained 6 μl 10× PCR buffer, 6 μl of 2 mM each dNTPs, 3.6 μl 25 mM MgCl2, 3 μl 10 pM forward primer, 3 μl 10 pM reverse primer, 9 U Taq DNA polymerase, and 3 μl (about 30 ng) of genomic DNA. Amplification conditions were as follows: 1 cycle of 94°C for 4 min; 29 cycles of 94°C for 45 s, 55°C for 60 s, 72°C for 1 min, followed by 1 cycle of 72°C for 8 min. PCR products were purified using agarose gel purification kit (QIAGEN, Hilden, Germany) following the protocol provided by the manufacturers. The purified PCR products were subjected to direct sequencing. Sequencing was conducted with amplification primers in an ABI 3730 DNA automated sequencer with the BigDye chemistry (Applied Biosystems, Foster City, CA). Sequences from 71 individuals with good quality were finally retained for further analysis.
The sequences were edited and compared in SeqManTM (DNASTAR). Chlorotypes were differentiated by sequence polymorphism and indel size variation across the trnT-trnF region following the previous study conducted by Blum et al. (2007). Indel size variation was scored as an increase or decrease in mononucleotide repeat number at two separate locations in the trnT-trnL region, and it was assumed that insertion/deletion events at the two locations were independent (Blum et al., 2007). Sequences representing all distinct chlorotypes were deposited in GenBank with accession numbers KJ499446–KJ499453.
Chlorotype diversity (h) in each population was calculated by Arlequin v. 3.5 (Excoffier et al., 2005), controlling for uneven sample sizes. Exact tests of population differentiation and pairwise F ST values of genetic distance based on chlorotype frequencies were also calculated. The analysis of molecular variance (AMOVA) was performed using Arlequin to assess the hierarchical distribution of molecular variation.
The chlorotypes detected in this study were classified according to Blum et al. (2007), or, alternatively, identified as new chlorotypes as might be the case if these chlorotypes were not reported in the previous study (Blum et al., 2007). Relationships between chlorotypes were examined via a chlorotype network constructed using the computer program network (Weir, 1996). A minimum spanning network was constructed to demonstrate relatedness between those newly identified chlorotypes and chlorotypes reported by Blum et al. (2007).
Microsatellite genotyping and analysis
For comparison with the chloroplast sequence data, a total of 92 samples from all eight populations were used for microsatellite genotyping. Recently, we have developed primers for 15 polymorphic SSR loci of S. alterniflora using the next-generation sequencing platform Illumina Genome Analyzer II (Guo et al., 2013). Seven disomic loci (referring to those that exhibit no more than two alleles per individual) among 15 polymorphic loci were scanned and analyzed in this study considering S. alterniflora is a polyploid. PCR reactions were conducted in 20 μl volume containing 2 μl 10× PCR buffer, 2 μl of 2 mM each dNTPs, 1.2 μl 25 mM MgCl2, 1 μl 10 pM forward primer, 1 μl 10 pM reverse primer, 3 U Taq DNA polymerase, and 1 μl (about 10 ng) of genomic DNA. PCR conditions were as follows: initial denaturation at 94°C for 5 min, followed by 34 cycles of 30 s at 94°C, 30 s at 60°C, and 45 s at 72°C, with a final extension cycle at 72°C for 10 min. Amplification products were electrophoresed through 8% polyacrylamide denaturing gels and visualized by silver staining. The band size was evaluated by comparison with a 20-bp DNA ladder.
The test for replicate genotypes was conducted before downstream analyses of genetic diversity and differentiation, in order to minimize repeated sampling of the same clone. We first identified multilocus genotypes (MLGs) using GenClone 2.0 (Arnaud-Haond & Belkhir, 2007), in which the genotypic resolution associated with each possible combination of analyzed loci was computed as the resulting number of distinct MLGs (Arnaud-Haond et al., 2005). Given individuals with the same MLG may also arise by chance from independent sexual events besides clonal reproduction, we employed GenClone to calculate the probability that observed multiple copies of the same MLG arose by independent sexual events (P sex), which is estimated from the probability of each genotype occurring in the pool of all individuals [P gen(F IS); Arnaud-Haond et al., 2007] taking into account deviations from the Hardy–Weinberg equilibrium (HWE). A P sex value lower than a chosen threshold (i.e., 0.01; Arnaud-Haond et al., 2007; Janko et al., 2012; Nibouche et al., 2014) supported the hypothesis that these identical individuals belong to the same clone. Those replicate genotypes, if identified, were screened out before the downstream analysis. While some MLG may represent distinct clones, others may differ by scoring errors and/or somatic mutations and form the so called ‘multilocus lineage’ (MLL). We identified MLLs using GenClone and following the standardized method proposed by Arnaud-Haond et al. (2007). P sex on the set of identical loci was used to estimate the likelihood that those slightly distinct MLGs would actually be derived from distinct reproductive events. To ascertain the uniqueness of MLGs with missing data (i.e., unamplified loci), case-by-case examination were performed on them after removing the missing loci from the entire dataset. These MLGs were either designated as being unique or were pooled with another MLG into a MLL according to the recalculated P sex estimates.
For each sample population, the average effective number of alleles (N e) (Kimura & Crow, 1964), observed (H O) and expected (H E) (Nei, 1973) heterozygosities and Shannon’s information index (I) (Lewontin, 1972) were calculated using POPGENE v. 1.32 (Yeh & Boyle, 1997). The coefficient of genetic differentiation (G ST) (Nei, 1973) was calculated by GENODIVE (Meirmans & Tienderen, 2004). Considering that S. alterniflora was introduced to China recently, we tested the presence of a bottleneck for each invasive population using BOTTLENECK (Piry et al., 1999). We used the heterozygosity excess method to test for the decrease in number of alleles relative to heterozygosity. During a bottleneck, the loss of number of alleles occurs more rapidly than the associated decrease in expected heterozygosity, so the observed gene diversity is higher than the expected equilibrium gene diversity (Chen et al., 2006). We adopted a Wilcoxon’s signed-rank test in BOTTLENECK, which is suitable for testing a small number of individuals per population with lower number polymorphic loci (Piry et al., 1999). This test was implemented under all three SSR (microsatellite) mutational models available—the infinite allele mutation model (IAM), the stepwise mutation model (SMM), and two-phased model of mutation (TPM) with 70% single step mutation and 30% multi-step mutation. Finally, we determined if there was a mode-shift, which is another sign of a bottlenecked population shown by a shift in allele frequency distribution, in each tested population. The distribution of allele frequency distribution in a population under mutation-drift equilibrium is expected to be approximately L-shaped, that is, a distribution with more alleles in the low frequency class (<0.1) than in intermediate frequency class (Nei et al., 1976; Luikart et al., 1998). A recent bottleneck often provokes a shift in allele frequency distribution toward more alleles being present at an intermediate frequency than a low frequency (Luikart et al., 1998).
Analysis of Molecular Variance (AMOVA) was conducted by Arlequin v. 3.5 to examine the hierarchical distribution of microsatellite genetic variation. The first AMOVA was arranged to determine if native and non-native populations can be differentiated. A second AMOVA was arranged to calculate the genetic variation distribution among populations within a region (native or non-native), and within populations. Nei’s unbiased measures of genetic identity and genetic distance (Nei, 1978) were calculated using POPGENE software. The significance of isolation by distance between populations in a region was tested by examining correlations between Nei’s unbiased genetic distance and geographical distances (in kilometers) over all pairs of populations. For this, Mantel tests (Mantel, 1967) employing 1000 permutations were performed on all introduced populations using the software Tools for Population Genetic Analysis (TFPGA) (Miller, 1997).
The genetic structure of the populations was further examined with two Bayesian clustering methods: InStruct (Gao et al., 2007) and STRUCTURE v.2.3.3 (Pritchard et al., 2000) based on microsatellite allelic variation, for further comparison with patterns of cpDNA chlorotype variation. We firstly used InStruct to assess genetic similarities among samples taken from China and native marshes. The program InStruct is an extension of STRUCTURE by eliminating the assumption of HWE within clusters and, instead, calculating expected genotype frequencies on the basis of inbreeding or selfing rates. The number of genetic clusters (K) was estimated and individuals sampled in the United States and China were fractionally assigned to the inferred groups. Initially, we modeled cluster assignments for K = 1–8 clusters. The distribution of posterior probabilities, lnP(K|X), and their variance were examined for 8 runs for each value of K. In all simulations, we found that a burn-in of 100,000 iterations and 100,000 Markov chain Monte Carlo iterations yielded consistent assignments of individuals to a given cluster across runs. For comparison and validation, we also ran STRUCTURE on the data for 8 independent chains for each K value (K = 1–8). Each chain was iterated 100,000 times after burn-in with 100,000 iterations. The ΔK statistic, based on the rate of change of log likelihood of data between successive K values, was used to select the optimal K following Evanno et al. (2005). We implemented the independent and correlated allele frequency models. Simulations were run using the admixture model without prior population information (Pritchard et al., 2000). Outputs of InStruct and STRUCTURE analyses were visualized using the software CLUMPP (Jakobsson & Rosenberg, 2007) and DISTRUCT (Rosenberg, 2004).
Individual assignment tests were performed using the program GeneClass v. 2 (Piry et al., 2004). This Bayesian procedure computes the likelihood of a genotype in a given population assuming an equal prior probability density to the allelic frequencies of each locus in each population. This method shows better assignment performance than frequentist methods or distance-based methods both in simulated or real populations (Cornuet et al., 1999; Arranz et al., 2001). In this study, we used this software to generate scores for the assignment of individuals from non-native populations to all sampled populations using Bayesian methods (Rannala & Mountain, 1997) and the resampling algorithm of Cornuet et al. (1999) set at 100,000 individuals. An individual was excluded from a given candidate population if its probability of belonging to a particular population was lower than 5%.
Results
cpDNA chlorotype diversity and distributions
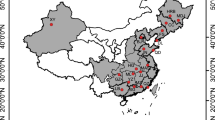
The partial trnT-trnF intergenic spacer region was sequenced from 71 individuals of S. alterniflora. The length of the total region was 1378–1382 bp in S. alterniflora. Base-pair polymorphism and indel variation across trnT-trnF regions provided a total of eight informative sites, two of which were novel discoveries. Correspondingly, eight distinct chlorotypes were identified, based on 42 previously reported chlorotypes (Blum et al., 2007), three of which were firstly discovered (Table 1). New informative sites and new chlorotypes were only found in native populations. The geographic distribution of these chlorotypes is shown in Fig. 1 and Table 1. A comparison of native and non-native populations revealed fewer chlorotypes in non-native populations (n = 3) compared with native populations (n = 7). More chlorotypes and polymorphic sites were detected in native populations, even though fewer individuals were examined compared with the introduced range.
To determine the location of those new chlorotypes, a network was constructed, including all 45 chlorotypes reported. We found that the chlorotypes detected in Chinese populations were all located in chlorotype group C (Blum et al., 2007), the most common group found across almost the entire range of sample locations in the United States. New chlorotypes 1 (NEW1) detected in NC exhibited a difference of one base-pair indel with chlorotype C6 and two base-pair indels with chlorotype C5. New chlorotypes 2 (NEW2) was found only in population GA, which differed from chlorotype C1 by only one nucleotide substitution (site 150), a new informative site first reported in S. alterniflora. New chlorotypes 3 (NEW3) was found only in population FL, which differed from chlorotype C4 by only one nucleotide substitution. For clarity, a subset of the chlorotype network, depicting the relationship between 14 relevant chlorotypes from this study is presented in Fig. 1c.

The distribution and frequency of chlorotypes among native (a) and non-native (b) S. alterniflora in eight sample locations. c Minimum spanning network of 14 group C trnT-trnF cpDNA chlorotypes exhibited by S. alterniflora genets sampled from native and non-native populations. White chlorotypes previously reported by Blum et al. (2007) but not found in this study. Chlorotypes presented in different hues are differentiated by ≥1 base-pair substitutions or indel size variation. The size of the circles representing different chlorotypes does not correspond to the relative abundance of each chlorotype
Chlorotype diversity had a range of 0.54–0.75 in native populations, which was higher than in non-native populations (0.50–0.68), except for the population TW in which chlorotype C4 was fixed. The differences between chlorotype diversities in native and non-native populations were not significant (Mann–Whitney U test: Z = −1.342, P = 0.180). However, we found that chlorotypes were unequally distributed across the native and introduced coasts (Table 1). Among all three chlorotypes (C1, C3, and C4) detected in Chinese populations, chlorotypes C3 and C4 were also found in two of the native populations (GA and NC). The most common chlorotype, C4, was found in 60% of the non-native individuals and only 21% of individuals sampled in the native range. However, chlorotype C1, which was detected in three of the non-native populations (FJ, JS and HK), was not found in any native populations in this study. In contrast, no chlorotype was shared between the FL population and any other populations.
Differences in the geographic distribution of cpDNA chlorotype groups were also reflected in estimates of population differentiation and the hierarchical distribution of genetic variation among sampled S. alterniflora. Approximately 65% of cpDNA variation captured in this study occurred among individuals within populations. Nearly 31% was partitioned among sample locations, and the remaining 4% occurred among geographic regions corresponding to native and introduced regions (Table 2). Most of the pairwise values of F ST based on chlorotype frequency differences were not significant (range, −0.0955 to 0.6250), particularly among comparisons of sites located in introduced regions (Bonferroni-corrected P < 0.05). Exact tests of population differentiation showed that nearly all sites were significantly different from FL, except FJ and SH. In contrast, the lack of significant differentiation was demonstrated between GA and all of the sample sites in the introduced regions.
Microsatellite genetic diversity
Genetic diversity of native and non-native S. alterniflora
Twenty-three alleles were produced from seven disomic microsatellite primer pairs. Between 2 and 5 alleles were detected per locus, with an average of 3.29 alleles per locus. None of the detected alleles were present in all individuals, showing 100% polymorphism of these loci in S. alterniflora. The test result for replicate genotypes showed that the probabilities that a shared/slightly differed genotype originated from a distinct seed P sex(F IS) were higher than 0.01, implying all individuals sampled in our study belong to different genets. Based on the seven disomic loci, average Nei’s expected heterozygosity was 0.42 within native and 0.38 within introduced areas; the observed heterozygosity range was 0.46–0.63 (average 0.53) and 0.21–0.51 (average 0.39) within native and non-native populations, respectively (Table 3). The Shannon’s information index at the population level had a range of 0.60–0.65 with a mean of 0.63 within the native region, and a range of 0.37–0.70 with a mean of 0.56 within the introduced region. These results showed no significant loss in genetic variation in the introduced range compared with the native range (Mann–Whitney U test: Z = −0.149, P = 0.881 for I, Z = −0.604, P = 0.546 for H E). Among all populations, JS exhibited the highest level of variability (N e = 1.92, H E = 0.46, I = 0.70), and TW showed the lowest variability (N e = 1.48, H E = 0.27, I = 0.37). The Bottleneck test found that three of the non-native populations showed excess heterozygosity indicative of a bottleneck (Table 4). Using the more conservative Wilcoxon sign-rank test, TW, JS, and HK populations showed excess heterozygosity under all models, as well as a mode shift. In contrast, FJ and SH populations were not bottlenecked under one or both tests.
Distribution of genetic variation
No significant genetic differentiation was found between populations of native and introduced regions. The level of genetic differentiation between these two regions was low, as revealed by G ST = 0.01. These results agreed with the genetic structure estimated by AMOVA, with no significant (P > 0.05) genetic variation existing between areas (Table 5). The majority of genetic variation was found among populations within regions and within populations, at 23.33 and 80.75% (P < 0.05), respectively. When the native and introduced ranges were analyzed separately, the majority of genetic variation (i.e., 79.86% or 74.57%) was found to be within populations. Each value of partitioned genetic variance was significant for both regions (P < 0.01 after Bonferroni correction, Table 5). Overall, pairwise F ST in the native range (0.21 ± 0.11 SD) and introduced range (0.23 ± 0.14 SD) were similar and indicated that populations were relatively highly structured (Table 6). A Mantel test showed no significant correlation between genetic differentiation and geographical distance (r = −0.4799, P = 0.9260) in the introduced region.
Admixture analysis and identification of sources
Bayesian clustering analysis by InStruct identified two distinct clusters when assigning groups (K = 2), which was the best grouping manner from K evaluation (ΔK = 97.28 when K = 2, Fig. 2a). However, most of the sampled populations were mixtures of individuals from the two clusters (with the membership probabilities of ≥0.80). The first cluster, cluster 1, was dominant in two populations from the native region (NC and GA) and three populations from the introduced region (FJ, SH, and JS). The second cluster, cluster 2, included individuals that came predominantly from the FL population from the native region and two populations from the introduced region (HK and TW). If K = 3 was considered, TW was separated from other Chinese populations; JS, SH, and FJ shared certain genetic composition traits with GA and NC, revealing relatively closer genetic relationships with GA and NC than with TW and HK (Fig. 2b).

Bayesian estimates of population structure based on microsatellite variation among genets sampled from native and non-native populations. a Estimation of the best grouping number based on ΔK. b Individual assignment by Instruct analyses
The simulations with independent and correlated allele frequency models using STRUCTURE yielded virtually identical clusters and proportional membership of individuals to each cluster. We presented results from InStruct, as calculated without the assumption of HWE within clusters, which are more likely to reflect the actual population structure. Individual assignment tests conducted using GeneClass2 preferentially assigned most individuals in non-native populations (except SH) to their own sampled populations (58.33–91.67%). In population SH, nine of the twelve individuals (75%) were primarily assigned to JS and eight of those individuals were secondarily assigned to SH. The percentage of individuals assigned primarily or secondarily to NC/GA was between 25 and 50% for all FJ, SH, JS, and HK individuals. No individuals from FJ, SH, and JS were identified as primary or secondary assignments to FL. In contrast, one third of HK individuals were identified as secondary assignments to FL. Interestingly, five of the twelve TW samples were primarily assigned to NC or GA instead of FL, while the remainders were primarily assigned to their own population and secondarily assigned to NC.
Discussion
Comparison of genetic variation and differentiation between the native and non-native populations
Our results showed that there was no significant loss in genetic variation in the introduced range compared with the native range, although three of the five populations of S. alterniflora in China showed signs of a bottleneck (Table 4). By comparison, the levels of genetic variation in native populations of S. alterniflora in our study were comparable to those found in previous studies (Richards et al., 2004; Travis & Hester, 2005; Blum et al., 2007; Novy et al., 2010). However, genetic variation in populations of S. alterniflora in China was much higher in this study than reported by Deng et al. (2007), which is the only research available to date on genetic variation of S. alterniflora in China. In their study, genetic variation was examined within and among three S. alterniflora populations in Jiangsu Province using amplified fragment length polymorphism (AFLP) markers. Although traditional techniques like AFLP, allozymes, and random amplified polymorphic DNA (RAPD) are also multilocus, none of them have the resolution and power of a multilocus microsatellite study (Selkoe & Toonen, 2006). Hedrick (1999) also pointed out that the information that highly variable loci give may be quite different from that obtained from traditional, less variable markers. Thus, it was considered that the lower H E value (0.0657) in their study compared with ours might largely result from sampling and/or molecular marker differences.
Theory predicts that introduced species will show lower levels of intra-population diversity and higher levels of population differentiation than their native counterparts (Brown & Marshall, 1981). However, on a case-by-case basis, empirical data show that the magnitude of these changes varies greatly (Stepien et al., 2002). In our study, relatively high intra-population diversity was observed in some non-native populations (Table 2), suggesting that S. alterniflora might have a relatively active, seed dispersal mechanism. Literature suggests that although the spread of S. alterniflora in established stands is achieved primarily through rhizomes to form dense ramets (Dai & Wiegert, 1996), the colonization of new areas is accomplished by long-distance seed dispersal driven by seedling recruitment (Seneca, 1974; Sayce et al., 1997; Ayres et al., 2004; Deng et al. 2006). Strong and Ayres (2013) indicated that S. alterniflora and its hybrids spread almost totally by seed during their invasions. Recent experimental work in China also demonstrated the seed dispersal of S. alterniflora (Zhang et al., 2012; Li et al., 2014). Our results support these data and are also consistent with that expected for colonies established from heterogeneous founding groups made up of members originating from different source populations, which is well documented for S. alterniflora in China (Xu & Zhuo, 1985; Lee et al., 2004).
Similar levels of population differentiation were observed for the non-native populations and native populations. Analysis of molecular variance of both nuclear and chloroplast loci indicated that genetic variation was greater within regions than among regions (Tables 2, 4). In addition, from analysis of isolation by distance and cluster analysis, there was no evidence of genetic grouping associated with geographical distribution for S. alterniflora in China (result not shown). These observations could be interpreted, to some extent, from the history of the development of S. alterniflora in China (Chung et al., 2004; Zhang, 2004). The deliberate human planting from a similar source of S. alterniflora to protect the coastline and accelerate sedimentation facilitated the early spread of S. alterniflora, which is expected to result in lower genetic variation among the populations than among naturally expanding populations (Wang et al., 2006; Deng et al., 2007).
Genetic grouping and source tracking based on chlorotypes and nuclear microsatellite data
Spartina alterniflora was introduced to China in 1979 from Morehead City (NC), Altamaha Estuary (GA), and Tampa Bay (FL) for ecological engineering purposes (Xu & Zhuo, 1985). The plants from these three regions were identified as different ecotypes differentiated by plant height: the tall form occurs in GA, the medium form in NC, and the short form in FL (Qin et al., 1985; Xu & Zhuo, 1985). In fact, it is difficult to distinguish the tall and medium forms because plant heights overlap between the two forms (An et al., 2007). This is largely consistent with the observation that the invasive populations of S. alterniflora in Europe, North America, and China have shown continuous variation in height among clones (D. R. Strong, pers. com.). Later studies, such as the work by Deng et al. (2007), recognized only the tall and short forms of S. alterniflora in China, and observed most colonies that have survived and expanded in China were the tall form. Their result implied that, using morphological criteria, the FL ecotype has significantly declined in population, even vanished in China; however, Deng et al. (2007) failed to mention the situation for the NC ecotype. In this study, all samples from China were the tall form, because we could not find typical medium or short forms in the field, using morphological criteria. Thus, to investigate the underlying genetic differentiation, we chose to compare three native populations from the source locations with typical plant heights, instead of three ecotypes to plants in China. We found that, using nuclear microsatellite data, the individuals in GA and NC were more genetically similar than individuals in FL whenever K = 2 and 3 (Fig. 2) in a Bayesian clustering analysis. The distribution of cpDNA haplotypes also distinguished those originating from the Gulf coast from those from the Atlantic coast (Table 1), which is largely consistent with the patterns of S. alterniflora variation observed in other studies that found evidence of genetic structure among populations of S. alterniflora (O’Brien & Freshwater, 1999; Travis & Hester, 2005; Blum et al., 2007). These results are suggestive of two genetic assemblages of S. alterniflora in China, corresponding to NC/GA and FL, instead of three ecotypes proposed by previous studies (Qin et al., 1985; Xu & Zhuo, 1985).
One of the goals in this study was to determine if invasive populations have undergone population admixture during colonization. Several methods were used to solve this problem, including the analysis of chlorotypes distribution and Bayesian clustering analysis using nuclear microsatellite data. However, the admixture hypothesis could not be tested clearly based on our data because we found two of source populations (GA and NC) are not genetically divergent on both SSR profiles and chlorotypes. In contrast, we detected the mixture of two distinct genetic lineages in all populations except TW. First, we found the chlorotypes are unequally distributed across the native and introduced coasts. Five of the eight chlorotypes identified were only distributed in native populations (Table 1). Chlorotype C1 was an exception, which was not found in any native populations although it was detected in three of the non-native populations. Blum et al. (2007) examined the distribution and structure of genetic variation among native S. alterniflora along the Atlantic, Gulf and Pacific coasts of North America. Only one individual with chlorotype C1 was found from South Carolina, which was not listed in the sources of the Chinese descendants (Xu & Zhuo, 1985). In their study, the rareness of C1 indicated its low abundance in its native range, which could explain its absence in current native populations, owing to the limited sampling in our study. However, the relatively high abundance of C1 in China suggested a likely chloroplast inheritance for those individuals in China from populations from NC. Considering the proximity of South Carolina and Morehead City (NC), the latter is likely one of the sources for the Chinese populations (Xu & Zhuo, 1985). In our study, another interesting observation of chlorotype distribution was that no chlorotypes were shared between FL population and any other non-native populations, although this population (FL) is close to one of the source populations located on the Gulf coast of FL (Xu & Zhuo, 1985). The discrepancy may be a result of unequal loss of source chlorotypes and/or limited sampling. Alternatively, it is possible those descended from introduced FL individuals decreased gradually due to lower capacity of reproduction, dispersal, and competition than high form (Deng et al., 2006), which would lead to loss of FL chlorotypes in current introduced populations. In fact, it has been reported that most of S. alterniflora were tall form in China and the short form (FL ecotype) nearly disappeared by the year 2000 (Deng et al., 2007; Qing et al., 2011) (Table 7).
However, Bayesian clustering analysis revealed different grouping patterns, with HK and TW having genetic components similar to FL but different from the NC, GA, and other Chinese populations when assigning two groups (K = 2), which was the best grouping manner by ΔK evaluation. A significant drop in ΔK was shown when K = 3, which shows HK and TW samples were still well distinguished with small degrees of shared genetic components (Fig. 2). In fact, Bayesian clustering analysis showed that the two best clusters in the introduced region correspond to populations from the Atlantic coast (NC and GA) and Gulf coast (FL) in the native region (Fig. 2), which might imply different sources for the current Chinese populations. It was unexpected that the genetic profiles of nuclear microsatellite data of HK and TW samples were more similar to FL, considering they are both tall forms, morphologically, and have no shared chlorotypes with FL. One possible interpretation is the genetic admixture of the two native genetic assemblages corresponded to NC/GA and FL, in the populations of HK and TW, respectively, in the introduced region. The assignment tests in this study, however, provided evidence for the genetic admixture of allopatric native populations only in HK populations, implying different genetic compositions for HK and TW. Upon careful inspection, we found that the similarity between FL and TW in Bayesian clustering analysis might be an artifact, owing to shared alleles that are commonly lost in these two populations, which was not observed between FL and HK. It was reported that the TW population might have derived from the mainland population across the Taiwan Strait and settled in Taipei only 10 years previously (Chen, 2009; Liao, 2012), which could account for its genetic uniformity in this study. Considering the natural settlement and expansion of S. alterniflora in HK and TW, these two populations with distinct (TW) and admixed (HK) genetic composition might warrant the most attention for coastal management, compared with other Chinese populations.
As the first comparative genetic study on native (US) and non-native (China) populations of S. alterniflora, our results provide new insights into the genetic diversity, the provenance, and post-introduction evolution of non-native populations. In the future, it will be important to test the ideas about genetic admixture using broader population analysis from wider geographical ranges and/or the use of additional loci with higher levels of polymorphism for further resolution on the evaluation of this species between native and non-native regions.
References
Amsellem, L., J. L. Noyer, T. Le Bourgeois & M. Hossaert-MaKey, 2000. Comparison of genetic diversity of the invasive weed Rubus alceifolius Poir. (Rosaceae) in its native range and in areas of introduction, using amplified fragment length polymorphism (AFLP) markers. Molecular Ecology 9: 443–455.
An, S. Q., B. H. Gu, C. F. Zhou, Z. S. Wang, Z. F. Deng, Y. B. Zhi, H. L. Li, L. Chen, D. H. Yu & Y. H. Liu, 2007. Spartina invasion in China: implications for invasive species management and future research. Weed Research 47: 183–191.
Anttila, C. K., R. A. King, C. Ferris, D. R. Ayres & D. R. Strong, 2000. Reciprocal hybrid formation of Spartina in San Francisco Bay. Molecular Ecology 9: 765–770.
Arnaud-Haond, S. & K. Belkhir, 2007. GENCLONE: a computer program to analyze genotypic data, test for clonality and describe spatial clonal organization. Molecular Ecology Notes 7: 15–17.
Arnaud-Haond, S., F. Alberto, S. Teixeira, G. Procaccini, E. A. Serrao & C. M. Duarte, 2005. Assessing genetic diversity in clonal organisms: low diversity or low resolution? Combining power and cost efficiency in selecting markers. Journal of Heredity 96: 434–440.
Arnaud-Haond, S., C. M. Duarte, F. Alberto & E. A. Serrao, 2007. Standardizing methods to address clonality in population studies. Molecular Ecology 16: 5115–5139.
Arranz, J. J., Y. Bayon & F. SanPrimitivo, 2001. Differentiation among Spanish sheep breeds using microsatellites. Genetics Selection Evolution 33: 529–542.
Ayres, D. R., D. Garcia-Rossi, H. G. Davis & D. R. Strong, 1999. Extent and degree of hybridization between exotic (Spartina alterniflora) and native (S. foliosa) cordgrass (Poaceae) in California, USA determined by random amplified polymorphic DNA (RAPDs). Molecular Ecology 8: 1179–1186.
Ayres, D. R., D. L. Smith, K. Zaremba, S. Klohr & D. R. Strong, 2004. Spread of exotic cordgrasses and hybrids (Spartina sp.) in the tidal marshes of San Francisco Bay, California. USA. Biological Invasions 6: 221–231.
Blum, M. J., J. Bando, M. Katz & D. R. Strong, 2007. Geographic structure, genetic diversity and source tracking of Spartina alterniflora. Journal of Biogeography 34: 2055–2069.
Bossdorf, O., H. Auge, L. Lafuma, W. E. Rogers, E. Siemann & D. Prati, 2005. Phenotypic and genetic differentiation between native and introduced plant populations. Oecologia 144: 1–11.
Brown, A. H. D. & D. R. Marshall, 1981. Evolutionary change accompanying colonization in plants. In Scudder, G. C. E. & J. L. Reveal (eds), Evolution Today. Hunt Institute for Botanical Documentation. Camegie-Mellon University Press, Pittsburgh, Pennsylvania: 351–363.
Chen, H. G., 2009. The invasion of Spartina alterniflora in Zhanghua coast. CDNEWS. http://www.cdnews.biz/cdnews_site/docDetail.jsp?coluid=108&docid=100773161. Accessed 20 May 2009.
Chen, J. Q. & C. H. Chung, 1990. Genecological studies of Spartina alterniflora. In Wang, Y. (ed.), Proceedings of the Fifth MICE Symposium for Asia and the Pacific. Nanjing University Press, Nanjing: 168–183.
Chen, Y. H., S. B. Opp, S. H. Berlocher & G. K. Roderick, 2006. Are bottlenecks associated with colonization? Genetic diversity and diapause variation of native and introduced Rhagoletis complete populations. Oecologia 149: 656–667.
Chung, C. H., R. Z. Zhuo & G. W. Xu, 2004. Creation of Spartina plantations for reclaiming Dongtai, China, tidal flats and offshore sands. Ecological Engineering 23: 135–150.
Cornuet, J. M., S. Piry, G. Luikart, A. Estoup & M. Solignac, 1999. New methods employing multilocus genotypes to select or exclude populations as origins of individuals. Genetics 153: 1989–2000.
Daehler, C. C. & D. R. Strong, 1996. Status, prediction and prevention of introduced cordgrass Spartina spp. invasion in Pacific estuaries. USA. Biological Conservation 78: 51–58.
Dai, T. & R. G. Wiegert, 1996. Ramet population dynamic and net aerial primary productivity of Spartina alterniflora. Ecology 77: 276–288.
Deng, Z. F., S. Q. An, Y. B. Zhi & C. F. Zhou, 2006. Preliminary studies on invasive model and outbreak mechanism of exotic species, Spartina alterniflora Loisel. Acta Ecologica Sinica 26: 2678–2686.
Deng, Z. F., S. Q. An, C. F. Zhou, Z. S. Wang, Y. B. Zhi, Y. J. Wang, S. H. Shi, L. Chen & C. J. Zhao, 2007. Genetic structure and habitat selection of the tall form Spartina alterniflora Loisel. in China. Hydrobiologia 583: 195–204.
Dlugosch, K. M. & I. M. Parker, 2008. Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Molecular Ecology 17: 431–449.
Doyle, J. J. & J. L. Doyle, 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19: 11–15.
Evanno, G., S. Regnaut & J. Goudet, 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology 14: 2611–2620.
Excoffier, L., G. Laval & S. Schneider, 2005. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online 1: 47–50.
Gao, H., S. Williamson & C. D. Bustamante, 2007. A markov chain monte carlo approach for joint inference of population structure and inbreeding rates from multilocus genotype data. Genetics 178: 1635–1651.
Guo, W. X., Y. L. Huang, Z. W. He, Y. B. Yan, R. C. Zhou & S. H. Shi, 2013. Development and characterization of microsatellite loci for smooth cordgrass, Spartina alterniflora (Poaceae). Application in Plant Sciences 1: 1200211.
Hedrick, P. W., 1999. Perspective: highly variable loci and their interpretation in evolution and conservation. Evolution 53: 313–318.
Hoos, P. M., A. W. Miller, G. M. Ruiz, C. V. Robert & J. B. Geller, 2010. Genetic and historical evidence disagree on likely sources of the Atlantic amethyst gem clam Gemma gemma (Totten, 1834) in California. Diversity and Distributions 16: 582–592.
Jakobsson, M. & N. A. Rosenberg, 2007. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806.
Janko, K., J. Kotusz, K. De Gelas, V. Slechtova, Z. Opoldusova, P. Drozd, L. Choleva, M. Popiolek & M. Balaz, 2012. PLoS ONE 7: e45384.
Jiang, F. X., B. S. Lu, C. X. Zhong & M. Zhao, 1985. Biological characteristics of three American species of Spartina and the nutrient analysis. Journal of Nanjing University (special issue for Research Advances in Spartina of China): 302–309.
Kimura, M. & J. F. Crow, 1964. The number of alleles that can be maintained in a finite population. Genetics 49: 725–738.
Lee, C. E., 2002. Evolutionary genetics of invasive species. Trends in Ecology & Evolution 17: 386–391.
Lee, P. L. M., P. M. Patel, R. S. Conlan, S. J. Wainwright & C. R. Hipkin, 2004. Comparison of genetic diversities in native and alien populations of hoary mustard (Hirschfeldia incana [L.] Lagreza-Fossat). International Journal of Plant Sciences 165: 833–843.
Lewontin, R. C., 1972. Testing the theory of natural selection. Nature 236: 181–182.
Li, J., S. Gao & Y. Li, 2006. Spatial and temporal variations in salt-marsh vegetation in Wanggang area, Jiangsu coast, based upon TM imagery analysis. Marine Sciences 30: 52–57.
Li, Z. J., W. Q. Wang & Y. H. Zhang, 2014. Recruitment and herbivory affect spread of invasive Spartina alterniflora in China. Ecology 95: 1972–1980.
Liao, S. H., 2012. Effects of Spartina alterniflora invasion on meiofauna in Kaomei Wetland. Dissertation, National Chung Hsing University.
Lombaert, E., T. Guillemaud, J. M. Cornuet, T. Malausa, B. Facon & A. Estoup, 2010. Bridgehead effect in the worldwide invasion of the biocontrol harlequin ladybird. PLoS One 5: e9743.
Luikart, G., F. W. Allendorf, J. M. Cornuet & W. B. Sherwin, 1998. Distortion of allele frequency distributions provides a test for recent population bottlenecks. Journal of Heredity 89: 238–247.
Mantel, N. A., 1967. The detection of disease clustering and a generalized regression approach. Cancer Research 27: 209–220.
McCauley, D. E., R. A. Smith, J. D. Lisenby & C. Hsieh, 2003. The hierarchical spatial distribution of chloroplast DNA polymorphism across the introduced range of Silene vulgaris. Molecular Ecology 12: 3227–3235.
Meirmans, P. G. & P. H. V. Tienderen, 2004. GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Molecular Ecology Notes 4: 792–794.
Miller, M. P., 1997. Tools for population genetic analysis (TFPGA) Version 1.3: A windows program for the analysis of allozyme and molecular population genetic data. Computer software distributed by the author.
Nei, M., 1973. Analysis of gene diversity in subdivided populations. PNAS 70: 3321–3323.
Nei, M., 1978. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590.
Nei, M., R. Chakraborty & P. A. Fuerst, 1976. Infinite allele model with varying mutation rate. Proceedings of the National Academy of Sciences of the United States of America 73: 4164–4168.
Nibouche, S., B. Fartek, S. Mississipi, H. Delatte, B. Reynaud & L. Costet, 2014. Low genetic diversity in Melanaphis sacchari aphid populations at the worldwide scale. PLoS ONE 9: e106067.
Novy, A., P. E. Souse, J. M. Hartman, L. Struwe, J. Honig, C. Miller, M. Alvarez & S. Bonos, 2010. Genetic variation of Spartina alterniflora in the New York metropolitan area and its relevance for marsh restoration. Wetlands 30: 603–608.
O’Brien, D. L. & D. W. Freshwater, 1999. Genetic diversity within tall form Spartina alterniflora Loisel. along the Atlantic and Gulf coast of the United States. Wetlands 19: 352–358.
Piry, S., G. Luikart & J. M. Cornuet, 1999. BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. Journal of Heredity 90: 502–503.
Piry, S., A. Alapetite, J. M. Cornuet, D. Paetkau, L. Baudouin & A. Estoup, 2004. CeneClass2: a software for genetic assignment and first-generation migrant detection. Journal of Heredity 95: 536–539.
Pritchard, J. K., M. Stephens & P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Provan, J., S. Murphy & C. A. Maggs, 2005. Tracking the invasive history of the green alga Codium fragile ssp. tomentosoides. Molecular Ecology 14: 189–194.
Qin, P., M. D. Jin & M. Xie, 1985. Community biomass among the three ecotypes of S. alterniflora in Luoyuan Bay, Fujian. Journal of Nanjing University (Special issue for research advances in Spartina: achievements of past 22 years): 226–236.
Qing, H., Y. H. Yao, Y. Xiao, F. Q. Hu, Y. X. Sun, C. F. Zhou & S. Q. An, 2011. Invasive and native tall forms of Spartina alterniflora respond differently to nitrogen availability. Acta Oecologica 37: 23–30.
Rannala, B. & J. L. Mountain, 1997. Detecting immigration by using multilocus genotypes. Proceedings of the National Academy of Sciences of the United States of America 94: 9197–9201.
Richards, C. L., J. L. Hamrick, L. A. Donovan & R. Mauricio, 2004. Unexpectedly high clonal diversity of two salt marsh perennials across a severe environmental gradient. Ecology Letters 7: 1155–1162.
Roderick, C. K. & M. Navajas, 2003. Genes in new environments: genetics and evolution in biological control. Nature Reviews Genetics 4: 889–899.
Rosenberg, N. A., 2004. DISTRUCT: a program for the graphical display of population structure. Molecular Ecology Notes 4: 137–138.
Sayce K., B. Dumbauld & J. Hidy, 1997. Seed dispersal in drift of Spartina alterniflora. In: Proceeding of the Second International Spartina Conference, Olympia, WA. Washington State University-Cooperative Extension, Pullman, WA.
Selkoe, K. A. & R. J. Toonen, 2006. Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecology Letters 9: 615–629.
Seneca, E. D., 1974. Germination and seedling response of Atlantic and Gulf coasts populations of Spartina alterniflora. American Journal of Botany 61: 947–956.
Stepien, C. A., C. D. Taylor & K. A. Dabrowska, 2002. Genetic variability and phylogeographical patterns of a nonindigenous species invasion: a comparison of exotic vs. native zebra and quagga mussel populations. Journal of Evolutionary Biology 15: 314–328.
Stiller, J. W. & A. L. Denton, 1995. One hundred years of Spartina alterniflora (Poaceae) in Willapa Bay, Washington: random amplified polymorphic DNA analysis of an invasive population. Molecular Ecology 4: 355–363.
Strong, D. R. & D. A. Ayres, 2013. Ecological and evolutionary misadventures of Spartina. Annual Review of Ecology, Evolution, and Systematic 44: 389–410.
Taberlet, P., L. Gielly, G. Pautou & J. Bouvet, 1991. Universal primers for amplification of three noncoding regions of chloroplast DNA. Plant Molecular Biology 17: 1105–1109.
Travis, S. E. & M. Hester, 2005. A space-for-time substitution reveals the long-term decline in genotypic diversity of a widespread salt plant, Spartina alterniflora, over a span of 1,500 years. Journal of Ecology 93: 417–430.
Travis, S. E., C. E. Proffitt, R. C. Lowenfeld & T. W. Mitchell, 2002. A comparative assessment of genetic diversity among differently-aged populations of Spartina alterniflora on restored versus natural wetlands. Restoration Ecology 10: 37–42.
Travis, S. E., C. E. Proffitt & K. Ritland, 2004. Population structure and inbreeding vary with successional stage in created Spartina alterniflora marshes. Ecological Applications 14: 1189–1202.
Verhoeven K. J. F., M. Macel, L. M. Wolfe & A. Biere, 2010. Population admixture, biological invasions and the balance between local adaptation and inbreeding depression. Proceedings of the Royal Society B-Biological Sciences 278: 2–8.
Wang, Q., S. Q. An, Z. J. Ma, B. Zhao, J. K. Chen & B. Li, 2006. Invasive Spartina alterniflora: biology, ecology and management. Acta Phytotaxonomica Sinica 44: 559–588.
Weir, B. S., 1996. Genetic Data Analysis II. Sinauer Associates, Sunderland, MA.
Xu, G. W. & R. Z. Zhuo, 1985. Preliminary studies of introduced Spartina alterniflora Loisel in China (I). Journal of Nanjing University (Suppl) 40(2): 212–225.
Yeh, F. C. & T. J. B. Boyle, 1997. Population genetic analysis of co-dominant and dominant markers and quantitative traits. Belgian Journal of Botany 129: 157.
Zhang, D., 2004. Plant Life History Evolution and Reproductive Ecology. Science Press, Beijing.
Zhang, Y. H., G. M. Huang, W. Q. Wang, L. Z. Chen & G. H. Lin, 2012. Interactions between mangroves and exotic Spartina in an anthropogenically disturbed estuary in southern China. Ecology 93: 588–597.
Zhi, Y. B., H. L. Li, S. Q. An, L. Zhao, C. F. Zhou & Z. F. Deng, 2007. Inter-specific competition: Spartina alterniflora is replacing Spartina anglica in coastal China. Estuarine, Coastal and Shelf Science 74: 437–448.
Acknowledgments
We thank Tian Tang, Weijing Li, Lu Lu, Xiaoting Fang, Bo Li, Shuqing An, Lu Xia, Zhenji Li, Chuan Tong, Donald R. Strong, Hui Guo, Jenny Q. Xiang, Yi Yu and Yupeng Zhao for their assistance in collecting or providing some plant materials. This study is supported by grants from the National Natural Science Foundation of China (41276107, 41130208, 31070290, 91331202, 31200466), the Natural Science Foundation of Guangdong Province (8151027501000089) and Chang Hungta Science Foundation of Sun Yat-Sen University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: K. W. Krauss
Rights and permissions
About this article

Cite this article
Guo, W., Qiao, S., Wang, Y. et al. Genetic diversity, population structure, and genetic relatedness of native and non-native populations of Spartina alterniflora (Poaceae, Chloridoideae). Hydrobiologia 745, 313–327 (2015). https://doi.org/10.1007/s10750-014-2117-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-014-2117-9