
Abstract
Iron deficiency or overload poses an increasingly complex issue in cardiovascular disease, especially heart failure. The potential benefits and side effects of iron supplementation are still a matter of concern, even though current guidelines suggest therapeutic management of iron deficiency. In this review, we sought to examine the iron metabolism and to identify the rationale behind iron supplementation and iron chelation. Cardiovascular disease is increasingly linked with iron dysmetabolism, with an increased proportion of heart failure patients being affected by decreased plasma iron levels and in turn, by the decreased quality of life. Multiple studies have concluded on a benefit of iron administration, even if just for symptomatic relief. However, new studies field evidence for negative effects of dysregulated non-bound iron and its reactive oxygen species production, with concern to heart diseases. The molecular targets of iron usage, such as the mitochondria, are prone to deleterious effects of the polyvalent metal, added by the scarcely described processes of iron elimination. Iron supplementation and iron chelation show promise of therapeutic benefit in heart failure, with the extent and mechanisms of both prospects not being entirely understood. It may be that a state of decreased systemic and increased mitochondrial iron levels proves to be a useful frame for future advancements in understanding the interconnection of heart failure and iron metabolism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Iron metabolism within the human body presents itself in physiology and pathology with increased interest in current research, aided by recent advancements in the understanding of the finely tuned mechanisms of ferroregulation. Iron (Fe) is a transitional metal, speculated to have been involved in the very formation of life on Earth [1]. The capacity of iron to accept or donate electrons by means of its polyvalent trait [2] catalyzes a plethora of redox reactions throughout biological systems. Being involved in oxygen transport, sequestration, and utilization, comprised in iron-sulfur clusters (ISC) [3], production of reactive oxygen species (ROS), and DNA synthesis, as well as in immunosurveillance [4], it is obvious that tight equilibrium must be maintained in order to avoid detrimental effects spawned by both iron overload and deficiency. Systemic regulation relies mainly on the peptide hormone hepcidin [5] in the hepcidin/ferroportin axis, whereas cellular regulation appears to be more complex. Depending on the iron-responsive protein/iron-responsive elements (IRP/IRE) as a way of iron sensing [6], cellular homeostasis is influenced by the hypoxia-sensing HIF/HRE (hypoxia-induced factor/hypoxia-responsive elements) [7] as well as by the recently discovered tissue-autonomous, cardiac hepcidin/ferroportin axis [8].
In cardiac pathology, iron homeostasis is dysregulated, with both deficiency and overload being cited as culprits [9, 10]. In this review, we examined the metabolic circuits iron participates in and the therapeutic opportunities iron homeostasis present in the treatment of cardiovascular disease.
Ferrokinetics
Iron absorption
The physiologic pathway of iron following ingestion takes part in the milieu of the duodenum. Because the ferrous iron (Fe2+) is readily oxidized in atmospheric conditions to its trivalent, insoluble state, Fe3+ requires reduction by metalloreductases (such as Dcytb [11]) or by reductants within the intestinal lumen (e.g., ascorbic acid), followed by ingress via a non-specific, divalent metal transporter DMT1 (SLC11A2) [12]. With regard to the organic sources of iron, proteolytic enzymes degrade protein-rich foods in order to release heme-abundant hemoglobin/myoglobin. Heme has been proposed to be internalized with the aid of either a low-affinity heme transporter, heme carrier protein 1 (HCP-1) [13], or by the heme-responsive gene 1 protein (HRG-1) [14]. Free Fe2+ from heme is released by catalytic removal of protoporphyrin by heme oxygenase 1 (HO-1) [15]. From here on, Fe can be either lost through shedding of the epithelial cells or be further conveyed at the basolateral membrane of the duodenal enterocyte. Efflux of ferrous iron is mediated by ferroportin (Fpn, SLC40A1 gene) [16]. Re-oxidation of Fe2+ by hephaestin [17] (a ceruloplasmin-like membrane protein), or serum ceruloplasmin itself, occurs to allow binding of Fe3+ to serum apo-transferrin. Serum transferrin (Tf) is a beta-1 globulin synthesized mainly from the liver which attaches two molecules of Fe3+ for transport within the circulatory torrent.
Cellular iron uptake
Cells that exhibit a high transferrin receptor (TfR) count on their surface membrane, such as erythropoietic cells from the bone marrow, are the predominant destination of holo-transferrin [18]. In the recent decades, two types of TfR have been characterized: TfR1 (coding gene TFRC), a transmembrane protein identified to be the same as CD71 antigen [19], and TfR2 (coding gene TFR2)—homologous with TfR1, but with additive function in the regulation of hepcidin, as shown by the development of hemochromatosis type 3 in individuals with TfR2 mutations [20]. The ligand-receptor complex formed is further engulfed within the cell by endocytosis and formation of an endosome. Acidification by vacuolar ATPase within the structure increases the dissociation constant of ferric iron which in turn prompts conformational changes of the Tf molecule with consequent release of Fe molecules. Interestingly enough, the depletion of vacuolar ATPase complexes and ensuing decrease in available intracellular iron are likely involved in the stimulation of hypoxia-inducible factor 1 (HIF1) [21], one of the regulatory pathways in cellular iron equilibrium. Non-transferrin bound iron (NTBI) can also permeate the cell directly by membrane DMT1 and via new metal transporters recently described. ZIP14 and ZIP8 (ZRT/IRT-like protein family) are members of the solute carrier family 39 (SLC39) with function as a zinc and iron membrane transport proteins [22,23,24] that are not regulated by the traditional IRE/IRP axis [25] (detailed below). From here on, the bivalent iron atoms, representing the labile pool of iron, are either chaperoned to different organelles by RNA-binding proteins [26,27,28] or stored within ferritin (Ft). Ft is a protein shell with a ferric hydroxyphosphate core, with two subunits types: H and L [19], owing to their heavy (21–23 kDa) and light (19–21 kDa) molecular weight [29]. Unlike heme, which is found mainly in animal products, Ft appears abundant in whole legumes [30]. Of note, the capacity of Ft to store iron has led to therapeutic use as a genetic iron chelator, with promising results [31]. One of the main sites of iron utilization by the cell is the mitochondrial apparatus [32], which has an array of iron-containing moieties, such as ISC, mitochondrial ferritin (MtFt), reductases, and oxidases, and is the main site of heme production [33].
Systemic ferroregulation
Iron homeostasis must be accomplished by way of uptake regulation, as there is no physiologically known control of excretion; proposed means of iron elimination are intestinal cells sloughing, skin desquamation, and blood loss [34]. Indeed, the pivotal role in iron balance is ascribed to hepcidin (Hep, HAMP gene), a 25 aminoacidic hormone secreted mainly by the liver [35]. Hepcidin downregulates iron uptake via induction of internalization and degradation of Fpn and/or DMT1, particularly acting within the gastrointestinal tract (via DMT1) and the reticuloendothelial system (via Fpn), sites of Fe absorption and recycling, respectively [36]. Studies concerning the mechanism of action of Hep within the enterocytes of the duodenum have suggested that brush border DMT1 is the target of internalization and subsequent degradation in the acute setting, rather than Fpn [37, 38]. It has been proposed that the low influx state driven by inhibition of DMT1 is the factor that decreases Fpn expression in the milieu of sustained Hep stimulation [37, 39]. Hep is transcriptionally upregulated by iron overload, inflammation, and obesity [40], and decreased synthetization is observed with hypoxia (via HIF), hypoferremia, increased erythropoietic drive (e.g., anemia) [41], and cell proliferation [42]. Mechanisms of regulation involve the BMP/SMAD (bone morphogenetic protein (BMP)/s-mother against decapentaplegic) pathway and JAK/STAT pathways [43]. Recently studied processes of regulation have suggested that leptin (an energy balance hormone) causes increased serum hepcidin levels [44]. Leptin disorders are closely associated with obesity and might supply a missing link between inflammation, cardiovascular disease, and metabolism [45].
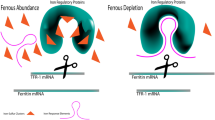
In the case of increased availability of iron (Fig. 1), Hep synthesis is augmented by the following process: increased concentration of holo-Tf binds to cell membrane TfR1, displacing the human hemochromatosis protein (HFE; high ferrum). HFE then translocates to TfR2, which in turn associates with hemojuvelin (HJV, a BMP co-receptor) which leads to phosphorylation of SMAD and induction of HAMP mRNA [43, 46]. Similarly, with inflammation, cytokines (such as IL-6, IL-1, tumor necrosis factor alpha (TNF-α) [47, 48]) interact with the JAK/STAT3 and activate transcription of Hep [49].

Hepcidin upregulation by iron and inflammation. Transferrin-bound iron becomes attached to the transferrin receptor 1, causing the formation of the HFE-TfR2-HJV-BMP6R complex, with the end result of Hep mRNA synthesis via the SMAD pathway. Inflammation, via cytokine IL-6, causes similar results, either via JAK-STAT pathway or by means of modulation of the BMP-SMAD. RBMP6, bone morphogenetic protein 6 receptor; HAMP, hepcidin antimicrobial peptide gene; HFE, human hemochromatosis protein; HJV, hemojuvelin; IL-6, interleukin 6; JAK, Janus kinases; RE, response elements; SMAD, s-mother against decapentaplegic; STAT, signal transducer and activator of transcription proteins; TfR transferrin receptor
Conversely, decreased iron storage (i.e., Ft), hypoxia, and anemia or increased cell proliferation suppress Hep secretion by transcriptional regulation (Fig. 2). Hypoxia inhibits Hep synthesis via hypoxia-inducible factors (HIFs), in order to coordinate the increased requirements of erythropoiesis with iron availability. Erythropoietic signaling by erythropoietin (EPO) indirectly downregulates Hep production by HIFs action, along with growth differentiation factor 15 (GDF15) [50] and twisted gastrulation factor 1 (TWSG1) [41], both released by bone marrow precursors of myeloid lineage. However, little is known on the exact mechanism of action.

Hepcidin downregulation. In the case of decreased iron availability, hypoxia, and anemia, hepcidin synthesis is inhibited by a molecular disruption of the TfR1-transferrin complex formation by competitive binding of the HFE protein. Hypoxia acts via hypoxia-inducible factors, in order to coordinate the increased requirements of erythropoiesis with iron availability. Erythropoietic signaling by erythropoietin indirectly downregulates Hep production by HIFs action, along with growth differentiation factor 15 and twisted gastrulation factor 1, both released by bone marrow precursors of myeloid lineage. However, little is known on the exact mechanism of action. Epo, erythropoietin; ERFO, erythroferrone; GDF15, growth differentiation factor 15; HAMP, hepcidin antimicrobial peptide gene; HFE, human hemochromatosis protein; HIF, hypoxia inducible factor; HJV, hemojuvelin; IL-6, interleukin 6; JAK, Janus kinases; RBMP6, bone morphogenetic protein 6 receptor; RE, response elements; SMAD, s-mother against decapentaplegic; STAT, signal transducer and activator of transcription proteins; TfR, transferrin receptor; TWSG1, twisted gastrulation factor 1
Cellular ferroregulation
Within the cell, intrinsic levels of iron control the regulation via iron regulatory proteins (IRP) 1/2 and iron-responsive elements (IRE), in a posttranscriptional fashion [6, 41]. Under conditions wherein iron is replete, IRP1 is found within the cytosol where it functions as an aconitase [51], whereas IRP2 undergoes proteasomal degradation [52]. Upon decreased iron availability, the IRPs bind to the untranslated regions (UTR) of target genes. Binding to the upstream 5′-UTR inhibits translation into proteins, while 3′-UTR interaction, located downstream from the protein transcript, will stabilize the mRNA and promote synthesis. Several genes involved in iron homeostasis/metabolism possess IREs either in the 5′-UTR or 3′-UTR of their mRNA transcript (Table 1) [6]. As a result, in low iron conditions, most cells will undergo enhanced iron uptake and modulation of cell development and division, while iron export, storage, and utilization are downregulated. IRP1-HIF2α interaction proves useful in elucidating mechanisms that regulate erythropoiesis in low oxygen and iron conditions in the liver and kidney (reviewed extensively in [52]). It has been proposed that the IRP/IRE pathway is implicated particularly in the iron handling by the mitochondria [53, 54], highlighting the iron-mitochondria connection in health and disease. Notably, the Ft proteins also appear to possess antioxidant properties (particularly subunit H) [55], as shown by the induced synthesis of Ft by inflammatory cytokines and oxidative stress [56, 57]. The molecular mechanism involves antioxidant response elements for the Ft mRNA, in addition to the IRP, connecting and complicating the iron metabolism—inflammation—oxidative stress interplay (Ft regulation reviewed in [58]).
Iron metabolism in the healthy and diseased cardiac myocytes
Previously described mechanisms of iron uptake also apply to cardiomyocytes, with Tf-bound Fe3+ entering the cell via endocytosis. Moreover, there are other pathways for ferrous iron. The cardiomyocytes possess L- and T-type calcium channels (LTCC, TTCC) which in the case of chronic heart failure (CHF) become altered [59]. Murine studies have indicated these channels as points of entry in the cardiac cells during iron overload [60], along with calcium channel blockers (CCB) decreasing iron load [61, 62]. Notably, the aforementioned ZIP14/8 carriers expressions are low in the cardiac tissue [63], suggesting minimal importance in iron trafficking within the cardiomyocyte. Upon being added to the cellular labile iron pool (LIP), Fe can be either deposited in the form of ferritin, used in metabolic cytosolic processes, or enter the mitochondria. In addition, ferritin H chain protein appears downregulated in the failing heart, possibly increasing the LIP and subsequent ROS formation [64]. The ingress of Fe into the mitochondrial apparatus is done by mitoferrin 1 and 2 [65]. Within the mitochondrion, iron contributes to the formation of heme, ISC, and MtFt [66], and oxidative phosphorylation [67]. Indeed, Fe and the cardiac mitochondria are of such paramount importance in the regulation of heart energetics that the regulation of local Hep and Fpn is done by cardiac transcriptional and translational modulation [8]. Lakhal-Littleton et al. [8] have shown that even in the case of physiologic systemic Fe levels, the cardiac iron homeostasis is determined in a paracrine and autocrine manner (via a local Hep/Fpn axis), apart from the systemic control of liver Hep/Fpn axis and the established cellular IRP/IRE system [54], further abetting the complexity of iron interplay in the heart. Seemingly contradictory, the cardiac level of Hep protein is downregulated in the case of systemic iron overload and downregulated during iron deficiency [8]. This is in contrast with the endocrine (systemic) model of Hep/Fpn, where high iron load causes increased Hep secretion as a way to inhibit iron absorption. The reason for this shift most likely relies on the fact that in the case of iron overload, decreased cardiac Hep expression will result in an upregulation of Fpn, therefore increasing iron egress capacity from the cardiomyocyte. Conversely, it can be appraised that in the case of systemic Fe deficiency, protection from organ shortage of the metallic element occurs via the limitation of iron export through cardiac Fpn [8].
Iron disequilibrium in heart diseases
CHF is associated with systemic iron deficiency (ID) in a very high proportion, typically between 10 and 50% of patients presenting some form of anemia or iron deficiency [68]. The etiology is most likely multifactorial, with iron sequestering as a result of malabsorption, gastrointestinal bleeding, and cachexia being the leading causes [9]. Although inflammation has an important role in ferrequilibrium, well-conducted studies [69] have failed to show any correlation between Hep and inflammatory status (i.e., IL-6, TNF-alpha), owing to the anti-inflammatory effects of Hep [70]. Interestingly, it appears that a nutritional iron intake disturbance is of less importance in myocardial ID development. Rather, myocytes dysregulation of iron is brought about by hemodynamic stress that is characteristic of HF [71]. Being cognizant that HF is a syndrome that retains inflammatory characteristics [72] and that iron metabolism is dysfunctional [69], one can surmise that processes within the intricate systemic and cellular meshwork of bioiron are altered, with adverse effects. As such, iron disequilibrium could prove an attractive target for cardiovascular therapies.
At the cellular milieu, HF patients exhibit a plethora of iron-mediated shifts in cell function, independent of systemic iron levels. In the cardiomyocytes of HF patients undergoing transplantation, Melenovsky et al. [73] found decreased iron content compared with controls, associated with decreased mitochondrial enzymatic (respiratory and ROS related) activities. Moreover, patients with the lowest myocardial iron content were most affected by the decrease in function. Congruent with this hypothesis, an in vitro investigation using iron-deficient human cardiomyocytes demonstrated a cellular iron status-dependent decrease of mitochondrial respiration and contractility, along with an increase in the quantitation of storage Ft mRNA [74]. In as much as this upregulation seems paradoxical, it can be appealing to further ascertain its roles (see above section) in the development of cardiac ID. Furthermore, it appears that the IRP/IRE regulation mechanism has a specific role in the development of HF brought about by iron deficiency. A transgenic murine study of cardiac IRP deletion with consequent ID expressed a diminution in respiratory activity of the myocytes mitochondria, especially under stress conditions [53], coupled with a downregulation of the membrane TfR1. Clinically relevant, the studies have shown that the administration of iron at least partially reversed the changes, suggesting an intrinsic role of iron deregulation to the development and changes of CHF patients.
Taken together, the pathophysiology of HF in respect to ID can be viewed as an effect of reduced myocardial iron content, which in turn causes the deficient activity of key enzymes of oxidative phosphorylation and impaired contractility. Thus, the energetical demands of the cardiac muscle cell cannot be met by dysfunctional mitochondria, which leads to the poor survival rates found in patients with ID and HF [69]. The etiological connection between HF and myocardial iron deficiency remains elusive, although the reduced expression of iron uptake via TfR1 [53], ZIP14, DMT1 [73, 75], a Ft mediated diminished availability of NTBI [74], and pleiotropic influence of the HIF/IRP/Hep regulation mechanisms [52, 76] could prove useful starting points in future endeavors.
In contrast, supplementary investigations have revealed that a state of iron disequilibrium (that is an imbalance rather than purely deficit of iron) could better explain the intracellular and extracellular metabolism of iron. To this end, multiple studies [10, 77,78,79,80,81] have concluded that an increase of iron content in the myocardial mitochondria of subjects with heart disease may be one of the reasons behind pathological disequilibrium, even though there have been little studies concerning intraorganellar iron quantitation and its effects in HF. It appears that even though decreased systemic iron availability is present in half of HF patients [82], at the cellular and mitochondrial level there might be an increase in Fe concentrations [78, 83], leading to ROS generation and oxidative damage. Additionally, in the acute and ischemic setting of heart diseases (i.e., myocardial infarction), both human and animal model researches have shown increased iron deposition in the cardiac cells [79, 84, 85], as well as increased Hep synthesis [86] associated with adverse cardiac effects.
Augmented iron levels in the HF patients were referred to as the probable cause of increased ROS via heme and non-heme bound iron in the mitochondrial compartment [83]. Likewise, it seems that in the iron-deficient setting, cardiac myocytes respond by an increase of Hep protein synthesis, followed by a Fpn-mediated decrease of iron egress [10, 87]. It also seems plausible that an increase of LTCC mediated iron absorption in the failing hearts may have a role, aside from the canonical Tf-bound iron [88]. Concurrently, ischemia/reperfusion models of HF have also been linked with decreased cellular viability and function, owing to ferroptosis by increased heme and non-heme iron contents within the mitochondria and augmented iron release by increased levels of HO-1, with chelators amending the degree of cardiomyocyte dysfunction [89]. Interestingly, the subcellular iron fraction was increased, while cytosolic levels were not significantly different from controls, suggesting mitochondrial iron deposition at the expense of the LIP and consequent dysfunction of the iron-dependent elements in the heart.
Indeed, one could hypothesize that the disparate relationship between systemic and cardiac intracellular iron levels, with diverging results regarding regulation of iron, transferrin, hepcidin, ferroportin [8, 74, 88], suggests a rather intimate relation between cardiac iron and development of HF, with both decrease of systemic and increase of intraorganellar iron as cause. Amplified heme and HO-1 levels in the cardiac mitochondria leading to increased unbound iron and production of ROS could prove a suitable theoretical model to explain such divergence between studies.
Therapeutic approaches
Defining iron deficiency and overload
In the most prominent randomized controlled trials (RCT) conducted to establish the potential benefits of iron supplementation in iron-deficient patients with CHF [90, 91], ID was defined based on Ft serum levels and/or serum saturation of Tf (TSAT) by way of a dichotomous cutoff: either a Ft < 100 ng/mL or Ft 100–300 ng/mL with a TSAT < 20% would serve as diagnostic criteria. The former is used to define absolute deficiency, while the latter suggests functional ID. Although arbitrarily chosen, the definitions were incorporated in an international consensus regarding ID in inflammatory diseases [92], signifying valid targets for treatment initiation and disease outcome monitoring. However, additional inquiries into the subject by Beverborg et al. [93] have steered to different propositions. Based on a number of 42 CHF patients undergoing coronary artery bypass grafting and bone marrow aspiration compared with 387 outpatients, the study found that the most reliable predictors of all-cause mortality were TSAT < 19.8% and serum iron levels < 13 μmol/L. While not an unequivocal research, it raises questions pertaining to the right treatment to the right patients. Similarly, another study [94] pinpointed low levels of iron and hemoglobin, but high values of Ft and vitamin B12 as culprits in total mortality of CHF patients, with only serum iron being attributed to cardiovascular-related mortality.
In the case of iron overload, the diagnosis issue is intertwined with multiple etiologies and even further complicated by the need of biochemical (TSAT, Ft, Tf), genetic, and imaging (MRI) tests. Detailed algorithms for defining hemochromatosis are beyond the scope of this paper and have been reviewed in depth elsewhere [95]. With regard to cardiac siderosis, the majority of investigations make extensive use of organ-level magnetic resonance imaging [96].
Supplementing iron
Due to the increased side effects and little clinical improvement of oral iron therapy [97], clinical trials have shifted towards intravenous supplementation in HF. Ferric carboxymaltose is the preferred preparation as opposed to other formulations, as it appears that it produces a more potent and precipitous increase in hematinic parameters [98]. Currently, there are clinical trials for a number of other forms of iron, such as iron protein succinylate (ClinicalTrials.gov Identifier: NCT03344523), ferric maltol (NCT03774615), and iron maltoside (NCT02642562).
A great focus has been put on evaluating and treating iron deficiency (ID) and the resulting iron-deficient anemia (IDA), as it appears that low iron availability correlates with deleterious effects, such as increased mortality and hospitalization and decreased quality of life [99]. The causative relation between heart failure and ID or IDA is likely multifactorial. Decreased dietary iron intake [100], multiple drug interactions and drug-induced bleeding [101], IL-6- and TNF-α-mediated inflammation [102], blunted erythropoietin production [103], and hemodilution [104] are the most frequently cited means of iron metabolism disruption.
Current guidelines in Europe and the USA suggest seeking and treating ID in patients with HF [105]. Multiple RCTs have shown that intravenous administration of carbohydrate-bound Fe results in improved quality of life and effort capacity and decreased hospitalization and symptoms [90]. Additionally, other organs such as the skeletal muscles seem to account for beneficial effects [106] of iron replenishment via an increase in cellular energy generation, as shown by current studies concerning HF patients [107, 108], again emphasizing the mitochondrial importance in iron metabolism. It is important to highlight that the decision to administer iron formulations is based on systemic (i.e., serum) iron parameters, using the Ganzoni formula [91]. Even though these trials were not powered for primary cardiovascular endpoints and survival duration, post-hoc analyses were able to infer that there is a potential benefit from IV iron replenishment regimens [99], with other RCTs currently underway to clarify such a vital issue [99]. However, another meta-analysis [90], which studied the effects of intravenous iron to patients with systolic heart failure, has failed to show any correlation between mortality and iron treatment.
Decreasing iron
Current treatment of hemochromatosis consists mainly of venesection, despite a lack of RCT to confirm the benefit. The European Association for the Study of the Liver recommends a target of serum Ft below 50 μg/L reached by regular phlebotomies started when the aforesaid parameter is above normal range [109]. However, in certain pathologies, such as hemoglobinopathies, this practice proves unfeasible. Chelation therapy has been made available in the last decades, and as such, three compounds are approved for therapy: deferoxamine, deferiprone (DFP), and deferasirox [110]. In patients with cardiac siderosis and β-thalassemia, DFP proved significantly superior to deferoxamine in regard to iron quantitation by T2* MRI and ventricular ejection fraction, while deferasirox compared with deferoxamine demonstrated non-inferiority [111]. Due to its lipophilic nature, DFP seems to be the only approved agent that is a mitochondria permeant chelator, ferrying intracellular and intramitochondrial iron [112]. In more recent studies, other xenobiotic chelators have shown increased efficacy. Eltrombopag, a thrombopoietin receptor agonist exhibited more potent chelation activity of cardiomyocytes in vitro than the licensed alternatives, along with reduced ROS formation [113]. Similarly, dexrazoxane, a drug approved for treatment of anthracycline cardiotoxicity, decreased cellular and organellar levels of iron [89, 114], as it seems reasonable that anthracyclines mediate cardiac dysfunction through increased mitochondrial iron deposition [115].
Effective iron release can occur, from a teleological point of view, in two ways: blocking the influx and removal of iron (i.e., chelation) from the labile iron pool. As it appears plausible in iron overload diseases (hemochromatosis, β-thalassemia) to inhibit cellular Fe ingress, blockers of iron transport (such as calcium channel blockers) [61, 62, 116] and of mitochondrial iron ingress (mitoferrin inhibitors) [89, 117] have been used in these pathologies with improvement in cardiac function, reduction of ROS, and cardioprotective effects. A series of studies have been employed to determine the efficiency of iron exclusion in managing iron disequilibrium present in cardiovascular disease (Table 2). The rationale behind iron removal is based on the increased formation of ROS (i.e., hydroxyl radicals), radical effects of iron itself, increased calcium uptake via LTCC [27, 60], and ferroptosis [24], facilitated by increased levels of iron within the mitochondria [119, 124,125,126]. Additionally, the low Fe export capacity of the mitochondria makes them more susceptible to the toxic effects of iron accumulation. The only known Fe2+/3+ transporter protein, the ATP-binding cassette B8 (ABCB8), is essential in mitochondrial ferrequilibrium, as indicated by the development of cardiomyopathy in the case of ABCB8 deletion [125]. Oxidative phosphorylation in the mitochondrion is the main site of ROS production [127]. With increased iron load, the consequent lipid peroxidation and organelle damage seem to be mediated by the augmentation of combined oxidative stress and unbound Fe [128].
ABCB8, ATP-binding cassette B8; Ang II, angiotensin II; EDTA, ethylenediaminetetraacetic acid; EF, ejection fraction; Ft, ferritin; HF, heart failure; HO-1, heme-oxygenase 1; I/R, ischemia/reperfusion; ROS, reactive oxygen species; Tf, transferrin
Conclusion
The iron metabolism issue in chronic inflammatory conditions is emerging as an important therapeutic and research subject, with a particular interest in the field of cardiovascular disease, cancer [129], and neurodegenerative disorders. As advancements in the understanding of ferrequilibrium unveil, theories regarding a compelling explanation in regard to contrasting findings of iron balance must accommodate both an increase and a decrease of iron availability (Fig. 3). Given the paucity of intramitochondrial iron levels quantifications and its effects in HF cardiomyocytes and an encouraging clinical response in patients receiving IV iron supplementation, it can be difficult to ascertain the magnitude of each end of the spectrum. Of note, the majority of the beneficial effects of iron chelation come from the use of DFP. Given the ability of shuttling iron between cellular compartments rather than simply eliminating the metal, it could prove one of the links of the high-low iron conundrum.

Iron disequilibrium model in heart disease. As compared with the physiologic modulation of iron metabolism, the pathologic shift and loss of bound and unbound iron from the circulatory system into the subcellular compartments cause increased formation of reactive oxygen species and even ferroptosis
Certainly, a few questions arise in view of the recent discrepancies that require addressing in future research endeavors: (1) What is the role of ferritins in HF? (2) Is the canonical transferrin-bound iron or other iron transport mechanisms that best explain the altered ferrequilibrium in the heart? (3) Are the current systemic markers for ID reliable and beneficial to direct iron replenishment therapies in HF patients? (4) What is the weighted effect of iron supplementation with respect to the heart versus other iron-deficient organs? and (5) Is iron treatment beneficial to ischemia/reperfusion caused HF?
It appears that even though a consistent proportion of patients with HF have a decreased systemic iron balance, recent developments have raised the issue of coexistent increased intracellular and intraorganellar iron levels, challenging the status quo of hypoferremia currently used in the clinical setting. While the systemic deficit is the main rationale behind IV iron supplementation in HF with proven symptomatic improvements, it may be that the increased mitochondrial and cardiomyocyte Fe deposits will benefit from chelation, removing the ROS generation substrate and alleviating the cells` oxidative stress and overall bioenergetic profile.
References
Camprubi E, Jordan SF, Vasiliadou R, Lane N (2017) Iron catalysis at the origin of life. IUBMB Life 69(6):373–381
Hohenberger J, Ray K, Meyer K (2012) The biology and chemistry of high-valent iron-oxo and iron-nitrido complexes. Nat Commun 3:720
Rouault TA (2015) Mammalian iron-sulphur proteins: novel insights into biogenesis and function. Nat Rev Mol Cell Biol [Internet] 16(1):45–55. https://doi.org/10.1038/nrm3909
Weiss G (2002) Iron and immunity: a double-edged sword. Eur J Clin Investig 32(SUPPL. 1):70–78
Nemeth E, Ganz T (2009) The role of hepcidin in iron metabolism. Acta Haematol 122(2–3):78–86
Anderson CP, Shen M, Eisenstein RS, Leibold EA (2012) Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta, Mol Cell Res 1823:1468–1483
Anderson SA, Nizzi CP, Chang YI, Deck KM, Schmidt PJ, Galy B, Damnernsawad A, Broman AT, Kendziorski C, Hentze MW, Fleming MD, Zhang J, Eisenstein RS (2013) The IRP1-HIF-2α axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab [Internet] 17(2):282–290. https://doi.org/10.1016/j.cmet.2013.01.007
Lakhal-Littleton S, Wolna M, Chung YJ, Christian HC, Heather LC, Brescia M et al (2016) An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. Elife. 5:1–25
Cohen-Solal A, Leclercq C, Deray G, Lasocki S, Zambrowski JJ, Mebazaa A, de Groote P, Damy T, Galinier M (2014) Iron deficiency: an emerging therapeutic target in heart failure. Heart Int 100(18):1414–1420. Available from. https://doi.org/10.1136/heartjnl-2014-305669
Kasztura M, Dziegała M, Kobak K, Bania J, Mazur G, Banasiak W et al (2017) Both iron excess and iron depletion impair viability of rat H9C2 cardiomyocytes and L6G8C5 myocytes. Kardiol Pol 75(3):267–275
McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E et al (2001) An iron-regulated ferric reductase associated with the absorption of dietary iron. Science (80- ) 291(5509):1755–1759
Conrad ME, Umbreit JN, Moore EG (1993) Regulation of iron absorption: proteins involved in duodenal mucosal uptake and transport. J Am Coll Nutr [Internet] 12(6):720–728. https://doi.org/10.1080/07315724.1993.10718365
Le Blanc S, Garrick MD, Arredondo M (2012) Heme carrier protein 1 transports heme and is involved in heme-Fe metabolism. AJP Cell Physiol [Internet] 302(12):C1780–C1785. Available from:. https://doi.org/10.1152/ajpcell.00080.2012
Hooda J, Shah A, Zhang L (2014) Heme, an essential nutrient from dietary proteins, critically impacts diverse physiological and pathological processes. Nutrients. 6(3):1080–1102
Staron R, Lipinski P, Lenartowicz M, Bednarz A, Gajowiak A, Smuda E et al (2017) Dietary hemoglobin rescues young piglets from severe iron deficiency anemia: duodenal expression profile of genes involved in heme iron absorption. PLoS One 12(7):1–22
Donovan A, Brownlie A, Zhou Y, Shepard J (2000) Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature [Internet] 403(6771):776–781. https://doi.org/10.1038/35001596
Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ (1999) Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 21(2):195–199
Ponka P, Lok CN (1999) The transferrin receptor: role in health and disease. Int J Biochem Cell Biol 31:1111–1137
Hoffbrand AV, Catovsky D, Tuddenham EGD, Green AR. Postgraduate haematology: sixth edition. 2010
Girelli D, Bozzini C, Roetto A, Alberti F, Daraio F, Colombari R, Olivieri O, Corrocher R, Camaschella C (2002) Clinical and pathologic findings in hemochromatosis type 3 due to a novel mutation in transferrin receptor 2 gene. Gastroenterology. 122(5):1295–1302
Miles AL, Burr SP, Grice GL, Nathan JA (2017) The vacuolar-ATPase complex and assembly factors, TMEM199 and CCDC115, control HIF1α prolyl hydroxylation by regulating cellular Iron levels. Elife. 6:e22693
Jenkitkasemwong S, Wang CY, MacKenzie B, Knutson MD (2012) Physiologic implications of metal-ion transport by ZIP14 and ZIP8. BioMetals. 25(4):643–655
Jenkitkasemwong S, Wang CY, Coffey R, Zhang W, Chan A, Biel T, Kim JS, Hojyo S, Fukada T, Knutson MD (2015) SLC39A14 is required for the development of hepatocellular iron overload in murine models of hereditary hemochromatosis. Cell Metab 22(1):138–150
Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y (2016) Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci [Internet] 41(3):274–286. https://doi.org/10.1016/j.tibs.2015.11.012
Wang CY, Jenkitkasemwong S, Duarte S, Sparkman BK, Shawki A, Mackenzie B, Knutson MD (2012) ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J Biol Chem 287(41):34032–34043
Philpott CC, Ryu MS, Frey A, Patel S (2017) Cytosolic iron chaperones: proteins delivering iron cofactors in the cytosol of mammalian cells. J Biol Chem 292(31):12764–12771
Coffey R, Ganz T (2017) Iron homeostasis: an anthropocentric perspective. J Biol Chem 292(31):12727–12734
Chaudhury A, Chander P, Howe PH (2010) Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: focus on hnRNP E1’s multifunctional regulatory roles. Rna 16:1449–1462
Bresgen N, Eckl PM (2015) Oxidative stress and the homeodynamics of iron metabolism. Biomolecules. 5(2):808–847
Theil EC. Ferritin iron minerals are chelator targets, antioxidants, and coated, dietary iron. In: Annals of the New York Academy of Sciences. 2010. p. 197–204
Zhu W, Li X, Xie W, Luo F, Kaur D, Andersen JK, Jankovic J, le W (2010) Genetic iron chelation protects against proteasome inhibition-induced dopamine neuron degeneration. Neurobiol Dis 37(2):307–313
Lill R, Dutkiewicz R, Freibert SA, Heidenreich T, Mascarenhas J, Netz DJ, Paul VD, Pierik AJ, Richter N, Stümpfig M, Srinivasan V, Stehling O, Mühlenhoff U (2015) The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron-sulfur proteins. Eur J Cell Biol 94(7–9):280–291
Hardison RC (1996) A brief history of hemoglobins: plant, animal, protist, and bacteria. Proc Natl Acad Sci [Internet] 93(12):5675–5679. Available from. https://doi.org/10.1073/pnas.93.12.5675
Wallace DF (2016) The regulation of iron absorption and homeostasis. Clin Biochem Rev [Internet] 37(2):51–62 Available from: http://www.ncbi.nlm.nih.gov/pubmed/28303071%0A http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC5198508%0A
Krause A, Neitz S, Mägert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, Adermann K (2000) LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett 480(2–3):147–150
Knutson MD (2017) Iron transport proteins: gateways of cellular and systemic iron homeostasis. J Biol Chem 292(31):12735–12743
Brasselagnel C, Karim Z, Letteron P, Bekri S, Bado A, Beaumont C (2011) Intestinal DMT1 cotransporter is down-regulated by hepcidin via proteasome internalization and degradation. Gastroenterol Int 140(4):1261–1271. https://doi.org/10.1053/j.gastro.2010.12.037
Mena NP, Esparza A, Tapia V, Valdés P, Núñez MT (2008) Hepcidin inhibits apical iron uptake in intestinal cells. Am J Physiol Liver Physiol [Internet] 294(1):G192–G198. Available from:. https://doi.org/10.1152/ajpgi.00122.2007
Lymboussaki A, Pignatti E, Montosi G, Garuti C, Haile DJ, Pietrangelo A (2003) The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. J Hepatol 39(5):710–715
Auguet T, Aragonès G, Berlanga A, Martínez S, Sabench F, Binetti J, Aguilar C, Porras JA, Molina A, del Castillo D, Richart C (2017) Hepcidin in morbidly obese women with nonalcoholic fatty liver disease. PLoS One 12(10):e0187065
Hentze MW, Muckenthaler MU, Galy B, Camaschella C (2010) Two to tango: regulation of mammalian iron metabolism. Cell. 142(1):24–38
Rishi G, Wallace DF, Subramaniam VN (2015) Hepcidin: regulation of the master iron regulator. Biosci Rep [Internet] 35(3):1–12. Available from:. https://doi.org/10.1042/BSR20150014
Papanikolaou G, Pantopoulos K (2017) Systemic iron homeostasis and erythropoiesis. IUBMB Life 69(6):399–413
Yamamoto K, Kuragano T, Kimura T, Nanami M, Hasuike Y, Nakanishi T (2018) Interplay of adipocyte and hepatocyte: leptin upregulates hepcidin. Biochem Biophys Res Commun 495(1):1548–1554
Zabeau L, Peelman F, Tavernier J (2015) Leptin: from structural insights to the design of antagonists. Life Sci 140:49–56
Schmidt PJ (2015) Regulation of iron metabolism by hepcidin under conditions of inflammation. J Biol Chem 290(31):18975–18983
Cavallaro F, Duca L, Pisani LF, Rigolini R, Spina L, Tontini GE et al (2017) Anti-TNF-mediated modulation of prohepcidin improves iron availability in inflammatory bowel disease, in an IL-6-mediated fashion. Can J Gastroenterol Hepatol 2017
Lee P, Peng H, Gelbart T, Wang L, Beutler E (2005) Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc Natl Acad Sci U S A [Internet] 102(6):1906–1910 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=548537&tool=pmcentrez&rendertype=abstract
Ganz T (2018) Iron and infection. Int J Hematol 107(1):7–15
Liu Q, Davidoff O, Niss K, Haase VH (2012) Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J Clin Invest 122(12):4635–4644
Sheftel AD, Lill R (2009) The power plant of the cell is also a smithy: the emerging role of mitochondria in cellular iron homeostasis. Ann Med 41:82–99
Shah YM, Xie L (2014) Hypoxia-inducible factors link iron homeostasis and erythropoiesis. Gastroenterol Int 146(3):630–642. https://doi.org/10.1053/j.gastro.2013.12.031
Haddad S, Wang Y, Galy B, Korf-Klingebiel M, Hirsch V, Baru AM et al (2017) Iron-regulatory proteins secure iron availability in cardiomyocytes to prevent heart failure. Eur Heart J 38(5):362–372
Galy B, Ferring-Appel D, Sauer SW, Kaden S, Lyoumi S, Puy H, Kölker S, Gröne HJ, Hentze MW (2010) Iron regulatory proteins secure mitochondrial iron sufficiency and function. Cell Metab 12(2):194–201
Zhao G, Arosio P, Chasteen ND (2006) Iron(II) and hydrogen peroxide detoxification by human H-chain ferritin. An EPR spin-trapping study. Biochemistry. 45(10):3429–3436
Miller LL, Miller SC, Torti SV, Tsuji Y, Torti FM (1991) Iron-independent induction of ferritin H chain by tumor necrosis factor. Proc Natl Acad Sci U S A [Internet] 88(11):4946–4950 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=51784&tool=pmcentrez&rendertype=abstract
Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, de Smaele E, Cong R, Beaumont C, Torti FM, Torti SV, Franzoso G (2004) Ferritin heavy chain upregulation by NF-κB inhibits TNFα-induced apoptosis by suppressing reactive oxygen species. Cell. 119(4):529–542
Mackenzie EL, Iwasaki K, Tsuji Y (2008) Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid Redox Signal [Internet] 10(6):997–1030. Available from. https://doi.org/10.1089/ars.2007.1893
Bond RC, Bryant SM, Watson JJ, Hancox JC, Orchard CH, James AF (2017) Reduced density and altered regulation of rat atrial L-type ca 2+ current in heart failure. Am J Physiol - Hear Circ Physiol [Internet] 312(3):H384–H391. Available from. https://doi.org/10.1152/ajpheart.00528.2016
Oudit GY, Sun H, Trivieri MG, Koch SE, Dawood F, Ackerley C, Yazdanpanah M, Wilson GJ, Schwartz A, Liu PP, Backx PH (2003) L-type Ca2+channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat Med 9(9):1187–1194
Ludwiczek S, Theurl I, Muckenthaler MU, Jakab M, Mair SM, Theurl M, Kiss J, Paulmichl M, Hentze MW, Ritter M, Weiss G (2007) Ca2+ channel blockers reverse iron overload by a new mechanism via divalent metal transporter-1. Nat Med [Internet] 13(4):448–454 Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17293870
Kumfu S, Chattipakorn S, Chinda K, Fucharoen S, Chattipakorn N (2012) T-type calcium channel blockade improves survival and cardiovascular function in thalassemic mice. Eur J Haematol 88(6):535–548
Nam H, Wang CY, Zhang L, Zhang W, Hojyo S, Fukada T, Knutson MD (2013) ZIP14 and DMT1 in the liver, pancreas, and heart are differentially regulated by iron deficiency and overload: implications for tissue iron uptake in iron-related disorders. Haematologica. 98(7):1049–1057
Omiya S, Hikoso S, Imanishi Y, Saito A, Yamaguchi O, Takeda T, Mizote I, Oka T, Taneike M, Nakano Y, Matsumura Y, Nishida K, Sawa Y, Hori M, Otsu K (2009) Downregulation of ferritin heavy chain increases labile iron pool, oxidative stress and cell death in cardiomyocytes. J Mol Cell Cardiol [Internet] 46(1):59–66. https://doi.org/10.1016/j.yjmcc.2008.09.714
Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, Gwynn B, Lambert AJ, Wingert RA, Traver D, Trede NS, Barut BA, Zhou Y, Minet E, Donovan A, Brownlie A, Balzan R, Weiss MJ, Peters LL, Kaplan J, Zon LI, Paw BH (2006) Mitoferrin is essential for erythroid iron assimilation. Nature. 440(7080):96–100
Nie G, Sheftel AD, Kim SF, Ponka P (2005) Overexpression of mitochondrial ferritin causes cytosolic iron depletion and changes cellular iron homeostasis. Blood. 105(5):2161–2167
Crielaard BJ, Lammers T, Rivella S (2017) Targeting iron metabolism in drug discovery and delivery. Nat Rev Drug Discov [Internet] 16(6):400–423. https://doi.org/10.1038/nrd.2016.248
Filippatos G, Farmakis D, Colet JC, Dickstein K, Lüscher TF, Willenheimer R et al (2013) Intravenous ferric carboxymaltose in iron-deficient chronic heart failure patients with and without anaemia: a subanalysis of the FAIR-HF trial. Eur J Heart Fail 15:1267–1276
Jankowska EA, Malyszko J, Ardehali H, Koc-Zorawska E, Banasiak W, Von Haehling S et al (2013) Iron status in patients with chronic heart failure. Eur Heart J 34(11):827–834
Pagani A, Nai A, Corna G, Bosurgi L, Rovere-Querini P, Camaschella C, Silvestri L (2011) Low hepcidin accounts for the proinflammatory status associated with iron deficiency. Blood. 118(3):736–746
Petrak J, Havlenova T, Krijt M, Behounek M, Franekova J, Cervenka L, Pluhacek T, Vyoral D, Melenovsky V (2019) Myocardial iron homeostasis and hepcidin expression in a rat model of heart failure at different levels of dietary iron intake. Biochim Biophys Acta, Gen Subj 1863(4):703–713
Shirazi LF, Bissett J, Romeo F, Mehta JL (2017) Role of inflammation in heart failure. Curr Atheroscler Rep 19(6)
Melenovsky V, Petrak J, Mracek T, Benes J, Borlaug BA, Nuskova H, Pluhacek T, Spatenka J, Kovalcikova J, Drahota Z, Kautzner J, Pirk J, Houstek J (2017) Myocardial iron content and mitochondrial function in human heart failure: a direct tissue analysis. Eur J Heart Fail [Internet] 19(4):522–530. Available from. https://doi.org/10.1002/ejhf.640
Hoes MF, Grote Beverborg N, Kijlstra JD, Kuipers J, Swinkels DW, Giepmans BNG, Rodenburg RJ, van Veldhuisen DJ, de Boer RA, van der Meer P (2018) Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur J Heart Fail 20(5):910–919
Naito Y, Sawada H, Oboshi M, Okuno K, Yasumura S, Okuhara Y, Eguchi A, Nishimura K, Soyama Y, Asakura M, Ishihara M, Tsujino T, Masuyama T (2017) Altered expression of intestinal duodenal cytochrome b and divalent metal transporter 1 might be associated with cardio-renal anemia syndrome. Heart Vessel 32(11):1410–1414
Lakhal-Littleton S, Robbins PA (2017) The interplay between iron and oxygen homeostasis with a particular focus on the heart. J Appl Physiol [Internet] 123(4):967–973. Available from. https://doi.org/10.1152/japplphysiol.00237.2017
Boddaert N, Sang KHLQ, Rötig A, Leroy-Willig A, Gallet S, Brunelle F et al (2007) Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood. 110(1):401–408
Sawicki KT, Shang M, Wu R, Chang HC, Khechaduri A, Sato T, Kamide C, Liu T, Naga Prasad SV, Ardehali H (2015) Increased heme levels in the heart lead to exacerbated ischemic injury. J Am Heart Assoc 4(8):e002272
Chang H-C, Wu R, Shang M, Sato T, Chen C, Shapiro JS, Liu T, Thakur A, Sawicki KT, Prasad SV, Ardehali H (2016) Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol Med [Internet] 8(3):247–267. Available from. https://doi.org/10.15252/emmm.201505748
Zou C, Liu X, Xie R, Bao Y, Jin Q, Jia X, Li L, Liu R (2017) Deferiprone attenuates inflammation and myocardial fibrosis in diabetic cardiomyopathy rats. Biochem Biophys Res Commun 486(4):930–936
Chang HC, Shapiro JS, Ardehali H (2016) Getting to the “heart” of cardiac disease by decreasing mitochondrial iron. Circ Res 119:1164–1166
Klip IT, Comin-Colet J, Voors AA, Ponikowski P, Enjuanes C, Banasiak W et al (2013) Iron deficiency in chronic heart failure: an international pooled analysis. Am Heart J [Internet] 165(4):575–582.e3. https://doi.org/10.1016/j.ahj.2013.01.017
Khechaduri A, Bayeva M, Chang H-C, Ardehali H (2013) Heme levels are increased in human failing hearts. J Am Coll Cardiol [Internet] 61(18):1884–1893 Available from: http://www.ncbi.nlm.nih.gov/pubmed/21959306%5Cn http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3739715/
Kali A, Kumar A, Cokic I, Tang RLQ, Tsaftaris SA, Friedrich MG, Dharmakumar R (2013) Chronic manifestation of postreperfusion intramyocardial hemorrhage as regional iron deposition: a cardiovascular magnetic resonance study with ex vivo validation. Circ Cardiovasc Imaging 6(2):218–228
Carberry J, Carrick D, Haig C, Ahmed N, Mordi I, McEntegart M, et al. Persistent iron within the infarct core after ST-segment elevation myocardial infarction. Implications for left ventricular remodeling and health outcomes. JACC Cardiovasc Imaging. 2017;
Simonis G, Mueller K, Schwarz P, Wiedemann S, Adler G, Strasser RH, Kulaksiz H (2010) The iron-regulatory peptide hepcidin is upregulated in the ischemic and in the remote myocardium after myocardial infarction. Peptides. 31(9):1786–1790
Lakhal-Littleton S.(2018) Mechanisms of cardiac iron homeostasis and their importance to heart function. Free Radic Biol Med [Internet]. ; Available from: https://linkinghub.elsevier.com/retrieve/pii/S0891584918313868
Jiri P, Tereza H, Matyas K, Matej B, Franekova J, Ludek C et al (2019) Myocardial iron homeostasis and hepcidin expression in a rat model of heart failure at different levels of dietary iron intake. Biochim Biophys Acta - Gen Subj [Internet]. https://doi.org/10.1016/j.bbagen.2019.01.010
Fang X, Wang H, Han D, Xie E, Yang X, Wei J et al (2019) Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci [Internet] 24:201821022 Available from: https://doi.org/10.1073/pnas.1821022116
Jankowska EA, Tkaczyszyn M, Suchocki T, Drozd M, von Haehling S, Doehner W, Banasiak W, Filippatos G, Anker SD, Ponikowski P (2016) Effects of intravenous iron therapy in iron-deficient patients with systolic heart failure: a meta-analysis of randomized controlled trials. Eur J Heart Fail [Internet] 18(7):786–795. Available from. https://doi.org/10.1002/ejhf.473
Anker SD, Comin Colet J, Filippatos G, Willenheimer R, Dickstein K, Drexler H, Lüscher TF, Bart B, Banasiak W, Niegowska J, Kirwan BA, Mori C, von Eisenhart Rothe B, Pocock SJ, Poole-Wilson PA, Ponikowski P, FAIR-HF Trial Investigators (2009) Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med [Internet] 361(25):2436–2448. Available from. https://doi.org/10.1056/NEJMoa0908355
Cappellini MD, Comin-Colet J, de Francisco A, Dignass A, Doehner W, Lam CSP et al (2017) Iron deficiency across chronic inflammatory conditions: international expert opinion on definition, diagnosis, and management. Am J Hematol 92(10):1068–1078
Beverborg NG, Klip IJT, Meijers WC, Voors AA, Vegter EL, Van Der Wal HH et al (2018) Definition of iron deficiency based on the gold standard of bone marrow iron staining in heart failure patients. Circ Heart Fail 11(2)
Cleland JGF, Zhang J, Pellicori P, Dicken B, Dierckx R, Shoaib A, Wong K, Rigby A, Goode K, Clark AL (2016) Prevalence and outcomes of anemia and hematinic deficiencies in patients with chronic heart failure. JAMA Cardiol 1(5):539–547
Adams PC (2015) Epidemiology and diagnostic testing for hemochromatosis and iron overload. Int J Lab Hematol 37(S1):25–30
Salerno M, Sharif B, Arheden H, Kumar A, Axel L, Li D et al (2017) Recent advances in cardiovascular magnetic resonance. Circ Cardiovasc Imaging 10(6)
Theresa MIC (2015) Iron therapy for the treatment of iron deficiency in chronic heart failure: intravenous or oral? Eur J Heart Fail 17(3):248–262
Rognoni C, Venturini S, Meregaglia M, Marmifero M, Tarricone R (2016) Efficacy and safety of ferric carboxymaltose and other formulations in iron-deficient patients: a systematic review and network meta-analysis of randomised controlled trials. Clinical Drug Investigation 36:177–194
Pope M, Kalra PR (2018) Iron deficiency in heart failure: to treat or not to treat? Curr Treat Options Cardiovasc Med [Internet] 20(8):65. Available from. https://doi.org/10.1007/s11936-018-0661-8
Hughes CM, Woodside JV, McGartland C, Roberts MJ, Nicholls DP, McKeown PP (2012) Nutritional intake and oxidative stress in chronic heart failure. Nutr Metab Cardiovasc Dis 22(4):376–382
Jankowska EA, Von Haehling S, Anker SD, MacDougall IC, Ponikowski P (2013) Iron deficiency and heart failure: diagnostic dilemmas and therapeutic perspectives. Eur Heart J 34(11):816–829
Shah R, Agarwal AK (2013) Anemia associated with chronic heart failure: current concepts. Clin Interv Aging 8:111–122
Opasich C, Cazzola M, Scelsi L, De Feo S, Bosimini E, Lagioia R et al (2005) Blunted erythropoietin production and defective iron supply for erythropoiesis as major causes of anaemia in patients with chronic heart failure. Eur Heart J 26(21):2232–2237
Alexandrakis MG, Tsirakis G (2012) Anemia in heart failure patients. ISRN Hematol [Internet] 2012:1–9 Available from: http://www.hindawi.com/journals/isrn/2012/246915/
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS et al (2016) ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 37(27):2129–2200m
Stugiewicz M, Tkaczyszyn M, Kasztura M, Banasiak W, Ponikowski P, Jankowska EA (2016) The influence of iron deficiency on the functioning of skeletal muscles: experimental evidence and clinical implications. Eur J Heart Fail 18(7):762–773
Melenovsky V, Hlavata K, Sedivy P, Dezortova M, Borlaug BA, Petrak J, Kautzner J, Hajek M (2018) Skeletal muscle abnormalities and iron deficiency in chronic heart failure. An exercise 31P magnetic resonance spectroscopy study of calf muscle. Circ Heart Fail 11(9):e004800
Charles-Edwards G, Amaral N, Sleigh A, Ayis S, Catibog N, Mcdonagh T et al (2019) Effect of iron isomaltoside on skeletal muscle energetics in patients with chronic heart failure and iron deficiency : the FERRIC-HF II randomized mechanistic trial. Circulation 44(0)
European Association for the Study of the Liver (2010) EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol [Internet] 53(1):3–22. https://doi.org/10.1016/j.jhep.2010.03.001
Brissot P (2016) Optimizing the diagnosis and the treatment of iron overload diseases. Expert Rev Gastroenterol Hepatol 10(3):359–370
Baksi AJ, Pennell DJ.(2014) Randomised controlled trials of iron chelators for the treatment of cardiac siderosis in thalassaemia major. Front Pharmacol. 5(Sep)
Sohn YS, Breuer W, Munnich A, Cabantchik ZI (2008) Redistribution of accumulated cell iron: a modality of chelation with therapeutic implications. Blood. 111(3):1690–1699
Vlachodimitropoulou E, Chen YL, Garbowski M, Koonyosying P, Psaila B, Sola-Visner M, Cooper N, Hider R, Porter J (2017) Eltrombopag: a powerful chelator of cellular or extracellular iron(III) alone or combined with a second chelator. Blood. 130(17):1923–1933
Xu X, Sutak R, Richardson DR (2007) Iron chelation by clinically relevant anthracyclines: alteration in expression of iron-regulated genes and atypical changes in intracellular iron distribution and trafficking. Mol Pharmacol 73:833–844
Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV et al (2014) Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest 124(2):617–630
Fernandes JL, Sampaio EF, Fertrin K, Coelho OR, Loggetto S, Piga A, Verissimo M, Saad ST (2013) Amlodipine reduces cardiac iron overload in patients with thalassemia major: a pilot trial. Am J Med [Internet] 126(9):834–837. https://doi.org/10.1016/j.amjmed.2013.05.002
Kumfu S, Chattipakorn S, Fucharoen S, Chattipakorn N (2012) Mitochondrial calcium uniporter blocker prevents cardiac mitochondrial dysfunction induced by iron overload in thalassemic mice. BioMetals. 25(6):1167–1175
Ishizaka N, Saito K, Mori I, Matsuzaki G, Ohno M, Nagai R (2005) Iron chelation suppresses ferritin upregulation and attenuates vascular dysfunction in the aorta of angiotensin II-infused rats. Arterioscler Thromb Vasc Biol 25(11):2282–2288
Zhang X, Lemastersn JJ (2013) Translocation of iron from lysosomes to mitochondria during ischemia predisposes to injury after reperfusion in rat hepatocytes. Free Radic Biol Med 63:243–253
Alpendurada F, Smith GC, Carpenter JP, Nair S V., Tanner MA, Banya W, et al.(2012) Effects of combined deferiprone with deferoxamine on right ventricular function in thalassaemia major. J Cardiovasc Magn Reson.
Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, Lindblad L, Lewis EF, Drisko J, Lee KL, TACT Investigators (2013) Effect of disodium EDTA chelation regimen on cardiovascular events in patients with previous myocardial infarction: the TACT randomized trial. JAMA - J Am Med Assoc 309(12):1241–1250
Pennell DJ, Porter JB, Piga A, Lai Y, El-Beshlawy A, Belhoul KM et al (2014) A 1-year randomized controlled trial of deferasirox vs deferoxamine for myocardial iron removal in β-thalassemia major (CORDELIA). Blood. 123:1447–1454
Pepe A, Lombardi M, Positano V, Cracolici E, Capra M, Malizia R, Prossomariti L, Marchi D, Midiri M, Maggio A (2006) Evaluation of the efficacy of oral deferiprone in β-thalassemia major by multislice multiecho T2. Eur J Haematol 76(3):183–192
Bayeva M, Gheorghiade M, Ardehali H (2013) Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol [Internet] 61(6):599–610. https://doi.org/10.1016/j.jacc.2012.08.1021
Ichikawa Y, Bayeva M, Ghanefar M, Potini V, Sun L, Mutharasan RK, Wu R, Khechaduri A, Jairaj Naik T, Ardehali H (2012) Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc Natl Acad Sci [Internet] 109(11):4152–4157. Available from. https://doi.org/10.1073/pnas.1119338109
Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, Cleland JGF, Colucci WS, Butler J, Voors AA, Anker SD, Pitt B, Pieske B, Filippatos G, Greene SJ, Gheorghiade M (2017) Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol 14(4):238–250
Chen Y-R, Zweier JL (2014) Cardiac mitochondria and reactive oxygen species generation [Internet]. Circ Res 114:524–537. Available from. https://doi.org/10.1161/CIRCRESAHA.114.300559
Gammella E, Recalcati S, Rybinska I, Buratti P, Cairo G (2015) Iron-induced damage in cardiomyopathy: oxidative-dependent and independent mechanisms. Oxidative Med Cell Longev 2015:1–10
Lui GYL, Kovacevic Z, Richardson V, Merlot AM, Kalinowski DS, Richardson DR (2015) Targeting cancer by binding iron: dissecting cellular signaling pathways. Oncotarget [Internet] 6(22):18748–18779 Available from: http://www.oncotarget.com/fulltext/4349
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lupu, M., Tudor, DV. & Filip, G.A. Influence of mitochondrial and systemic iron levels in heart failure pathology. Heart Fail Rev 24, 647–659 (2019). https://doi.org/10.1007/s10741-019-09788-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-019-09788-z