
Abstract
Breast cancer is a frequent cancer among women. The current study investigated the biological functions of Nek2 in breast cancer and its possible mechanism. The mRNA expression of Nek2 in breast epithelial cells and eight breast cancer cell lines was detected by qRT-PCR. Silencing Nek2 was transfected into MDA-MB-231 and MCF7 cells to examine its roles in the viability, migration, invasion, cell colony, apoptosis and cell cycle of the breast cancer cells by performing CCK-8, wound scratch, Transwell, clone formation and flow cytometry assays, respectively. The expressions of related genes were detected using qRT-PCR and Western blot. MAPK pathway agonist IGF (insulin-like growth factor-1) was added into MDA-MB-231 and MCF7 cells and then cell viability was examined. Nek2 expression was frequently up-regulated in breast cancer cell lines, and silencing Nek2 significantly inhibited the viability, cell migration, invasion and clone formation, promoted cell apoptosis of MDA-MB-231 and MCF7 cells, and arrested cell cycle in G0/G1 phase. Furthermore, knocking down Nek2 decreased the mRNA and protein expressions of Bcl-2, CyclinB1 and CyclinD1, and increased Bax and p27 expressions. Moreover, knocking down Nek2 inhibited the phosphorylation of ERK and p38, and almost completely reversed the expression of p-ERK increased by IGF, but Nek2 knockdown had no obvious effect on p-p38. The inhibitory effect of Nek2 silencing on the cell viability was mainly realized by the inhibition of ERK/MAPK signaling. Nek2 plays an important role in the regulation of the progression of breast cancer in vitro probably through regulating the ERK/MAPK signaling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer is a frequent cancer among women and ranks second among the causes for the cancer-related death in women (Fahad Ullah 2019). According to the estimation by the American Cancer Society, there will be 276,480 cases of breast cancer and 42,170 cases of death in patients with breast cancer in 2020 in the US (Siegel et al. 2020). According to the degree of diffusion, breast cancer can be classified into orthotopic breast cancer and invasive breast cancer (Cedolini et al. 2014; Ward et al. 2015), the former of which has two main types, namely, Ductal Carcinoma In Situ (DCIS) and Lobular Carcinoma In Situ (LCIS) (Badve and Gökmen-Polar 2019; Wen and Brogi 2018). Additionally, breast cancer can also be classified into the four main intrinsic molecular subtypes, such as Luminal A, Luminal B, HER2-enriched, and TNBC (Triple Negative Breast Cancer) (Fisusi and Akala 2019; Tsang and Tse 2020). At present, mastectomy is widely applied in the treatment for breast cancer (Zehra et al. 2020). However, the metastasis remains as the major obstacle in the management of breast cancer, and metastasis in the advanced stages of breast cancer could decrease the survival of patients to approximately 5 years (Peart 2017; Tian et al. 2017). Early detection and early diagnosis are the keys to the treatment of breast cancer, with the focus from cellular pathology to molecular pathology (Lorenzoni 2019). The application of molecular biological techniques contributes to the discovery of some oncogenes in breast cancer, which facilitates the diagnosis of early breast cancer (Liu et al. 2018; Suarez-Cabrera et al. 2018; Ye et al. 2018).
As the center of the microtubule tissue of human cells, the centrosome is closely related to the tumorigenesis. It has been showed that the damaged cells in the centrosome affect the movement of cell because they cannot accurately reposition the spindle of the body during the morphological changes of cells, thus hindering the smooth progress of cell cycle (Ye et al. 2005). The regulation of centrosomal kinases on the centrosome has been widely studied recently (Kazazian et al. 2017; Sdelci et al. 2012). The families of centrosomal kinases consist of Aurora protein kinase family, Polo-like Kinases (PLK) family, Cyclin-Dependent Kinase 5 Regulatory subunit-Associated protein 2 (CDK5RAP2) and Never in mitosis gene A (NIMA) Related Kinases family (Li and Li 2006). NIMA-related expressed kinase (NEK), which is a kind of NIMA-associated protein kinase in mammalian cells, is originally the cell cycle mutant NIMA discovered by Morris in his study of Aspergillus nidulans (Morris 1975). Nek2 has the highest homology with NIMA and is the most representative member of Neks family (Fry et al. 1995; Schultz et al. 1994). Nek2 plays an important role in cell mitosis. It was also found that the expression of Nek2 was increased in many tumors, such as prostate cancer, Ewing’s tumor, colorectal cancer, breast cancer, malignant peripheral neurilemmoma, lymphoma, ovarian cancer, and cholangiocarcinoma. Moreover, Nek2 expression is closely correlated with the degree of malignancy and the prognosis of the tumors (Kokuryo et al. 2016; Liu et al. 2019; Lu et al. 2015; Marina and Saavedra 2014; Zeng et al. 2015).
We aimed to explore the roles of Nek2 in the regulation of the progression of breast cancer and the possible mechanism.
Materials and methods
Cell culture
Normal breast epithelial cells (MCF-10A cells) and eight breast cancer cell lines (BT474, BT20, MCF7, Hs578T, MDA-MB-231, T47D, MDA-MB-453 and ZR75-1 cells) were purchased from American Type Culture Collection (ATCC, Manassas, USA). The cells were cultured in RPMI-1640 medium (Sigma, USA) containing 10% fetal bovine serum, 100 U/ml streptomycin and penicillin (Gibco, Thermo Fisher, USA) at 37 °C with 5% CO2.
After the cells adhered to the plate wall, 120 ng/ml IGF (IGF-1, PeproTech, Rocky Hill, NJ, USA) was added into medium to incubate with the cells. The medium with 120 ng/ml IGF was refreshed once a day.
Cell transfection
The cells were seeded in 6-well plates (1.0 × 105) for 24 h (h). Nek2 silencing sequence (siNEK2, Cat No.:143612) and negative control (NC, Cat No.:12935111) were synthesized by Invitrogen (USA). The transient transfection was performed using Lipofectamine 3000 (Invitrogen, USA) according to manufacturer’s protocol. siNek2 or NC and Lipofectamine 3000 were respectively added into Opti-MEM medium. Lipofectamine/silencing RNA mixtures were cultured at room temperature for 10 min (min) and then added into Opti-MEM medium. After culture for 6 h, the medium was replaced by RPMI 1640 medium containing 10% FBS.
Cell counting kit-8 (CCK-8)
MDA-MB-231 and MCF7 cells were respectively placed into 96-well plates at a density of 5 × 103 cells per well for 24 h. Then cells were administrated and incubated for 24, 48 or 72 h. 10 μl CCK-8 solution was subsequently added into each well and incubated the cells at 37 °C for another 1 h. The viability of cell was determined using a microplate reader (Thermo Fisher, USA) at 450 nm.
Wound Scratch assay and transwell assay
After the transfection, the cells (8 × 104) were seeded in 6-well plates at 37 °C overnight. A wound was drawn using the 100 μl sterile pipette tip in the center of the plates. The cells were gently washed with PBS 3 times, the suspended cells were then removed and the serum-free medium was added into the cells. Cell migration was observed under an inverted microscope at 48 h, and the scratch area was measured using Image J software. Migration rates were calculated as indicated: (distance 0 h − distance 48 h)siNEK2/(distance 0 h − distance 48 h)Control × 100%.
After the treatment with transfection and digestion, the cells were resuspended in serum-free medium and were then seeded at a density of 1 × 104 cells/well in the upper chamber coated with matrigel. In the lower 24-well chamber, RPMI1640 medium containing 10% fetal bovine serum was added and used to incubate with the cells for 60 h at 37 °C with 5% CO2. The cells in the lower chamber were fixed with 1% formaldehyde for 10 min at 25 °C and stained with 0.01% crystal violet for another 5 min. The invasion of cells was counted at 200 × magnification.
Plate clone formation assay
The cells were divided into control group, NC group (cells were transfected with negative control) and siNEK2 group (cells were transfected with siNek2 sequence), and respectively incubated in the dishes at the density of 3.0 × 102 cells/dish at 37 °C with 5% CO2. After culture for about 10 days, the cells were washed with PBS twice and stained with crystal violet for 15 min, followed by washing with deionized water. The number of colonies containing 50 cells was counted under a microscope.
Flow cytometry
The cells were digested with 0.25% trypsin and centrifuged at 2000 r/min for 5 min. Apoptosis assay was performed by Annexin V-FITC. The cells were washed twice using washing buffer, and the suspension was cultured with Annexin V-FITC and propidium iodide (PI, Cayman Chemical, Canada) in the dark at 25 °C for 20 min. The binding buffer was subsequently added into each well, and the samples were subjected to the detection with flow cytometry within 1 h.
After transfection for 48 h, the cells were washed twice with PBS and fixed in ethanol at 4 °C for 30 min. After being centrifuged at 1000 r/min for 5 min, the cells were washed and resuspended in PBS with RNase and PI at 37 °C for 30 min.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Trizol regent (Invitrogen, USA) was used to extract RNAs from the cultured cells according to the manufacturer’s protocol. GoScript™ Reverse Transcription kit (Promega, USA) was used to conduct the reverse transcription of RNAs into cDNAs. The qRT-PCR was performed by SYBR Fast qPCR Mix (Invitrogen, USA) for detecting the expressions of Nek2, Bcl-2, Bax, Cyclin B1, Cyclin D1 and p27. The samples were run under the following cycling parameters: Nek2, 95 °C for 10 min, 95 °C for 15 s (sec), followed by 40 cycles of 53 °C for 30 s and 72 °C for 30 s; Bcl-2, 94 °C for 5 min, 94 °C for 30 s, followed by 35 cycles of 55.9 °C for 30 s and 72 °C for 59 s; Bax, 94 °C for 5 min, 94 °C for 30 s, followed by 35 cycles of 50 °C for 30 s and 72 °C for 59 s; Cyclin B1, Cyclin D1 and p27, 95 °C for 5 min, 94 °C for 30 s, followed by 40 cycles of 54 °C for 30 s and 72 °C for 30 s. The sequences for the primers are listed in Table 1. All the primers were commercially purchased from Sangon Biotech (Shanghai, China). For the amplification of the products, 2% agarose gels were used for electrophoresis. The 2−ΔΔCT method was used to calculate the relative expressions of genes.
Western blotting analysis
RIPA lysis buffer (Beyotime, Shanghai, China) was used to isolate and extract the proteins from the cultured cells, and the contents of proteins were detected by BCA protein quantification (Thermo Fisher, USA). The proteins were loaded on 12% SDS-PAGE gel for the separation and then transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, USA). The membranes were blocked in 5% non-fat milk, and TBS, Tween-20. The proteins were incubated with the following primary antibodies: rabbit anti-Nek2 antibody (ab227958, 1:500, Abcam, USA) anti-Bcl-2 antibody (ab182858, 1:2000, Abcam, USA), anti-Bax antibody (ab77566, 1:1500, Abcam, USA), anti-CyclinB1 antibody (ab181593, 1:2000, Abcam, USA), anti-CyclinD1 antibody (ab16663, 1:200, Abcam, USA), anti-p27 antibody (sc-1641, 1:200, Santa Cruz Biotechnology, USA), anti-ERK antibody (ab184699, 1:1000, Abcam, USA), anti-p-ERK antibody (ab201015, phospho T202, phospho T185, 1:1000, Abcam, USA), anti-p38 antibody (ab170099, 1:1000, Abcam, USA), anti-p-p38 antibody (sc-7973, Tyr 182, 1:1000, Santa Cruz Biotechnology, USA), and anti-GAPDH antibody (ab181602, 1:10,000, Abcam, USA). After the incubation at 4 °C overnight, the blots were washed with TBST, and then incubated with secondary antibody HRP-conjugated goat anti-Rabbit IgG (Protein tech, USA). Enhanced chemiluminescence (ECL; Thermo Fisher, USA) was used to detect the signal. The density of the blots was read by the Quantity one software version 2.4 (Bio-Rad, USA).
Statistical analysis
Statistical analysis was detected by Prism Graphpad version 6.0 software. All the data were presented as mean ± standard deviation (SD). Differences were determined using one-way analysis of variance (ANOVA) following Tukey multiple comparison. A P < 0.05 was considered as statistically significant.
Results
Up-regulation of Nek2 expression in breast cancer cells and transfection efficiency of Nek2 in MDA-MB-231 and MCF7 cells
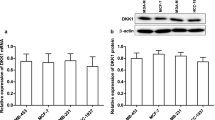
Eight breast cancer cell lines (BT20, BT474, Hs578T, MCF7, MDA-MB-231, MDA-MB-453, T47D and ZR75-1) and normal breast epithelial cells (MCF-10A cells) were used for the detection of Nek2 expression by qRT-PCR. The result showed that Nek2 was high-expressed in all of breast cancer cells compared with MCF-10A (P < 0.01, Fig. 1a). Highly metastatic MDA-MB-231 and low metastatic MCF7 were used as research breast cancer cells in the following experiments. The Nek2 silencing plasmid was respectively transfected in MDA-MB-231 (Fig. 1b) and MCF7 (Fig. 1c) cells. Compared with control or NC group, mRNA and protein expressions of Nek2 were significantly down-regulated in siNEK2 group (P < 0.01).

Nek2 was highly expressed in breast cancer cells and silencing Nek2 inhibited cell viability. a The mRNA expressions of Nek2 in normal breast epithelial cells (MCF-10A cells) and eight breast cancer cell lines (BT20, BT474, Hs578T, MCF7, MDA-MB-231, MDA-MB-453, T47D and ZR75-1) were detected by qRT-PCR. (*compared with MCF-10A, *P < 0.05; **P < 0.01). b The transfection efficiency of Nek2 on MDA-MB-231 cells was detected by qRT-PCR and western blot. c The transfection efficiency of Nek2 on MCF7 cells was detected by qRT-PCR and western blot. d The effects of silencing of Nek2 on cell viability were detected by CCK-8 in MDA-MB-231 cells at 24, 48 and 72 h. E The effects of silencing of Nek2 on cell viability were detected by CCK-8 in MCF7 cells at 24, 48 and 72 h. GAPDH served as an internal control. Data were expressed as mean ± SD from three independent experiments (^compared with control, #compared with NC, ^/#P < 0.05; ^^/##P < 0.01)
Nek2 silencing inhibited cell proliferation, migration and invasion of MDA-MB-231 and MCF7 cells
To explore the effects of Nek2 silencing on breast cancer, a series of biological functional experiments were performed. The results showed that the viabilities of MDA-MB-231 (Fig. 1d) and MCF7 (Fig. 1e) cells were significantly decreased at 48 h and 72 h after transfection of silencing Nek2 (P < 0.01). As shown in Fig. 2, Nek2 silencing noticeably inhibited the cell migration (P < 0.01, Fig. 2a) and invasion (P < 0.01, Fig. 2b) of MDA-MB-231 cells. Similarly, the migration (P < 0.01, Fig. 2c) and invasion (P < 0.01, Fig. 2d) of MCF7 were significantly suppressed in Nek2 silencing group as compared with control or NC group. It might be not perfect to use cells with visible gaps at the start of the assay; however, it might not affect the detection of the wound healing effect (Jonkman et al. 2014).

The effects of silencing of Nek2 on the migration and invasion in MDA-MB-231 and MCF7 cells. a The migration of cell was detected by wound scratch assay and observed under an inverted microscope after MDA-MB-231 cells were transfected with siNek2. b The invasion of cell was detected by Transwell assay after MDA-MB-231 cells were transfected with siNek2, and the cells were stained with crystal violet. C Cell migration was performed by wound scratch assay and observed under an inverted microscope after MCF7 cells were transfected with siNek2. D Cell invasion was detected by Transwell assay after MCF7 cells were transfected with siNek2, and the cells were stained with crystal violet. Data were expressed as mean ± SD from three independent experiments (^compared with control, #compared with NC, ^/#P < 0.05; ^^/##P < 0.01)
Nek2 silencing attenuated cell clone formation, induced cell apoptosis and arrested cell cycle of MDA-MB-231 and MCF7 cells
The effects of Nek2 silencing on cell clone formation, apoptosis and cell cycle were detected. The clone formation of MDA-MB-231 cells was inhibited by silencing Nek2 (P < 0.01, Fig. 3a). Also, silencing Nek2 remarkably increased cell apoptosis (P < 0.01, Fig. 3b). As for cell cycle, silencing of Nek2 significantly enhanced G0/G1 phase of cycle (P < 0.01, Fig. 3c) but obviously reduced G2/M phase of cycle (P < 0.01). For MCF7 cells, silencing of Nek2 showed similar effects as those on MDA-MB-231 cells, and specifically, silencing of Nek2 inhibited cell clone formation (P < 0.01, Fig. 3d), promoted cell apoptosis (P < 0.01, Fig. 3e) and arrested cell cycle at G0/G1 phase (P < 0.01, Fig. 3f).

The effects of silencing of Nek2 on the cell clone formation, apoptosis and cell cycle of MDA-MB-231 and MCF7 cells. a The colony number was observed to assess the effect of silencing of Nek2 on the clone formation in MDA-MB-231 cell. b The apoptosis of cell was detected by flow cytometry after MDA-MB-231 cells were transfected with siNek2. c Cell cycle was detected by flow cytometry after MDA-MB-231 cells were transfected with siNek2. d The colony number was observed to assess the effect of Nek2 silencing on the clone formation in MCF7 cell. e The apoptosis of cell was detected by flow cytometry after MCF7 cells were transfected with siNek2. f Cell cycle was detected by flow cytometry after MCF7 cells were transfected with siNek2. Data were expressed as mean ± SD from three independent experiments (^compared with control, #compared with NC, ^/#P < 0.05; ^^/##P < 0.01)
Nek2 silencing regulated the expressions of related genes in MDA-MB-231 and MCF7 cells
Moreover, qRT-PCR and Western blot were performed to detect the expressions of related genes. Silencing Nek2 significantly decreased the mRNA and protein expressions of Bcl-2, CyclinB1 and CyclinD1 (P < 0.01, Fig. 4a and b), and noticeably increased the expressions of Bax and p27 both at mRNA and protein levels in MDA-MB-231 cells (P < 0.01). In MCF7 cells, Bcl-2, CyclinB1 and CyclinD1 expressions were obviously down-regulated by silencing Nek2 (P < 0.01, Fig. 4c and d). Moreover, silencing Nek2 also increased the mRNA and protein expressions of Bax and p27 (P < 0.01).

The effects of silencing of Nek2 on the expressions of apoptosis and cyclin-associated proteins genes in MDA-MB-231 and MCF7 cells. a qRT-PCR was performed to detect the mRNA expressions of Bcl-2, Bax, CyclinB1, CyclinD1 and p27 after MDA-MB-231 cells were transfected with siNek2. b Western blot was used to assess the protein expressions of Bcl-2, Bax, CyclinB1, CyclinD1 and p27 after MDA-MB-231 cells were transfected with siNek2. c qRT-PCR was performed to detect the mRNA expressions of Bcl-2, Bax, CyclinB1, CyclinD1 and p27 after MCF7 cells were transfected with siNek2. d Western blot was used to assess the protein expressions of Bcl-2, Bax, CyclinB1, CyclinD1 and p27 after MCF7 cells were transfected with siNek2. GAPDH served as an internal control. Data were expressed as mean ± SD from three independent experiments (^compared with control, #compared with NC, ^/#P < 0.05; ^^/##P < 0.01)
Nek2 silencing suppressed cell proliferation via reversing the activation of ERK/MAPK signaling in MDA-MB-231 and MCF7 cells
Furthermore, we explored the effect of Nek2 silencing and IGF on ERK/MAPK signaling in breast cancer cells. In MDA-MB-231 cells, the phosphorylation ratio of p-ERK/ERK and p-p38/p38 were detected. Moreover, significant elevated protein ratios of p-ERK/ERK (P < 0.01, Fig. 5a, b) and p-p38/p38 were induced by IGF, an activator of ERK/MAPK signaling, compared with control. Silencing of Nek2 greatly decreased the protein ratios of p-ERK/ERK and p-p38/p38 (P < 0.01), and greatly reversed the increased protein ratios of p-ERK/ERK and p-p38/p38 (P < 0.01) induced by IGF. Then, CCK-8 assay was performed to detect the effects of Nek2 silencing and IGF on the viability of breast cancer cells. We found that the cell viability was decreased at 48 h and 72 h in silencing Nek2 group (P < 0.01, Fig. 5c). Moreover, the IGF-promoted cell viability was reversed by silencing Nek2 (P < 0.01). In MCF7 cells, silencing of Nek2 had similar effects on the protein expressions of ERK/MAPK signaling and the viability. Noticeably, silencing Nek2 expression could down-regulate the protein expressions of p-ERK/ERK and p-p38/p38 (P < 0.01, Fig. 5d, e). In addition, silencing Nek2 expression reversed the increased activation of ERK/MAPK by IGF and cell viability induced by IGF (P < 0.01, Fig. 5f).

Silencing of Nek2 suppressed cell proliferation via reversing the activation of ERK/MAPK signaling in MDA-MB-231 and MCF7 cells. a Western blot was performed to detect the effects of Nek2 silencing and IGF on the expression of ERK/MAPK signaling-related proteins in MDA-MB-231 cells. b Relative proteins levels (p-ERK/ERK and p-p38/p38) were shown as bar diagrams. c The effects of Nek2 silencing and IGF on cell viability was detected by CCK-8 assay at 24, 48 and 72 h. d Western blot was performed to detect the effects of Nek2 silencing and IGF on the expression of related protein of ERK/MAPK signaling in MCF7 cells. e Relative proteins levels (p-ERK/ERK and p-p38/p38) were shown as bar diagrams. f The effects of Nek2 silencing and IGF on cell viability were detected by CCK-8 assay at 24, 48 and 72 h. Data were expressed as mean ± SD from three independent experiments (^compared with control, #compared with NC, &compared with IGF, ^/#/&P < 0.05; ^^/##/&&P < 0.01)
Discussion
Centrosome replication is a contributor to carcinogenesis and the protein kinases regulating centrosome in tumor cells are abnormally controlled, and abnormally expressed Nek2 could cause CIN and aneuploidy, which would eventually lead to the tumorigenesis (Zhou et al. 2013). It has been reported that Nek2 overexpression enhances the centrosome amplification in her2 + breast cancer model (Harrison Pitner and Saavedra 2013). It was also found that Nek2 is high-expressed in human tumor cell lines and tissues, such as gastric cancer (Li et al. 2019; Ouyang et al. 2019), cervical cancer (Xu et al. 2020) and hepatocellular carcinoma (Deng et al. 2019). The up-regulation of Nek2 is involved in the transformation of malignant cell and the progression of tumor (Takahashi et al. 2014; Zhang et al. 2018; Zhou et al. 2013). As for breast cancer, under the condition of increased positive expression of Nek2, the tumor cells penetrate the basement membrane and are not confined to ducts or lobules, suggesting that Nek2 may play a role in the invasion and metastasis of breast cancer (Hayward et al. 2004). Nek2 serves as one of the five genes that constructs molecular grade index (MGI) to provide prognostic information for patients with breast cancer (Marina and Saavedra 2014). The cytoplasmic expression of Nek2 in invasive ductal carcinoma (IDC) is positively correlated with the grade and size of the tumor (Wang et al. 2011). Therefore, Nek2 may promote tumorigenesis both in vitro and in vivo. In various human breast cancer cell lines, Nek2 knockdown induced aneuploidy and cell cycle arrest that led to cell death (Cappello et al. 2014). In our study, Nek2 was found significantly high-expressed in several breast cancer cell lines.
In addition, we used two different types of breast cancer cell lines to systematically examine the biological effects of Nek2 silencing. Highly metastatic MDA-MB-231, which is a cell line of trip-negative breast cancer, is a subtype of breast cancer without the expressions of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) (Heuser et al. 2018; Kong et al. 2016). MDA-MB-231 is characterized by the high invasiveness and malignancy that impose great difficulties for the treatment. MCF7 cell is usually studied for its sensitivity to hormone, such as ER and PR (Holliday and Speirs 2011; Razak et al. 2019). Silencing of Nek2 in these two breast cancer cell lines could inhibit cell viability, migration, invasion, cell clone formation, and promote cell apoptosis and arrest G0/G1 phase of cell cycle. The effect of hormones (ER, PR and HER2) on the rested proteins needs to be further explored and elucidated. Nek2 is associated with cell cycle, and the expression of Nek2 is lower in G1 phase but will be increased after entering the S phase, and decreased upon cell entry into mitosis (Kokuryo et al. 2019). Cappello demonstrated that knockout of Nek2 in breast cancer cells could induce aneuploidy, cell cycle arrest, and Caspase-dependent and non-dependent cell death (Cappello et al. 2014). Lai also reported that down-regulation of Nek2 induced hepatocellular carcinoma cell arrest in the G2/M phase by retarding the S phase of cell cycle (Lai et al. 2017). The results are not in consistence with those in our study, which might be explained by the possible involvement of intratumor heterogeneity.
Cyclin D1, which is a rate-limiting regulator of G1 progression, promotes cells to enter S phase, thereby increasing cell proliferation (Ramos-García et al. 2017; Tchakarska and Sola 2020). Cyclin B1 is high-expressed in many malignant tumors, including in breast cancer and cervical cancer, and knockdown of cyclin B1 inhibits the proliferation of cancer cell (Androic et al. 2008). p27 is a negative regulator of cell cycle, and its protein binds tightly to Cyclin/CDKs complex in cell cycle to inhibit serine/threonine kinase activity of the protein, promoting cell cycle arrest in G1 phase from entering S phase to synthesize histone and the enzyme for DNA replication, thus inhibiting DNA replication (Roy and Banerjee 2015). In this study, silencing of Nek2 suppressed the expressions of Bcl-2, Cyclin B1 and Cyclin D1, promoted Bax and p27 expressions. The effects of Nek2 silencing on the regulation of the expressions of related genes in our study were in accordance with the biological functions in breast cancer cells. The lack of analysis on CDK4 and CDK6 phosphorylation, and cell cycle inhibitors was a limitation of this study.
ERK/MAPK pathway is an important signaling pathway that mediates cellular responses, and their hyperactivation plays a major role in the development and progression of cancers (Guo et al. 2020). As a class of threonine/serine protein kinases, ERK/MAPK pathway participates in regulating gene expressions, and transduces signals from outside cell into the nucleus and then activates transcription factors via MAPKK-MAPKK-MAPK (conserved cascade of three reactions) (Eblen 2018; Guo et al. 2020). Shah reported that DCIS could switch to invasive breast cancer by activating ERK/MAPK signaling (Shah et al. 2018). Also, DNA-dependent protein kinase catalytic subunit promotes breast cancer cell growth via p38MAPK-AP1 signaling (Zhang et al. 2019). Insulin-like growth factor-1 (IGF-1), which is a MAPK pathway agonist, mediates epithelial-mesenchymal transition (EMT) of breast, lung and gastric cancer cells, and thus, an IGF-treated cell model was established in MCF7 and MDA-MB-231 cells (Cevenini et al. 2018). Nek2 knockdown inhibited the phosphorylation of ERK and the proliferation in gastric cancer cells (Fan et al. 2019), and Nek2 could be a carcinogenic factor and regulated the proliferation and apoptosis of human hepatoma cells via MAPK signal pathway (Zhang et al. 2016). In consistence with the previous research, our findings demonstrated that silencing of Nek2 could inhibit the phosphorylation of ERK and p38 in breast cancer cells. In addition, silencing of Nek2 almost reversed the ratios of p-ERK/ERK and p-p38/p38 which were increased by IGF, the activator of MAPK signaling, indicating that the inhibitory effect of Nek2 silencing on breast cancer cell viability was related to ERK/MAPK pathway. This study may provide a new target gene for the treatment of breast cancer and enrich the regulatory mechanism of Nek2 in the development of breast cancer.
Collectively, it was demonstrated in this work that Nek2 plays an important role in the regulation of the progression of breast cancer in vitro. Nek2 expression was up-regulated in a number of breast cancer cells and silencing of Nek2 could inhibit cell proliferation, invasion and metastasis via regulating the ERK/MAPK signaling.
Data availability
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.
References
Androic I et al (2008) Targeting cyclin B1 inhibits proliferation and sensitizes breast cancer cells to taxol. BMC Cancer 8:391. https://doi.org/10.1186/1471-2407-8-391
Badve SS, Gökmen-Polar Y (2019) Ductal carcinoma in situ of breast: update 2019. Pathology 51:563–569. https://doi.org/10.1016/j.pathol.2019.07.005
Cappello P et al (2014) Role of Nek2 on centrosome duplication and aneuploidy in breast cancer cells. Oncogene 33:2375–2384. https://doi.org/10.1038/onc.2013.183
Cedolini C et al (2014) Type of breast cancer diagnosis, screening, and survival. Clin Breast Cancer 14:235–240. https://doi.org/10.1016/j.clbc.2014.02.004
Cevenini A, Orrù S, Mancini A, Alfieri A, Buono P, Imperlini E (2018) Molecular signatures of the insulin-like growth factor 1-mediated epithelial-mesenchymal transition in breast, lung and gastric cancers. Int J Mol Sci. https://doi.org/10.3390/ijms19082411
Deng L, Sun J, Chen X, Liu L, Wu D (2019) Nek2 augments sorafenib resistance by regulating the ubiquitination and localization of β-catenin in hepatocellular carcinoma. J Exp Clin Cancer Res 38:316. https://doi.org/10.1186/s13046-019-1311-z
Eblen ST (2018) Extracellular-regulated kinases: signaling from Ras to ERK substrates to control biological outcomes. Adv Cancer Res 138:99–142. https://doi.org/10.1016/bs.acr.2018.02.004
Fahad Ullah M (2019) Breast cancer: current perspectives on the disease status. Adv Exp Med Biol 1152:51–64. https://doi.org/10.1007/978-3-030-20301-6_4
Fan WD, Chen T, Liu PJ (2019) NIMA related kinase 2 promotes gastric cancer cell proliferation via ERK/MAPK signaling. World J Gastroenterol 25:2898–2910. https://doi.org/10.3748/wjg.v25.i23.2898
Fisusi FA, Akala EO (2019) Drug combinations in breast cancer therapy. Pharm Nanotechnol 7:3–23. https://doi.org/10.2174/2211738507666190122111224
Fry AM, Schultz SJ, Bartek J, Nigg EA (1995) Substrate specificity and cell cycle regulation of the Nek2 protein kinase, a potential human homolog of the mitotic regulator NIMA of Aspergillus nidulans. J Biol Chem 270:12899–12905
Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL (2020) ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med 19:1997–2007. https://doi.org/10.3892/etm.2020.8454
Harrison Pitner MK, Saavedra HI (2013) Cdk4 and nek2 signal binucleation and centrosome amplification in a her2+ breast cancer model. PLoS ONE 8:e65971. https://doi.org/10.1371/journal.pone.0065971
Hayward DG, Clarke RB, Faragher AJ, Pillai MR, Hagan IM, Fry AM (2004) The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res 64:7370–7376. https://doi.org/10.1158/0008-5472.can-04-0960
Heuser VD et al (2018) Formin proteins FHOD1 and INF2 in triple-negative breast cancer: association with basal markers and functional activities. Breast Cancer Basic Clin Res 12:1178223418792247. https://doi.org/10.1177/1178223418792247
Holliday DL, Speirs V (2011) Choosing the right cell line for breast cancer research. Breast Cancer Res 13:215. https://doi.org/10.1186/bcr2889
Jonkman JE, Cathcart JA, Xu F, Bartolini ME, Amon JE, Stevens KM, Colarusso P (2014) An introduction to the wound healing assay using live-cell microscopy. Cell Adhes Migr 8:440–451. https://doi.org/10.4161/cam.36224
Kazazian K et al (2017) Plk4 promotes cancer invasion and metastasis through Arp2/3 complex regulation of the actin cytoskeleton. Cancer Res 77:434–447. https://doi.org/10.1158/0008-5472.can-16-2060
Kokuryo T et al (2016) Nek2 siRNA therapy using a portal venous port-catheter system for liver metastasis in pancreatic cancer. Cancer Sci 107:1315–1320. https://doi.org/10.1111/cas.12993
Kokuryo T, Yokoyama Y, Yamaguchi J, Tsunoda N, Ebata T, Nagino M (2019) NEK2 is an effective target for cancer therapy with potential to induce regression of multiple human malignancies. Anticancer Res 39:2251–2258. https://doi.org/10.21873/anticanres.13341
Kong Y et al (2016) Platycodin D, a metabolite of Platycodin grandiflorum, inhibits highly metastatic MDA-MB-231 breast cancer growth in vitro and in vivo by targeting the MDM2 oncogene. Oncol Rep 36:1447–1456. https://doi.org/10.3892/or.2016.4935
Lai XB et al (2017) NIMA-related kinase 2 regulates hepatocellular carcinoma cell growth and proliferation. Oncol Lett 13:1587–1594. https://doi.org/10.3892/ol.2017.5618
Li JJ, Li SA (2006) Mitotic kinases: the key to duplication, segregation, and cytokinesis errors, chromosomal instability, and oncogenesis. Pharmacol Ther 111:974–984. https://doi.org/10.1016/j.pharmthera.2006.02.006
Li Y et al (2019) NEK2 promotes proliferation, migration and tumor growth of gastric cancer cells via regulating KDM5B/H3K4me3. Am J Cancer Res 9:2364–2378
Liu B, Qi X, Zhang X, Gao D, Fang K, Guo Z, Li L (2018) Med19 is involved in chemoresistance by mediating autophagy through HMGB1 in breast cancer. J Cell Biochem. https://doi.org/10.1002/jcb.27406
Liu XL, Liu HM, Han N, Li FH, Sun F, Fan DM, Xu Q (2019) PCAT1 promotes the proliferative and migratory potentials of ovarian cancer via targeting NEK2. Eur Rev Med Pharmacol Sci 23:8239–8248. https://doi.org/10.26355/eurrev_201910_19133
Lorenzoni C, Vilajeliu A, Carrilho C, Castillo P, Barreales S, Ismail MR, Sidat M, Augusto O, Garcia-Basteiro AL, Menendez C, Ordi J (2019) Gynaecological malignancies at a tertiary care centre in Mozambique. Eur J Gynaecol Oncol 40:295–299. https://doi.org/10.12892/ejgo4447.2019
Lu L, Zhai X, Yuan R (2015) Clinical significance and prognostic value of Nek2 protein expression in colon cancer. Int J Clin Exp Pathol 8:15467–15473
Marina M, Saavedra HI (2014) Nek2 and Plk4: prognostic markers, drivers of breast tumorigenesis and drug resistance. Front Biosci (landmark Edition) 19:352–365
Morris NR (1975) Mitotic mutants of Aspergillus nidulans. Genet Res 26:237–254
Ouyang Y et al (2019) CircRNA circPDSS1 promotes the gastric cancer progression by sponging miR-186–5p and modulating NEK2. J Cell Physiol 234:10458–10469. https://doi.org/10.1002/jcp.27714
Peart O (2017) Metastatic breast cancer. Radiol Technol 88:519m–539m
Ramos-García P, Gil-Montoya JA, Scully C, Ayén A, González-Ruiz L, Navarro-Triviño FJ, González-Moles MA (2017) An update on the implications of cyclin D1 in oral carcinogenesis. Oral Dis 23:897–912. https://doi.org/10.1111/odi.12620
Razak NA et al (2019) Cytotoxicity of eupatorin in MCF-7 and MDA-MB-231 human breast cancer cells via cell cycle arrest, anti-angiogenesis and induction of apoptosis. Sci Rep 9:1514. https://doi.org/10.1038/s41598-018-37796-w
Roy A, Banerjee S (2015) p27 and leukemia: cell cycle and beyond. J Cell Physiol 230:504–509. https://doi.org/10.1002/jcp.24819
Schultz SJ, Fry AM, Sutterlin C, Ried T, Nigg EA (1994) Cell cycle-dependent expression of Nek2, a novel human protein kinase related to the NIMA mitotic regulator of Aspergillus nidulans. Cell Growth Differ Mol Biol J Am Assoc Cancer Res 5:625–635
Sdelci S et al (2012) Nek9 phosphorylation of NEDD1/GCP-WD contributes to Plk1 control of gamma-tubulin recruitment to the mitotic centrosome. Curr Biol 22:1516–1523. https://doi.org/10.1016/j.cub.2012.06.027
Shah S, Brock EJ, Jackson RM, Ji K, Boerner JL, Sloane BF, Mattingly RR (2018) Downregulation of Rap1Gap: a switch from DCIS to invasive breast carcinoma via ERK/MAPK activation. Neoplasia (new York, NY) 20:951–963. https://doi.org/10.1016/j.neo.2018.07.002
Siegel RL, Miller KD, Jemal A (2020) Cancer statistics, 2020. CA Cancer J Clin 70:7–30. https://doi.org/10.3322/caac.21590
Suarez-Cabrera C et al (2018) The Ras-related gene ERAS is involved in human and murine breast cancer. Sci Rep 8:13038. https://doi.org/10.1038/s41598-018-31326-4
Takahashi Y et al (2014) Up-regulation of NEK2 by microRNA-128 methylation is associated with poor prognosis in colorectal cancer. Ann Surg Oncol 21:205–212. https://doi.org/10.1245/s10434-013-3264-3
Tchakarska G, Sola B (2020) The double dealing of cyclin D1. Cell Cycle (georgetown, Tex) 19:163–178. https://doi.org/10.1080/15384101.2019.1706903
Tian Q et al (2017) Recent perspectives of management of breast cancer metastasis - an update. J BUON off J Balkan Union Oncol 22:295–300
Tsang JYS, Tse GM (2020) Molecular classification of breast cancer. Adv Anat Pathol 27:27–35. https://doi.org/10.1097/pap.0000000000000232
Wang S et al (2011) Abnormal expression of Nek2 and beta-catenin in breast carcinoma: clinicopathological correlations. Histopathology 59:631–642. https://doi.org/10.1111/j.1365-2559.2011.03941.x
Ward EM et al (2015) Cancer statistics: breast cancer in situ. CA Cancer J Clin 65:481–495. https://doi.org/10.3322/caac.21321
Wen HY, Brogi E (2018) Lobular carcinoma in situ. Surg Pathol Clin 11:123–145. https://doi.org/10.1016/j.path.2017.09.009
Xu T et al (2020) Targeting NEK2 impairs oncogenesis and radioresistance via inhibiting the Wnt1/β-catenin signaling pathway in cervical cancer. J Exp Clin Cancer Res 39:183. https://doi.org/10.1186/s13046-020-01659-y
Ye X, Niu Y, Fang ZY (2005) Correlations of centrosome abnormality and genomic instability to tumor. Ai Zheng = Aizheng = Chin J Cancer 24:1290–1292
Ye L et al (2018) Overexpression of CDCA7 predicts poor prognosis and induces EZH2-mediated progression of Triple-Negative Breast. Cancer Int J Cancer. https://doi.org/10.1002/ijc.31766
Zehra S, Doyle F, Barry M, Walsh S, Kell MR (2020) Health-related quality of life following breast reconstruction compared to total mastectomy and breast-conserving surgery among breast cancer survivors: a systematic review and meta-analysis. Breast Cancer (tokyo, Japan) 27:534–566. https://doi.org/10.1007/s12282-020-01076-1
Zeng YR et al (2015) Overexpression of NIMA-related kinase 2 is associated with progression and poor prognosis of prostate cancer. BMC Urol 15:90. https://doi.org/10.1186/s12894-015-0085-7
Zhang MX et al (2016) Effect of silencing NEK2 on biological behaviors of HepG2 in human hepatoma cells and MAPK signal pathway. Tumour Biol J Int Soc Oncodev Biol Med 37:2023–2035. https://doi.org/10.1007/s13277-015-3993-y
Zhang Y et al (2018) NEK2 promotes hepatocellular carcinoma migration and invasion through modulation of the epithelial-mesenchymal transition. Oncol Rep 39:1023–1033. https://doi.org/10.3892/or.2018.6224
Zhang Y et al (2019) High expression of PRKDC promotes breast cancer cell growth via p38 MAPK signaling and is associated with poor survival. Mol Genet Genom Med 7:e908. https://doi.org/10.1002/mgg3.908
Zhou W et al (2013) NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell 23:48–62. https://doi.org/10.1016/j.ccr.2012.12.001
Funding
This work was supported by the Special Research Fund for Central Universities, Peking Union Medical College [Grant No. 3332020026].
Author information
Authors and Affiliations
Contributions
Substantial contributions to conception and design: ZX, MZ. Data acquisition, data analysis and interpretation: XW, JL, GL, KF, XW. Drafting the article or critically revising it for important intellectual content: ZX, MZ. Final approval of the version to be published: All authors. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of the work are appropriately investigated and resolved: All authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xing, Z., Zhang, M., Wang, X. et al. Silencing of Nek2 suppresses the proliferation, migration and invasion and induces apoptosis of breast cancer cells by regulating ERK/MAPK signaling. J Mol Histol 52, 809–821 (2021). https://doi.org/10.1007/s10735-021-09979-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10735-021-09979-9