
Abstract
Leclercia adecarboxylata QDSM01 is a plant growth-promoting bacterium isolated from the rhizosphere soil of maize. However, the relevant molecular mechanisms remain obscure. This study investigated the effect of Leclercia adecarboxylata QDSM01 on the seedling growth of maize, rice and soybean and uncovered the bacterium’s biochemical characterization (biofilm, plant hormone secretion, nitrogen generation, phosphorus and potassium solubilization). In-depth analysis of the functional genes related to the biochemical characterization was conducted by complete genome sequence. Strain QDSM01 significantly promoted the seedling growth of maize and rice, but not soybean. The strain QDSM01 was identified as Leclercia adecarboxylata based on a comparison of 16 S rRNA gene sequences and complete genome sequence. The complete genome sequence indicated the strain comprised a 4,461,951 bp chromosome and 5 plasmids. Moreover, antiSMASH analysis revealed a total of four secondary metabolite gene clusters, consisting of putative non-ribosomal peptide synthase (NRPS), terpenoids, thiopeptides and arylpolyene. These gene clusters play an important role in promoting plant growth and resistance. Furthermore, the genes involved in biofilm formation, quorum sensing, chemotaxis, motility, indole-3-acetic acid production (IAA), siderophore, nitrogen generation, solubilization and uptake of phosphate and potassium were identified. Meanwhile, in vitro experiments were also performed to confirm these functions. In addition, the strong IAA production ability of strain QDSM01 was observed than other strains of Leclercia adecarboxylata. These results suggest that Leclercia adecarboxylata QDSM01 can serve as a biofertilizer that improves plant growth. This study will be helpful for further studies of Leclercia adecarboxylata on the mechanisms of plant growth promotion.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Currently, global agriculture is facing enormous challenges from global population growth and climate change, and beneficial microorganisms are emerging as an important alternative for sustainable crop production (Andrés–Barrao et al. 2017). Plant growth promoting rhizobacteria (PGPR) are direct association with plant radicles, and PGPR promotes plant growth through the availability of essential nutrients (nitrogen, phosphate and potassium), iron mobilization as well as synthesis of plant hormones (Abdelaal et al. 2021; Bhise and Dandge 2019; Danish et al. 2020; Shilev 2020). Moreover, PGPR interacts with plants to promote seed germination, radicle-shoot growth and tolerance to stress (Sandilya et al. 2022). Consequently, the PGPR considerably influences plant health.
Leclercia adecarboxylata is a member of the Enterobacteriaceae family, which is widely distributed in nature and has been reported from blood, food, water and other environmental sources (Naveed et al. 2014; Snak et al. 2021). Leclercia adecarboxylata is metabolically diverse and can produce phytohormones, synthesize extra-cellular enzymes, degrade hydrocarbons and solubilize minerals (Kang et al. 2019). Moreover, such traits have been reported to mediate salinity stress (Kang et al. 2019), drought stress (Danish et al. 2020), and heavy metal stress (Danish et al. 2019; Kang et al. 2021), and recent studies have shown that Leclercia adecarboxylata have a role in promoting plant growth (Kumawat et al. 2019; Snak et al. 2021). However, the relevant molecular mechanisms remain obscure.
Strain QDSM01 was isolated from maize growing in the northeast black soil zone of China. The present study aims to investigate the effect of QDSM01 on maize, rice and soybean growth and to analyze the molecular mechanism of strain QDSM01 promoting plant growth through the entire genome sequence, nutrient transformation, biofilm formation and phytohormone secretion.
Materials and methods
Materials
Leclercia adecarboxylata QDSM01 is a plant growth-promoting bacterium isolated from the rhizosphere soil of maize, and stored in China Center for Type Culture Collection at Wuhan University (CCTCC NO. M 20,211,653).
Surface sterilization and germination of seeds and the inoculation of bacteria on seedlings
The seeds of maize, soybean and rice were Kangui No.1, Zhonghuang No.9, and Longjing 46, respectively. The seeds were surface-sterilized by dipping in 5% (vol/vol) sodium hypochlorite for 3 min and then in 70% (vol/vol) ethanol for another 3 min, followed by three rinses with sterile distilled water. The seeds were incubated in a light incubator (Thermo Scientific, USA) until the buds were 1 cm long.
The bacterial strain was inoculated in liquid Luria–Bertani (LB) medium and shaken at 30 °C overnight. Then, the activated QDSM01 was transferred into fresh liquid LB medium and shaken at 30 °C for another 8 h. Cells were collected by centrifuging and resuspended in 1 × PBS buffer. The cell suspension was diluted to 105 (T1), 106 (T2), and 107 (T3) cells per mL, respectively. Moreover, the surface-sterilized and germinated maize seeds with primary radicles of 1–2 cm were soaked in the suspensions for 1.0 h, and equal amounts of sterilized PBS buffer solution were used as a control (C). In plant growth promotion tests, fully dissolved Murashige and Skoog (MS) solid medium was injected into the test tubes at 15 mL per tube, sterilized and stored; then, the germinated seeds with radicles of 1–2 cm were put on the surface of agar by sterile tweezers. The germinated seeds inoculated with a bacterial suspension were grown under controlled environmental conditions in a plant growth chamber at 28 °C, 70% relative humidity, and a 16 h light/8 h dark cycle (Niu et al. 2017). The seedling radicle lengths, radicle node number, radicle tip number, radicle connections and radicle bifurcation number were analyzed by root analysis system GXY-A (Zhejiang Top Cloud-agri Technology CO.,LTD, China).
For experiments with bacteria in physical contact with plant roots, the germinated seeds with radicles of 1–2 cm were sown in sterile Petri-dishes (100 × 100 × 15 mm) on MS medium, then, inoculated with 100 µL of the inoculum at 106 cells per mL in physical contact with their roots and equal amounts of sterilized PBS buffer solution were used as a control (Dahmani et al. 2020). After 48 h at 28 °C, 70% relative humidity, and a 16 h light/8 h dark cycle, the plants and their growth media in the plates were transferred to sterilized PBS buffer solution and vortex shaken for 30 min, after which the bacterial density was performed.
Genome sequence of Leclercia adecarboxylata QDSM01
The genomic DNA of strain QDSM01 was extracted using the Wizard® Genomic DNA Purification Kit (Promega, USA), and DNA was subsequently quantified using a TBS-380 fluorometer (Turner BioSystems Inc., Sunnyvale, CA). For library construction, the DNA was sheared into 400–500 bp fragments using a Covaris M220 Focused Acoustic Shearer. Sequencing libraries were individually prepared using the NEXTflex™ Rapid DNA-Seq Kit (BIOO SCIENTIFIC, USA). Briefly, the 5′ prime ends were first end-repaired and phosphorylated. Next, the 3′ ends were A-tailed and ligated to sequencing adapters. The third step is to enrich the adapters-ligated products using PCR. Before sequencing, all libraries were quantified by using 0.45 × volumes of Agencourt AMPure XP beads (Beckman Coulter Genomics, MA). The prepared libraries were then paired-end Illumina sequencing (2 × 150 bp) at Shanghai Majorbio Biopharm Technology Co., Ltd using an Illumina HiSeq X Ten machine (de Souza et al. 2019).
The complete genome of QDSM01 was assembled by Unicycler (v0.4.8). Then, the plasmid and coding sequence (CDS) of strain QDSM01 were predicted using PlasFlow (https://github.com/smaegol/PlasFlow) and Glimmer (http://ccb.jhu.edu/software/glimmer/index.shtml), respectively. Genome annotation of the CDS was accomplished using NR, Swiss-Prot, Pfam, COG, GO, and KEGG databases. The tRNA, rRNA, sRNA, gene islands (GIs) and prophage regions were predicted in the strain QDSM01 genome using tRNAscan-SE (v2.0, http://trna.ucsc.edu/software/), Barrnap (https://github.com/tseemann/barrnap), Infernal (http://eddylab.org/infernal/), and IslandViewer (v1.2) (Dong et al. 2022). After BLAST-searched against the National Center for Biotechnology Information (NCBI) database (https://blast.ncbi.nlm.nih.gov/Blast.cgi), MEGA 6 software was used to construct a phylogenetic tree using the phylogenomic neighbor-joining (NJ) method.
Comparative analysis of Leclercia adecarboxylata QDSM01 with other Leclercia adecarboxylata
Whole genome sequences of Leclercia adecarboxylata were downloaded from the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/). Average nucleotide identity (ANI) is a measure of nucleotide-level genomic similarity and was carried out using EzBioCloud (http://www.ezbiocloud.net/tools/ani). ANI for the 12 Leclercia adecarboxylata genomes that determine whether the genomes in a pair belong to the same species, only the subset of high-quality genome pairs was utilized and an ANI cutoff of ≥ 96% was used to define species. dDDH analysis was carried out by the type strain genome server (https://tygs.dsmz.de/). Biosynthetic gene clusters for secondary metabolites were analyzed using antiSMASH (https://antismash.secondarymetabolites.org/#!/start).
Determination of plant growth promoting traits
The bacterial strain was grown overnight in liquid LB medium, and 1 mL of the resulting suspension was inoculated into 100 mL of the phosphorus-soluble medium, potassium-soluble medium, and nitrogen-fixing medium in 250 mL conical flasks, separately. After 5 days, the culture filtrate was collected by centrifugation at 12,000 rpm for 10 min and used for quantitative analysis. The contents of soluble phosphorus, potassium ion, and nitrogen in the bacterial culture filtrate were determined using the molybdenum blue method, flame photometry (FP6431, Shanghai Yidian Analytical Instruments Co., China), and carbon and nitrogen analyzer (multi N/C 2100 S, Analytik Jena AG, Germany), respectively. Moreover, the production of IAA and siderophore were qualitatively analyzed in the liquid LB medium containing (l%) tryptone and MKB growth medium, respectively. After 2 days, the IAA contents in the culture filtrates were measured based on the chromogenic reaction of Salkowski’s reagent with IAA (Wang et al. 2018). The siderophore production was measured based on the liquid version of the CAS (chrome azurol S) assay (Gu et al. 2020). The mediums in this study were listed in Supplementary Table 1.
Quantification of biofilm formation
The strain was inoculated in liquid LB medium and incubated for 24 h at 30 °C at 180 rpm, and then diluted to 0.8 at OD600 nm in sterile water. The 20 µL bacterial cultures were then added to the wells of the 96-wells plates containing fresh medium in a total volume of 160 µL. The plates were sealed with Parafilm and incubated at 30 °C for 48 h without shaking. The floating cells were washed with sterilized water and then stained for 20 min in 180 µL of 1% (W/V) crystal violet aqueous solution. The cells were washed again with sterilized water three times, and then 200 µL of 96% ethanol was used to release the crystal violet absorbed by the biofilm. Finally, absorbance values at OD590nm were measured with a plate spectrophotometer to quantify the thickness of the biofilm (Berendsen et al. 2018).
Moreover, one-way ANOVA followed by Tukey’s HSD test was performed to detect the significant differences among all the experimental treatments, with p < 0.05 denoting significance.
Results
Plant-beneficial activities of Leclercia adecarboxylata QDSM01
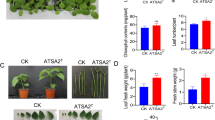
Test tube seedling experiments were performed to evaluate the effects of QDSM01 on the growth of maize, soybean, and rice, the most important and widely-planted food crops in northeast China. To avoid the confounding influence on seedlings, equal amounts of sterilized PBS buffer solution were used as a control. The results revealed that QDSM01 significantly promoted the growth of maize and rice plants (Figs. 1 and 2). Briefly, QDSM01 significantly promoted shoot height, radicle length, connections, radicle node, radicle tip and bifurcation. In Fig. 1a and b, compared to the C group, T1 and T2 had the best effect on maize plant development, followed by T3; For radicle length (Fig. 1c), the growth-promoting effect was T2, T3 > C > T1; The cell concentration with the best growth-promoting effect on connections (Fig. 1d), the numbers of node, radicle tip, and bifurcation (Fig. 1e–g) was T2 and T3, followed by T1 when compared with the C group. Compared to the C group, T2 had the best effect on maize plant development, followed by T1 and T3.

The effects of QDSM01 on the seedling growth of maize. a Photos showing maize plants inoculated with various concentration of Leclercia adecarboxylata QDSM01. b Seedling height. c Radicle length. d Radicle connections. e Radicle node number. f Radicle tip number. g Radicle bifurcation number. C represents the control group, T1, T2, and T3 treated with 105, 106, and 107 bacterial solution concentrations, respectively. Significance test was performed using one-way ANOVA followed by Tukey’s HSD test. Different letters indicate statistically significant (p < 0.05) differences

The effects of QDSM01 on the seedling growth of rice. a Photos showing rice plants inoculated with various concentration of Leclercia adecarboxylata QDSM01. b Seedling height. c Radicle length. d Radicle connections. e Radicle node number. f Radicle tip number. g Radicle bifurcation number. C represents the control group, T1, T2, and T3 treated with 105, 106, and 107 bacterial solution concentrations, respectively. Significance test was performed using one-way ANOVA followed by Tukey’s HSD test. Different letters indicate statistically significant (p < 0.05) differences
In Fig. 2a and b, compared to the control, the significant growth-promoting effect on rice seedling height was T3, while T1 and T2 were not significantly different from the C and T3; For the length, connections, and node number of radicle (Fig. 2c and e), the cell concentration with the best growth-promoting effect was T1, followed by T2 and T3 when compared with the C group; In Fig. 2f, g, the cell concentration with the best growth-promoting effect on the numbers of radicle tip and bifurcation was T2 and T3, followed by T1 when compared with the C group.
In Fig. 3a and b and f, there was no significant change in soybean seedling height and radicle tip number with the addition of QDSM01 strain; For the length, connections, and node number of radicle (Fig. 3c and e), the cell concentration with the best growth-promoting effect was T3, followed by T2 and T1 when compared with the C group; In Fig. 3g, the cell concentration with the best growth-promoting effect on the number of bifurcation was T3, followed by T1 and T2 when compared with the C group. Therefore, the QDSM01 strain has a significant growth-promoting effect on graminaceous crops and a weak growth-promoting ability on legumes. This outstanding plant-promoting performance indicates that QDSM01 can be regarded as an ideal PGPR agent for graminaceous crops.

The effects of Leclercia adecarboxylata QDSM01 on the seedling growth of soybean. a Photos showing soybean plants inoculated with various concentration of Leclercia adecarboxylata QDSM01. b Seedling height. c Radicle length. d Radicle connections. e Radicle node number. f Radicle tip number. g Radicle bifurcation number. C represents the control group, T1, T2, and T3 treated with 105, 106, and 107 bacterial solution concentrations, respectively. Significance test was performed using one-way ANOVA followed by Tukey’s HSD test. Different letters indicate statistically significant (p < 0.05) differences
In Supplementary Fig. 1, after 60 h of physical contact between strain QDSM01 and plant roots (rice and maize), maize and rice significantly increased the cell number of strain QDSM01, while maize had a better effect than rice.
Genomic analysis of Leclercia adecarboxylata QDSM01
In general, 16 S rRNA gene sequences are highly conserved among the same bacterial species and are frequently used to identify and classify microorganisms. Phylogenetic analysis based on 16 S rRNA gene sequences showed that the strain QDSM01 was clustered together with Leclercia adecarboxylata LJ-16 (KX959963.1:38-1369) (Fig. 4a). The result suggested that strain QDSM01 could be assigned to the genus Leclercia. To identify the species of strain QDSM01, the phylogenomic NJ tree was constructed based on whole-genome sequences. The comparative phylogenomic analysis indicated that strain QDSM01 showed the highest homology with Leclercia adecarboxylata (Fig. 4b). Moreover, ANI and dDDH analyses between strain QDSM01 and Leclercia adecarboxylata were ≥ 98.31% and ≥ 90.3%, respectively, which indicated that the strain QDSM01 was finally identified as Leclercia adecarboxylata (Table 1).

Phylogenetic analysis, circular map and COG category of the Leclercia adecarboxylata QDSM01 genome. a-b 16 S rRNA sequence and genome based phylogenetic analysis performed using MEGA 6.0. c 1st circle: a marker of genome size; 2nd and 3rd circle: CDS on the positive and negative chains. Different colors indicate the functional classification of COG; 4th circle: rRNAs and tRNAs; 5th circle: GC content. The outward red part indicates that GC content in this region is higher than the average GC content in the whole genome, while blue shows the opposite result; 6th circle: GC skew. In general, the GC skew of the leading chain is > 0, and the GC skew of the trailing lag chain is < 0. It can also assist in determining the starting point and end point of replication
To explore the genetic potential of traits associated with plant colonization, the genomic features associated with strain QDSM01 was sequenced using Illumina MiSeq (300-bp paired-end) technology. The general features of the QDSM01 genome are summarized in Table 2; Fig. 4c. The single circular chromosome (4,461,951 bp) with a GC content of 55.7% encodes 4734 coding sequences (CDS), 87 tRNA genes, 25 rRNA operons, 150 sRNA. Gene island was predicted by Islander and identified 12 GIs in the strain QDSM01 genome. Moreover, the classification of QDSM01 genes into clusters of orthologous groups (COGs) assigned 4377 CDSs to twenty COG groups (92.46%). The most abundant COG category was “Carbohydrate transport and metabolism” (n = 402), followed by “Amino acid transport and metabolism” (n = 358) (Fig. 4d). The QDSM01 strain has five plasmids, P1, P2, P3, P4, and P5, with sequence sizes of 118,035 bp, 116,089 bp, 22,558 bp, 4346 bp and 2508 bp, and the GC contents were 55.15, 50.82, 49.36, 55.45 and 51.56% (Supplementary Table 2). Furthermore, three prophage regions were predicted in the QDSM01 genome. The three intact prophage genome sizes are 51.9 (55.7 GC%), 34.8 (55.7 GC%), and 33.9 kb (55.7 GC%) coding 61, 40, and 42 CDS, respectively (Supplementary Table 3).
Genome mining for the synthesis of bioactive compounds
In Fig. 5 and Supplementary Tables 4, the antiSMASH analysis revealed a total of 4 secondary metabolite BGCs composed of putative NRPS (Non-Ribosomal Peptide Synthetase), terpenes, thiopeptide and arylpolyene. The cluster of NRPS encoding for turnerbactin exhibit 30% similarity with BGC0000451. Turnerbactin, a novel triscatecholate siderophore, is responsible for iron regulation and uptake and the genes of tonB, dhbC and the homologs of entABCDEF for turnerbactin biosynthesis were found. The cluster of terpenes encoding for carotenoid exhibit 100% similarity with BGC0000640. Carotenoids are yellow to red colored pigments which originate from the terpenoid biosynthetic pathway, and the four genes (idi, crtI, crtB, and crtZ) for carotenoids biosynthesis were showed. Thiopeptide encoding for O-antigen exhibit 14% similarity with BGC0000781 and we found three genes (rpsA, wzzB, and wzxC) for O-antigen biosynthesis. The cluster of arylpolyene encoding for surfactin exhibit 94% similarity with BGC0002008 and the biosynthesis genes were plsC, mdcC, fadD, waaE, tesC, lolA, fabB, fabG, and fabF.

Graphical representation of key Biosynthetic Gene Clusters (BGCs) in Leclercia adecarboxylata QDSM01 genome. Organization of putative novel BGCs coding for NRPS, terpenes, thiopeptide and arylpolyene
Gene mining associated with colonization of Leclercia adecarboxylata QDSM01
In a liquid culture without shaking, QDSM01 forms robust pellicles at the liquid-air interface (Fig. 6a), and the absorbance values at OD590nm was 2.5 (Fig. 6b). In this study, the genome of QDSM01 contains a complete set of genes implicated in biofilm–formation, including three genes related to polysaccharide biosynthesis, 13 genes related to the type VI secretion system and 32 genes for other functions (Table 3 and Supplementary Table 5).

Quantification associated with plant-growth-promoting factors of Leclercia adecarboxylata QDSM01. a Photos showing biofilm formation. b Quantification of biofilm formation. c Quantification of IAA production. d Quantification of siderophore. e Nitrogen generation, phosphorus-solubilization and potassium-solubilization
We identified 14 genes involved in chemotaxis and motility (Table 3 and Supplementary Table 5). This signal transduction system consists of a set of conserved proteins, which includes cheA, cheB, cheR, cheV, cheW, cheY, cheZ, motA, and motB, and a set of chemoreceptors known as methyl-accepting chemotaxis protein (tsr, trg, mcp, tap, and tar). Flagellin is the most important structural protein that is part of flagella and is essential for the mobility of these bacteria. In this study, 19 genes (fliA, fliC, fliD, fliS, fliT, fliE, fliF, fliG, fliH, fliI, fliJ, fliK, fliL, fliM, fliNY, fliOZ, fliP, fliQ, and fliR) are involved in flagellum movement.
We identified 20 genes involved in quorum sensing in Table 3 and Supplementary Table 5. QDSM01 contained six genes (secB, secE, secA, yajC, secG, and secY) associated with preprotein translocase. The lsr operon (lsrACDBFG) consisting of six genes, was identified in QDSM01. lsrB encodes a ligand-binding protein, lsrC and lsrD each encodes a transmembrane protein, and lsrA encodes a cytoplasmic protein responsible for ATP hydrolysis during transport. Eight transcriptional regulators of the LuxR family were identified in QDSM01.
Quantification and gene mining associated with plant-growth-promoting factors of Leclercia adecarboxylata QDSM01
In Fig. 6c, the average yield of IAA in QDSM01 culture was 47.78 µg/mL. In Table 4 and Supplementary Tables 6, we found some of the trp cluster (trpA, trpB, trpCF, trpE, and trpGD) genes involved in tryptophan biosynthesis, and the genes catalyzing decarboxylation (ddc) and oxidation (aldH) were searched in bacterial genomes.
In Fig. 6d, siderophores production in QDSM01 culture was 27.27% on average. In Table 4 and Supplementary Tables 6, siderophore translocation through the bacterial outer membrane is performed by an energy transducing complex with proteins tonB, exbB, and exbD. The iron (III)-hydroxamate ABC transporter cluster fhuCDB, is responsible of the transport of ferrichrome and other Fe3+-hydroxamate compounds. Furthermore, ferrous iron uptake protein efeU was also found.
In Fig. 6e, nitrogen generation capacity in QDSM01 was 13723.33 µg/mL on average. In Table 4 and Supplementary Tables 6, the genome of QDSM01 contains a gene essential for nitrite reduction pathways (nasA). Furthermore, nitrate and nitrite transport-related genes nrtABC and narGHIJKLQX were also detected.
In Fig. 6e, the average capacities of phosphorus-solubilization and potassium-solubilization in QDSM01 were 26.86 and 2.5 µg/mL, respectively. In Table 4 and Supplementary Tables 6, The gcd is a critical gene in the biosynthetic pathway of gluconic acid (GA). Genes related to phosphonate transport (phnC, phnD, and phnE) and degradation (phnG, phnH, phnI, phnJ, phnK, and phnL) were found in QDSM01. Moreover, genes related to potassium transport (kdpA, kdpB, and kdpC), potassium-efflux system (kefB, kefC, kefF, and kefG), and potassium uptake system (trkA, trkH, and kup) were found in QDSM01.
Discussion
The plant growth-enhancing potential of PGPR is well recognized, with extensive research has examined this phenomenon. In our study, QDSM01 inoculation significantly improved almost all plant growth parameters of maize and rice seedlings, including total biomass, radicle length, radicle tip number and other radicle metrics. PGPR has been reported to promote plant growth via various mechanisms including nitrogen generation, phosphate solubilization, biofilm formation and phytohormone production and also by the synthesis of antimicrobial secondary metabolite (Iqbal et al. 2021). Therefore, we performed gene mining and quantification of the above pathways.
Biosynthetic gene clusters (BGCs) are operonic sets of microbial genes that synthesize specialized metabolites with diverse functions, including siderophores and antibiotics (Crits-Christoph et al. 2020). The O-antigen is required for Gram-negative LPS effective symbioses, and it confers the resistance to host defense mechanisms such as opsonization and phagocytosis (Lulamba et al. 2021), and also plays an important role in radicle tip colonization (Santoyo et al. 2021). Surfactin plays an important role in the suppression of powdery mildew of cucurbits caused by the fungal plant pathogen Podosphaera fusca (Romero et al. 2007). As a versatile lipopeptide, surfactin could stimulate biofilm formation by inducing potassium leakage and the subsequent activation of downstream genes, and serve as a signal to induce plant resistance (López et al. 2009; Zhang et al. 2015). Therefore, except to its ability to promote plant growth, QDSM01 may also play an important role in the resistance to plant diseases.
The ability of PGPR to effectively colonize the plant radicle surface is a prerequisite for stimulating plant growth and colonization capacity is related to the ability to form biofilms (Santoyo et al. 2021). Polysaccharides are important components of the biofilm matrix, and VpsM and VpsN are essential for polysaccharide synthesis and biofilm formation (Fong et al. 2010). RpoS not only modulates the expression of type VI secretion system but also regulates flagellum formation by positively controlling, and affecting the formation of biofilm by regulating the synthesis of exopolysaccharides (Guan et al. 2015). Moreover, the adhesion step is crucial for biofilm formation and involves the flagellin protein fliC, the flagellar cap protein fliD, and flagellar movement (Bouteiller et al. 2021) .
Bacterial chemotactic signals are detected by methyl-receptive chemotactic proteins (MCPs) then communicated to the flagellar motor via a series of chemotactic (Che) proteins (Sharma et al. 2018; Tunchai et al. 2017). In this study, QDSM01 possesses four MCPs (tsr, tar, trg, and tap) and seven Che proteins (cheA, cheB, cheR, cheV, cheW, cheY, and cheZ). The proteins fliG, fliM, and fliN bind to the bacterial flagellar switch complex and determine whether the bacteria swim, modulating chemotactic movement toward nutrients (Qiao et al. 2022).
The function of Autoinducer-2 (AI-2) which acts as the signal molecule of LuxS-mediated quorum sensing (QS), is regulated through the lsr operon (Zuo et al. 2019). In Table 3 and Supplementary Tables 5, a lsr operon consisting of six genes (lsrACDBFG) was identified in QDSM01. One of the best studied is sdiA, which is involved in detecting and responding to N-acetyl homotypic lactone (NAHL, QS signaling) in the environment. Additionally, the luxS gene encoding S-ribosylhomocysteine lyase, which generates autoinducer-QS signaling, regulates population density-dependent gene expression (Shankar et al. 2012).
The ability of bacteria to produce IAA depends on the availability of IAA precursors and the uptake of bacterial IAA by plants (Jung et al. 2017). In Fig. 6c, the average yield of IAA in QDSM01 culture was 47.78 µg/mL, and its IAA yield was 9 times and 18 times higher than that of Leclercia adecarboxylata MO1 (9.815 µg/mL) and Leclercia adecarboxylata Palotina (2.600 µg/mL), proving that QDSM01 is a high IAA-producing strain (Kang et al. 2019; Snak et al. 2021). In Table 4 and Supplementary Tables 6, we searched for the genes involved in two different IAA biosynthetic pathways (IPyA, and TAM pathways). In the “IPyA pathway”, tryptophan is converted to indole-3-pyruvate (IPyA) by a transamination, which is then decarboxylated to indole-3-acetaldehyde dehydrogenase (IAAld) by indole-3-pyruvate decarboxylase (IPDC). In the last step, IAAld is oxidized to IAA by an indole-3-acetaldehyde dehydrogenase (ALDH). Genes encoding decarboxylation (ipdC) and oxidation (aldH) are rate-limiting for IAA biosynthesis through this pathway, and thus were used to determine the presence of this pathway in individual genomes (Cueva-Yesquén et al. 2021; de Souza et al. 2019). In QDSM01 genome, we also found some of the trp cluster (trpA, trpB, trpCF, trpE, and trpGD) genes involved in tryptophan biosynthesis. These genes may play a role in the synthesis of tryptophan used in multiple biological processes including IAA biosynthesis (Kang et al. 2016). As for the “TAM pathway”, tryptophan is converted to tryptamine (TAM), by decarboxylation, which is then converted to IAAld by oxidation. Genes catalyzing decarboxylation (ddc) and oxidation (aldH) are critical in this pathway and were searched in bacterial genomes (de Souza et al. 2019).
Iron is the fourth most abundant element in the earth’s crust, but in aerobic (oxidant) conditions and neutral pH, it is almost insoluble for plants (Cueva–Yesquén et al. 2021). Siderophores are low-molecular-weight secondary metabolites produced by PGPR (and other microbes) in iron deficiency states to bind iron and supply it to the bacterial cells (Santoyo et al. 2021). In Fig. 6d, siderophores production in QDSM01 culture was 27.27%. In Table 4 and Supplementary Tables 6, siderophore translocation through the bacterial outer membrane is performed by an energy transducing complex with proteins tonB, exbB, and exbD. The iron (III)-hydroxamate ABC transporter cluster fhuCDB, is responsible of the transport of ferrichrome and other Fe3+-hydroxamate compounds. Furthermore, ferrous iron uptake protein efeU was also found (Suarez et al. 2019). Moreover, the NRPS cluster is also involved in cell motility and biofilm formation, both of which are directly dependent on iron concentration in various bacteria (Wang et al. 2021). Additionally, turnerbactin encoded by NRPS cluster, a novel triscatecholate siderophore, is responsible for iron regulation and uptake. The biosynthesis of turnerbactin requires homologs of entABCDEF. These genes are clustered alongside genes required for siderophore export and uptake (Reitz et al. 2017).
Nitrogen can be captured and fixed in soil by diazotrophic bacteria, which are responsible for the fixation of atmospheric nitrogen into ammonia (Santoyo et al. 2021). In Fig. 6e, nitrogen generation capacity in QDSM01 was 13723.33 µg/mL on average. In Table 4 and Supplementary Tables 6, the genome of QDSM01 lacks the genes responsible for the nitrogen fixation (nif) main component. However, the strain contains a gene essential for nitrite reduction pathways (nasA). Furthermore, nitrate and nitrite transport-related genes nrtABC and narGHIJKLQX were also detected (Iqbal et al. 2021).
In soil, phosphorus-solubilizing bacteria and potassium-solubilizing bacteria can transform insoluble phosphorus and potassium in soil into soluble substances that can be easily absorbed by plants (Chen et al. 2022; Santoyo et al. 2021). In Fig. 6e, the average capacities of P-solubilization and K-solubilization in QDSM01 were 26.86 and 2.5 µg/mL, respectively. One of the mechanisms by which bacteria improve plant acquisition of phosphorus and potassium is by producing and releasing gluconic acid (GA). GA acts by solubilizing poorly soluble minerals making them available to plants. In Table 4 and Supplementary Tables 6, the gcd is a critical gene in the biosynthetic pathway of GA and has been widely documented in many P-solubilizing bacteria K-solubilizing bacteria (de Souza et al. 2019; Etesami et al. 2017). Genes related to phosphonate transport (phnC, phnD, and phnE) and degradation (phnG, phnH, phnI, phnJ, phnK, and phnL) were found in robust colonizers of the genera Ensifer, Variovorax, Burkholderia, and Agrobacterium, as well as in the non-robust colonizers of the genera Pantoea, Enterobacter, and Un. Bradyrhizobiaceae (de Souza et al. 2019). For K+-transport, KdpABC complex transporters play versatile roles in K+ acquisition and transport (Yang et al. 2022), and the genes of kefB, kefC, kefF, and kefG are associated with efflux system (Naveed et al. 2014). Moreover, potassium uptake system-related genes (trkA, trkH, and kup) were also found in strain QDSM01 (Wang and Sun 2017).
Conclusion
In this study, after phylogenetic analysis and ANI analysis, the strain QDSM01 was identified as Leclercia adecarboxylata. Moreover, the utilization of QDSM01 to plant growth promotion is highlighted by their potential for IAA production, nitrogen generation, phosphate solubilization, potassium solubilization, biofilm formation, and siderophores as demonstrated in the present experiments using maize and rice as models. Meanwhile, QDSM01 harbored many genes related to biofilm formation, quorum sensing, chemotaxis, motility, IAA production, siderophore, nitrogen generation, solubilization and uptake of phosphate and potassium. Overall, our study generated molecular evidences for the growth-promoting properties of QDSM01 and thus paved way for its large-scale application in agriculture.
Data availability
The genome sequences and associated data for Leclercia adecarboxylata QDSM01 reported in this study were deposited in NCBI under the BioProject accession number PRJNA841055.
References
Abdelaal K, AlKahtani M, Attia K, Hafez Y, Király L, Künstler A (2021) The role of plant growth-promoting bacteria in alleviating the adverse effects of drought on plants. Biology 10:520. https://doi.org/10.3390/biology10060520
Andrés-Barrao C, Lafi FF, Alam I, de Zélicourt A, Eida AA, Bokhari A et al (2017) Complete genome sequence analysis of enterobacter sp. SA187, a plant multi-stress tolerance promoting endophytic bacterium. Front Microbiol 8:2023. https://doi.org/10.3389/fmicb.2017.02023
Berendsen RL, Vismans G, Yu K, Song Y, de Jonge R, Burgman WP et al (2018) Disease-induced assemblage of a plant-beneficial bacterial consortium. ISME J 12:1496–1507. https://doi.org/10.1038/s41396-018-0093-1
Bhise KK, Dandge PB (2019) Mitigation of salinity stress in plants using plant growth promoting bacteria. Symbiosis 79:191–204. https://doi.org/10.1007/s13199-019-00638-y
Bouteiller M, Dupont C, Bourigault Y, Latour X, Barbey C, Konto-Ghiorghi Y et al (2021) Pseudomonas flagella: generalities and specificities. Int J Mol Sci 22:3337. https://doi.org/10.3390/ijms22073337
Chen YF, Yang H, Shen ZZ, Ye JR (2022) Whole-genome sequencing and potassium-solubilizing mechanism of Bacillus aryabhattai SK1-7. Front Microbiol 12:722379. https://doi.org/10.3389/fmicb.2021.722379
Crits-Christoph A, Bhattacharya N, Olm MR, Song YS, Banfield JF (2020) Transporter genes in biosynthetic gene clusters predict metabolite characteristics and siderophore activity. Genome Res 31:239–250. https://doi.org/10.1101/gr.268169.120
Cueva-Yesquén LG, Goulart MC, de Angelis A, Nopper Alves D, Fantinatti-Garboggini M (2021) Multiple plant growth-promotion traits in endophytic bacteria retrieved in the vegetative stage from passionflower. Front Plant Sci 11:2282. https://doi.org/10.3389/fpls.2020.621740
Dahmani MA, Desrut A, Moumen B, Verdon J, Mermouri L, Kacem M et al (2020) Unearthing the plant growth-promoting traits of Bacillus megaterium RmBm31, an endophytic bacterium isolated from root nodules of Retama monosperma. Front Plant Sci 11:124. https://doi.org/10.3389/fpls.2020.00124
Danish S, Kiran S, Fahad S, Ahmad N, Ali MA, Tahir FA et al (2019) Alleviation of chromium toxicity in maize by Fe fortification and chromium tolerant ACC deaminase producing plant growth promoting rhizobacteria. Ecotoxicol Environ Saf 185:109706. https://doi.org/10.1016/j.ecoenv.2019.109706
Danish S, Zafar-ul-Hye DM, Mohsin F, Hussain M (2020) ACC-deaminase producing plant growth promoting rhizobacteria and biochar mitigate adverse effects of drought stress on maize growth. PLoS ONE 15:e0230615. https://doi.org/10.1371/journal.pone.0230615
de Souza RSC, Armanhi JSL, Damasceno NdB, Imperial J, Arruda P (2019) Genome sequences of a plant beneficial synthetic bacterial community reveal genetic features for successful plant colonization. Front Microbiol 10:1779. https://doi.org/10.3389/fmicb.2019.01779
Dong X, Tu C, Xie Z, Luo Y, Zhang L, Li Z (2022) The genome of Bacillus velezensis SC60 provides evidence for its plant probiotic effects. Microorganisms 10:767. https://doi.org/10.3390/microorganisms10040767
Etesami H, Emami S, Alikhani HA (2017) Potassium solubilizing bacteria (KSB): mechanisms, promotion of plant growth, and future prospects—a review. J Soil Sci Plant Nutr 17:897–911. https://doi.org/10.4067/S0718-95162017000400005
Fong JCN, Syed KA, Klose KE, Yildiz FH (2010) Role of Vibrio polysaccharide (vps) genes in VPS production, biofilm formation and Vibrio cholerae pathogenesis. Microbiol 156:2757–2769. https://doi.org/10.1099/mic.0.040196-0
Gu S, Wei Z, Shao Z, Friman VP, Cao K, Yang T et al (2020) Competition for iron drives phytopathogen control by natural rhizosphere microbiomes. Nat Microbiol 5:1002–1010. https://doi.org/10.1038/s41564-020-0719-8
Guan J, Xiao X, Xu S, Gao F, Wang J, Wang T et al (2015) Roles of RpoS in Yersinia pseudotuberculosis stress survival, motility, biofilm formation and type VI secretion system expression. J Microbiol 53:633–642. https://doi.org/10.1007/s12275-015-0099-6
Iqbal S, Ullah N, Janjua HA (2021) In vitro evaluation and genome mining of Bacillus subtilis strain RS10 reveals its biocontrol and plant growth-promoting potential. Agriculture 11:1273. https://doi.org/10.3390/agriculture11121273
Jung BK, Khan AR, Hong S-J, Park G-S, Park Y-J, Kim H-J et al (2017) Quorum sensing activity of the plant growth-promoting rhizobacterium Serratia glossinae GS2 isolated from the sesame (Sesamum indicum L.) rhizosphere. Ann Microbiol 67:623–632. https://doi.org/10.1007/s13213-017-1291-1
Kang S, Asaf S, Kim S-J, Yun B-W, Lee I-J (2016) Complete genome sequence of plant growth-promoting bacterium Leifsonia xyli SE134, a possible gibberellin and auxin producer. J Biotechnol 239:34–38. https://doi.org/10.1016/j.jbiotec.2016.10.004
Kang SM, Shahzad R, Bilal S, Khan AL, Park YG, Lee KE et al (2019) Indole-3-acetic-acid and ACC deaminase producing Leclercia adecarboxylata MO1 improves Solanum lycopersicum L. growth and salinity stress tolerance by endogenous secondary metabolites regulation. BMC Microbiol 19:80. https://doi.org/10.1186/s12866-019-1450-6
Kang S, Shahzad R, Khan MA, Hasnain Z, Lee K-E, Park H-S et al (2021) Ameliorative effect of indole-3-acetic acid and siderophore-producing Leclercia adecarboxylata MO1 on cucumber plants under zinc stress. J Plant Interact 16:30–41. https://doi.org/10.1080/17429145.2020.1864039
Kumawat KC, Sharma P, Singh I, Sirari A, Gill BS (2019) Co-existence of Leclercia adecarboxylata (LSE-1) and Bradyrhizobium sp. (LSBR-3) in nodule niche for multifaceted effects and profitability in soybean production. World J Microbiol Biotechnol 35:172. https://doi.org/10.1007/s11274-019-2752-4
López D, Fischbach MA, Chu F, Losick R, Kolter R (2009) Structurally diverse natural products that cause potassium leakage trigger multicellularity in Bacillus subtilis. Proc Natl Acad Sci 106:280–285
Lulamba TE, Green E, Serepa-Dlamini MH (2021) Genome assembly and annotation of Photorhabdus heterorhabditis strain ETL reveals genetic features involved in pathogenicity with its associated entomopathogenic nematode and anti-host effectors with biocontrol potential applications. Gene 795:145780. https://doi.org/10.1016/j.gene.2021.145780
Naveed M, Ahmed I, Khalid N, Mumtaz AS (2014) Bioinformatics based structural characterization of glucose dehydrogenase (gdh) gene and growth promoting activity of Leclercia sp. QAU-66. Brazilian J Microbiol 45:603–611. https://doi.org/10.1590/s1517-83822014000200031
Niu B, Paulson Joseph N, Zheng X, Kolter R (2017) Simplified and representative bacterial community of maize roots. Proc Natl Acad Sci 114:E2450–E2459
Qiao Y, Feng L, Jia R, Luo Y, Yang Q (2022) Motility, biofilm formation and associated gene expression in Vibrio parahaemolyticus impaired by co-culture with live Ulva fasciata. J Appl Microbiol 132:101–112. https://doi.org/10.1111/jam.15175
Reitz ZL, Sandy M, Butler A (2017) Biosynthetic considerations of triscatechol siderophores framed on serine and threonine macrolactone scaffolds. Metallomics 9:824–839. https://doi.org/10.1039/c7mt00111h
Romero D, de Vicente A, Rakotoaly RH, Dufour SE, Veening J-W, Arrebola E et al (2007) The iturin and fengycin families of lipopeptides are key factors in antagonism of Bacillus subtilis toward Podosphaera fusca. Mol Plant Microbe Interact 20:430–440. https://doi.org/10.1094/MPMI-20-4-0430
Sandilya SP, Jeevan B, Subrahmanyam G, Dutta K, Vijay N, Bhattacharyya N et al (2022) Co-inoculation of native multi-trait plant growth promoting rhizobacteria promotes plant growth and suppresses alternaria blight disease in castor (Ricinus communis L). Heliyon 8:e11886. https://doi.org/10.1016/j.heliyon.2022.e11886
Santoyo G, Urtis-Flores CA, Loeza-Lara PD, Orozco-Mosqueda MD, Glick BR (2021) Rhizosphere colonization determinants by plant growth-promoting rhizobacteria (PGPR). Biology 10:475. https://doi.org/10.3390/biology10060475
Shankar M, Ponraj P, Ilakiam D, Rajendhran J, Gunasekaran P (2012) Genome sequence of the plant growth-promoting bacterium Enterobacter cloacae GS1. J Bacteriol 194:4479–4479. https://doi.org/10.1128/JB.00964-12
Sharma G, Khatri I, Subramanian S (2018) Comparative genomics of myxobacterial chemosensory systems. J Bacteriol 200:e00620–e00617. https://doi.org/10.1128/JB.00620-17
Shilev S (2020) Plant-growth-promoting Bacteria mitigating soil salinity stress in plants. Appl Sci 10:7326. https://doi.org/10.3390/app10207326
Snak A, Vendruscolo ECG, Santos MFd, Fiorini A, Mesa D (2021) Genome sequencing and analysis of plant growth-promoting attributes from Leclercia adecarboxylata. Genet Mol Biology 44:e20200130. https://doi.org/10.1590/1678-4685-gmb-2020-0130
Suarez C, Ratering S, Hain T, Fritzenwanker M, Schnell S (2019) Complete genome sequence of the plant growth-promoting bacterium Hartmannibacter diazotrophicus strain E19T. Int J Genomics. https://doi.org/10.1155/2019/7586430
Tunchai M, Hida A, Oku S, Nakashimada Y, Tajima T, Kato J (2017) Identification and characterization of chemosensors for d-malate, unnatural enantiomer of malate, in Ralstonia pseudosolanacearum. Microbiology 163:233–242. https://doi.org/10.1099/mic.0.000408
Wang H-l, Sun L (2017) Comparative metagenomics reveals insights into the deep-sea adaptation mechanism of the microorganisms in Iheya hydrothermal fields. World J Microbiol Biotechnol 33:86. https://doi.org/10.1007/s11274-017-2255-0
Wang X, Wang C, Li Q, Zhang J, Ji C, Sui J et al (2018) Isolation and characterization of antagonistic bacteria with the potential for biocontrol of soil-borne wheat diseases. J Appl Microbiol 125:1868–1880. https://doi.org/10.1111/jam.14099
Wang X, Zhang M, Loh B, Leptihn S, Ahmed T, Li B (2021) A novel NRPS cluster, acquired by horizontal gene transfer from algae, regulates siderophore iron metabolism in Burkholderia seminalis R456. Int J Biol Macromol 182:838–848. https://doi.org/10.1016/j.ijbiomac.2021.04.051
Yang L, Muhammad I, Chi YX, Wang D, Zhou XB (2022) Straw return and nitrogen fertilization to maize regulate soil properties, microbial community, and enzyme activities under a dual cropping system. Front Microbiol 13:823963. https://doi.org/10.3389/fmicb.2022.823963
Zhang N, Yang DQ, Wang DD, Miao YZ, Shao JH, Zhou X et al (2015) Whole transcriptomic analysis of the plant-beneficial rhizobacterium Bacillus amyloliquefaciens SQR9 during enhanced biofilm formation regulated by maize root exudates. BMC Genomics 16:685. https://doi.org/10.1186/s12864-015-1825-5
Zuo J, Yin H, Hu J, Miao J, Chen Z, Qi K et al (2019) Lsr operon is associated with AI-2 transfer and pathogenicity in avian pathogenic Escherichia coli. Vet Res 50:109. https://doi.org/10.1186/s13567-019-0725-0
Funding
This work was supported by the Key Research and Development Projects in Heilongjiang, China (GA21B007); and the Outstanding Youth Foundation in Heilongjiang Province, China (JQ2023D001).
Author information
Authors and Affiliations
Contributions
ZW and WX conceived and designed the experiments; WC performed the experiments and analyzed the data; YH participated in the collection of samples and the planning and coordination of the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors have no relevant financial or nonfinancial interests to disclose.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Communicated by Xiue Wang
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, W., Wang, Z., Xu, W. et al. Genome sequence of Leclercia adecarboxylata QDSM01 with multiple plant growth promoting properties. Plant Growth Regul 102, 445–459 (2024). https://doi.org/10.1007/s10725-023-01071-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10725-023-01071-4