
Abstract
Evolution of genus is accompanied by ecological diversification. The majority of species grow in open, sunny, rather dry sites in arid and moderately humid climates. However, Allium species have adapted for many other ecological niches. Classical approaches for the identification of Allium cultivars are based on morphological traits. The assessment of these traits is difficult and their evaluation can be subjective considering that most of these cultivars are closely related. Hence, this study of Internal Transcribed Spacer (ITS) sequencing and four barcoding regions, matK, rbcL, trnH-psbA, trnL and Inter Simple Sequence Repeats (ISSR) were researched in onion, Allium cepa L. (Alliaceae) collected from three different cultivation sites. The results established noticeable hereditary divergence among the three cultivars. In ITS and matK, BDUT 1453, BDUT 1454 and BDUT 1455 were independent of each other and formed three clusters. In rbcL, BDUT 1453 formed an independent cluster from the cluster of BDUT 1454 and BDUT 1455. But in trnH-psbA, BDUT 1454 formed an independent cluster and BDUT 1453 and BDUT 1455 were closely placed whereas trnL showed all the three forming a cluster wherein BDUT 1453 and BDUT 1454 were placed closely in a sub-cluster. In ISSR, BDUT 1454 and BDUT 1455 formed a single cluster and BDUT 1453 diverged from it. Even though the tested cultivars belong to the same species they showed genetic divergence among themselves.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The identification of crop cultivars holds paramount importance in safeguarding and harnessing biodiversity; however, this process might encounter delays stemming from insufficient taxonomic expertise (Chase and Fay 2009; Pathirana and Carimi 2022). In addition to whole-plant identification, there are instances where, identifying a cultivar from roots, seeds, pollen, or even from plant mixtures sampled across diverse ecosystems proves valuable. Nonetheless, conventional morphological methods may prove intricate or unfeasible in such scenarios (CBOL plant working group 2009).
Root and bulb vegetables (RBV) are often considered as orphan crops— playing a vital role in regional food security— majorly belonging to the families Apiaceae and Amaryllidaceae (formerly Alliaceae) (Bhasi et al. 2010). Alliaceae consists of common food crops like Onion, Garlic and Leek which are used in large volume in many parts of the world. However, the number of dedicated Plant Scientists focusing on these RBV crops remains limited – potentially as few as 10–25 full-time equivalent scientists engaged in academic and government initiatives worldwide. Much of the research effort emanates from breeders, geneticists, taxonomists, plant pathologists, and plant physiologists. A comparable level of dedication is also exhibited by seed companies globally (Brooks and Vest 1985; Frey 1999). Despite the relatively modest size of the scientific community, there has been a remarkable accretion of genomic information. This is notably due to endeavours aimed at expanding molecular genetic maps, elucidating taxonomic relationships, and investigating molecular aspects of gene expression. This compact RBV scientific community is getting more curious as the recent outcomings of genomic data from different domains of plant science facilitating the data accessibility (Bhasi et al. 2010).
Historically, systematic and phylogenetic analyses of these pungent plants primarily relied on morphological traits (Gurcharan 2004). However, the past few decades have witnessed constant breakthroughs in molecular biology, particularly genetics, presenting a new array of tools suitable for unravelling their relationships and elucidating the unique chemical profiles of associated cultivars (Gounaris et al. 2002; Labra et al. 2004). There were several modern molecular techniques employed for the assessment of genomic profiling of Allium species. Some of the significant markers include Expressed Sequence Tags (ESTs) using Cleavage amplified polymorphisms (CAPs) and Single-Stranded Conformation polymorphisms (SSCP) markers (McCallum et al. 2001), Intron Length Polymorphic (ILP) markers (Jayaswall et al. 2019; Khade et al. 2022), Potential Intron Polymorphism (PIP) markers (Jayaswall et al. 2024), R-gene derived molecular markers (Herlina et al. 2019) and Single-nucleotide polymorphism (SNP) markers (Fujito et al. 2021).
Discrete markers like Random Amplified Polymorphic DNA (RAPD), Amplified Fragment Length Polymorphism (AFLP), and hypervariable DNA regions (such as SSRs in the microsatellite regions) have commonly been employed as molecular methodologies for such investigations (Friesen and Klaas 1998; Labra et al. 2004; Trindade 2007; Baldwin et al. 2012; Mallor et al. 2014; Jayaswall et al. 2022; Raj et al. 2022; Chalbi et al. 2023). Still — despite their demonstrated effectiveness — these markers face limitations in discriminating between diverse species or cultivars (Trindade 2007; Azizi et al. 2009). Moreover, their applicability across a broad spectrum of taxa is often constrained, as they have evolved within specific genera or species (Novak 2008; Segarra-Moragues and Gleiser 2009). However, homozygosity must be factor to be considered while genotypic classification especially in the in-bred cultivars invariant to the molecular markers (Moon et al. 2023).
The progress in sequencing and computational technologies has elevated DNA sequences to a prime source of innovative insights, enhancing our understanding of evolutionary and genetic relationships. The impacts of sequence analysis are now noticeable across nearly all domains of Biological Sciences, spanning from developmental research to epidemiology (Tibayrenc 2005). However, two distinct branches of biology have pioneered the tools and applications used to explore biological relationships through DNA sequences: molecular phylogenetics and population genetics. These fields address different tiers of organizational complexity. Molecular phylogenetics traditionally delves into evolutionary relationships among broader clades, whereas population genetics focuses on variations within and among populations of individual species. In contrast, DNA barcoding occupies an intermediary role, aiming for comprehensive species coverage while emphasizing their identification rather than relational aspects.
DNA barcoding represents a relatively recent technique that has been developed to offer swift, accurate, and automated species identification by utilizing standardized DNA sequences as tags (Hebert et al. 2003; Taberlet 2007). The origins of this approach trace back to the seminal work of Hebert et al. (2003), who demonstrated that a collection of 200 closely related lepidopteran species could be distinguished with 100% accuracy using the mitochondrial gene cytochrome c oxidase subunit I (COI). While COI proved less effective in plants, several other genetic loci have been proposed as potential plant barcodes, including Internal Transcribed Spacer (ITS) (Kress et al. 2005; Chase et al. 2007), rbcL (Newmaster et al. 2006; Kress and Erickson 2007; Hollingsworth et al. 2009; Anvarkhah et al. 2013), psbA-trnH (Chase et al. 2007; Kress and Erickson 2007; Lahaye et al. 2008; Chen et al. 2010; Gao et al. 2010; Fu et al. 2011), and matK (Chase et al. 2007; Hollingsworth et al. 2009; Zarei et al. 2020).
In the present context, barcoding has evolved into a dependable technique for species identification (Vijayan and Tsou 2010; Singh et al. 2021). The fundamental principle underlying barcoding involves comparing sequence data from an unknown sample (the specimen under study) to a reference sequence obtained from a voucher specimen. The barcode sequence of each unknown specimen is then matched against a reference barcode sequence library, established from individuals with known attributes. A species is confirmed if its sequence closely corresponds to one within the barcode library. Alternatively, novel documentation might lead to the proposal of a new barcode sequence for a known species, or it may even contribute to the recognition of a previously undiscovered species (Hajibabaei et al. 2007).
The assessment of genetic diversity through molecular markers plays a pivotal role in comprehending genome structure, characterizing and preserving genetic variations within plant germplasm, pinpointing genes linked to significant traits, and formulating effective breeding strategies for crop enhancement (Hayden et al. 2010). The utilization of markers and the recognition of polymorphic nucleotide sequences dispersed across the genome have opened up fresh avenues for appraising diversity and discerning inter- and intra-species genetic relationships (Gostimsky et al. 2005).
Numerous molecular markers are at our disposal for probing genetic diversity. Among these, SSR (Tautz 1989; Becker and Heun 1994), RAPD (Williams et al. 1990), AFLP (Vos 1995), and inter simple sequence repeat( (ISSR) (Zietkiewciz et al. 1994) have stood out as the most influential. However, these methods have faced significant limitations, such as the RAPD’s poor reproducibility, the high costs associated with AFLP, and the necessity to possess flanking sequences for designing specific primers in the case of SSR markers. In comparison, ISSR markers have successfully surmounted many of these challenges (Reddy et al. 2002). They offer the distinct advantages of being relatively cost-effective, showcasing high levels of polymorphism, and exhibiting strong reproducibility (Peng et al. 2006).
The ISSR, representing a relatively recent category of molecular markers, relies on the presence of short DNA sequences organized in tandem repeats. Notably, these inter-repeat regions display substantial polymorphism in their sizes, even among closely related genotypes, owing to the absence of evolutionary functional constraints within these non-functional domains (Rizkalla et al. 2012).
The objective of this study is to elucidate the relationships among three distinct cultivars of A. cepa (onion) through multiple approaches. These include analyzing ITS sequences, employing DNA barcodes through primers like ITS, matK, rbcL, trnH-psbA, and trnL, as well as incorporating seven ISSR markers.
Materials and methods
To assess the appropriate degree of sequence divergence within the plant genome, Allium cepa specimens collected from three different cultivation sites: Surandai (BDUT 1453) and Alankulam (BDUT 1454) in the Tirunelveli District, and Vilathikulam (BDUT 1455) in the Tuticorin District of Tamil Nadu, India. All three specimens, representing traditional cultivars of their respective localities, were cultivated in sites closely resembling their natural habitats. Specimens were collected from the field with due permissions from the farmers. For genetic analysis, fresh young roots (each weighing 200 mg) were obtained from the selected Allium plants. DNA isolation was conducted by employing the CTAB method (Doyle and Doyle 1987). To ensure DNA of high quality, the isolated genetic material underwent purification utilizing a spin column kit. Subsequently, the concentration of purified DNA for each sample was quantified based on the intensity of ethidium bromide-stained bands.
To compare the performance of various DNA markers, each sample underwent analysis using both ITS and four candidate DNA barcoding genomic regions. These include five DNA loci amplified with universal primers: Internal Transcribed Spacer (ITS) gene (White et al. 1990), rbcL gene (Bafeel et al. 2011), matK gene (Costion et al. 2011), PsbA gene (Jabbes et al. 2011), and trnL gene amplifications (Table 1).
For ISSR analysis, primers were constructed according to the method outlined by Jabbes et al. (2011). Seven distinct ISSR primers were employed for the amplification of the cultivars: ISSR8US, ISSR9, ISHY 1b, ISHY 2, ISHY 3, ISHY 4, and ISSR a (Table 2). Consider that the annealing temperature varied for each specific primer (Jabbes et al. 2011). Each PCR reagent mixture comprised 10 µl of Taq pre-mix, 4 µl of water, 1.5 µl of the forward primer, 1.5 µl of the reverse primer, and 3 µl of DNA.
The PCR Thermal Cycler Program was performed using the Eppendorf ProS, Hamburg, Germany. For DNA barcoding and ITS markers, the PCR thermal cycle involved one initial cycle of 5 min at 94 °C, followed by 35 cycles comprising 30 s at 94 °C, 30 s at 58 °C, and 60 s at 72 °C, with a final cycle of 10 min at 72 °C.
Regarding the PCR programs for amplifying ISSR markers, 40 cycles of denaturation at 94 °C for 30 s, annealing at a temperature determined by the specific primer used (Table 2), followed by a 2 min amplification step at 72 °C. The final amplification set with the last cycle for 10 min at 72 °C. Subsequently, all PCR products were subjected to electrophoresis on a 1.2% agarose gel in 1X TAE buffer at 60 V for a duration of 1.30 h, and visualized using a UV transilluminator. Additionally, DNA barcoding and ITS PCR products underwent purification prior to sequencing. The Sanger dideoxy method was employed for DNA sequencing of the PCR products. The generated data were imported and aligned using Molecular Evolutionary Genetics Analysis (MEGA v5.2.2).
AF-Total amplified fragments; PF- Number of polymorphic amplicons; % P- Percentage of polymorphism.
Basic sequence statistics, like nucleotide frequencies, the transition/transversion (ns/nv) ratio, and variability within distinct sequence regions, were computed using the MEGA software. The sequence data were subjected to analysis through both phenetic and cladistic methods. The phenetic approach involved employing the neighbor-joining method (NJ), while the cladistic method utilized the maximum parsimony method (MP).
The ISSR marker index was calculated to assess the efficacy of each primer in identifying polymorphic loci (P) within the cultivars. Subsequently, the computation of Shannon index (I) (Lewontin 1972) and Nei’s standard genetic distance (D) (Nei 1972) took place, followed by the construction of a dendrogram using POPGENE v32 Software. For a Bayesian analysis of the ISSR data, the Structure v3.2.2 Software was utilized.
Results
The ITS region of A. cepa was analyzed to determine the phylogenetic relationships among the various cultivars of Allium species. The lengths of the ITS regions in the evaluated cultivars were 666 bp in BDUT 1453, 651 bp in BDUT 1454, and 663 bp in BDUT 1455. Similarly, for the matK region, all three cultivars shared different lengths: 684 bp in BDUT 1453, 818 bp in BDUT 1454, and 822 bp in BDUT 1455. Likewise, the trnH-psbA region were 646 bp in BDUT 1453, 653 bp in BDUT 1454, and 645 bp in BDUT 1455. In the same way, the rbcL region exhibited different lengths of 531 bp in BDUT 1453, 530 bp in BDUT 1454, and 534 bp in BDUT 1455. Remarkably, the trnL region demonstrated a smaller size compared to the other barcoding regions examined in this study, featuring a consistent length of 246 bp with all three cultivars.
Interestingly, the ITS phylogeny indicated that the three distinct Allium cultivars were failed to coalesce into a single clade. Employing the phenetic method for ITS sequence analysis, BDUT 1453 exhibited a close relationship with A. fistulosum gi338191570, while the cultivar BDUT 1454 formed an out-group, branching away from the cluster encompassing A. cepa gi21627887 to A. altaicum gi133919855. Conversely, the cultivar BDUT 1455 demonstrated a close relationship with A. altaicum gi259186340 (Fig. 1).

NJ (Phenetic method) tree based on Internal Transcribed Spacer (ITS) region of Allium species
Among the cultivars studied, BDUT 1453 and BDUT 1454 displayed a close relationship, evidenced by a pairwise distance of 5.308 between them. Both these cultivars had diverged from their near common ancestor, BDUT 1455. Notably, the pairwise distance between BDUT 1454 and 1455 was 6.22, while that between BDUT 1453 and BDUT 1455 was 8.44.
In the context of the cladistic method applied to ITS sequence analysis, the cultivar BDUT 1455 exhibited a pronounced affinity with A. altaicum gi259186340. Conversely, the cultivar BDUT 1454 diverged from the cluster that encompassed A. cepa gi444237454. Intriguingly, BDUT 1453 emerged from the cluster formed by A. fistulosum gi256596112 and A. cepa gi256596111 (Fig. 2). Remarkably, the cultivars clustered together, suggesting a significant genetic divergence, when subjected to both the neighbour-joining (NJ) and maximum parsimony (MP) methods.

MP (Cladistic method) tree based on Internal Transcribed Spacer (ITS) region of Allium species.
In this present study, the nucleotide frequencies calculated were as follows: A = 20.56%, T/U = 33.33%, C = 20.56%, and G = 25.56%. Both the neighbor-joining (NJ) and maximum parsimony (MP) methods highlighted substantial genetic divergence among the three tested onion cultivars of Allium.
The shape parameter for the distinct gamma distribution was estimated to be approximately 200.0000. Substitution patterns and rates were determined using the Tamura-Nei model (+ G) (Tamura and Nei 1993). To account for variations in evolutionary rates across sites, a discrete gamma distribution was applied with 5 categories (+ G). The mean evolutionary rates within these categories were identified as 0.90, 0.96, 1.00, 1.04, and 1.10 substitutions per site.
Likewise, the nucleotide frequencies were distributed as follows: A = 20.56%, T/U = 33.33%, C = 20.56%, and G = 25.56%. The computation resulted in a maximum Log likelihood value of − 245.115. Furthermore, the estimated Transition/Transversion bias (R) was calculated to be 0.33 (Kimura 1980). The nucleotide frequencies were characterized as follows: A = 25.00%, T/U = 25.00%, C = 25.00%, and G = 25.00%. The maximum Log likelihood value for this particular computation was − 249.497.
The phylogenetic relationships of barcoding gene sequences, matK, rbcL, trnH-psbA, and trnL, were determined through both phenetic and cladistic methods of phylogenetic analysis. During the matK analysis, the nucleotide frequencies were distributed as follows: A = 32.81%, T/U = 40.09%, C = 14.52%, and G = 12.58%. The corresponding computation yielded a maximum Log likelihood value of -2690.657. Additionally, the Transition/Transversion bias (R) was estimated to be 0.74, with the maximum Log likelihood value of − 2921.083.
During the matK region analysis, BDUT 1453 exhibited divergence from A. cyaneum gi379323965. BDUT 1454 diverged from a common ancestor shared by A. scorodoprasum var. viviparum gi519670102 and A. monanthum gi519670092. BDUT 1455’s evolutionary path led from a comprehensive cluster encompassing a majority of other Allium species (Fig. 3).

NJ (Phenetic method) tree based on matK region of Allium species
Similar to the ITS sequence analysis, the phylogenetic relationship among the three tested onion cultivars remained consistent. The pairwise distance between BDUT 1453 and 1454 was calculated at 3.74, while the distance between BDUT 1455 and 1454 was 6.42. The pairwise distance between BDUT 1453 and 1455 was 6.28.
In the MP method applied to matK sequence analysis, BDUT 1453 exhibited divergence from A. cepa gi387865328. The evolutionary trajectory of BDUT 1454 stemmed from a cluster containing A. oleraceum gi3790415782 and A. condensatum gi379323423. On the other hand, BDUT 1455 demonstrated divergence from a cluster featuring some Allium species, while also branching out from another cluster of different Allium species (Fig. 4). Notably, the shape parameter for the discrete Gamma Distribution was identical to the ITS sequence analysis. During this study, the analysis of matK using both the NJ and MP methods revealed a discrepancy in terms of species genetic divergence. Despite this difference, the phylogenetic trees generated by both methods showcased distinct topologies.

MP (Cladistic method) tree based on matK region of Allium species
Examining the rbcL region of the tested cultivars revealed that BDUT 1454 and BDUT 1455 shared a close relationship, forming a cohesive cluster in both methods of phylogenetic analysis. The calculated pairwise distance between them amounted to a mere 0.01.
The analysis of rbcL using NJ method revealed that BDUT 1454 and 1455 diverged from the common cultivar BDUT 1453 which was out grouped from A. cepa gi387865420 (Fig. 5). On the other hand, MP method produced a result where BDUT 1453 formed a cluster with A. cepa gi478430773, while the other two cultivars clustered together (Fig. 6). In terms of pairwise distances, the measurement between BDUT 1453 and 1455 amounted to 5.82, while the distance between BDUT 1453 and 1454 stood at 5.84.

NJ (Phenetic method) tree based on rbcL region of Allium species

MP (Cladistic method) tree based on rbcL region of Allium species
The shape parameter for the distinct gamma distribution was estimated as 47.6767. The nucleotide frequencies for the four bases were recorded as follows: A = 29.56%, T/U = 29.37%, C = 21.57%, and G = 19.50% with a corresponding maximum Log likelihood value of -1482.077. Moreover, the estimated Transition/Transversion bias (R) was calculated was 0.51, with the maximum Log likelihood value of − 1501.008.
During the trnH-psbA region analysis, BDUT 1454 diverged from BDUT 1455 and aligned with A. sikkimense gi379323442 to form a cluster. Meanwhile, the other cultivar, BDUT 1453, formed a cluster with A. carinatum gi406033480, exhibiting divergence in the NJ method (Fig. 7).

NJ (Phenetic method) tree based on trnH-psbA region of Allium species
In the MP method of analysis, BDUT 1453 and 1455 were observed to cluster together, exhibiting a pairwise distance of 3.05. This cluster diverged from the other Allium cultivar, which in turn formed a cluster with BDUT 1454 (Fig. 8). The calculated pairwise distances were 2.51 between BDUT 1453 and 1455, and 2.76 between BDUT 1453 and 1454. Through trnH-psbA sequence analysis, it became evident that all three onion cultivars displayed genetic divergence in both the neighbor-joining (NJ) and maximum parsimony (MP) methods. Notably, both BDUT 1454 and 1455 formed a unified cluster, stemming from the cultivar BDUT 1453.

MP (Cladistic method) tree based on trnH-psbA region of Allium species
In the context of trnH-psbA analysis, the maximum Log likelihood corresponding to the shape parameter for the discrete Gamma Distribution was − 73.982. For the trnH-psbA region, the nucleotide frequencies were as follows: A = 25.00%, T/U = 48.33%, C = 13.33%, and G = 13.33%. This calculation yielded a maximum Log likelihood value of − 73.982. Moreover, the estimated Transition/Transversion bias (R) was found to be 0.00, with the maximum Log likelihood value of − 81.567.
The analysis of the trnL sequence revealed that BDUT 1453 and 1454 clustered together, and both cultivars exhibited divergence from BDUT 1455 in both the NJ method (Fig. 9) and the MP method (Fig. 10) of phylogenetic analysis. Notably, the pairwise distance was calculated to be 0.012 between BDUT 1453 and 1455, 0.016 between BDUT 1453 and 1454, and 0.029 between BDUT 1454 and 1455. For the discrete Gamma Distribution, the estimated value of the shape parameter was − 366.703. In terms of nucleotide frequencies, the four bases were recorded as follows: A = 40.51%, T/U = 24.39%, C = 14.77%, and G = 20.33%. This calculation produced a maximum Log likelihood value of -366.703. Furthermore, the estimated Transition/Transversion bias (R) was found to be 0.75, with the maximum Log likelihood value of -385.227.

NJ (Phenetic method) tree based on trnL region of Allium species

MP (Cladistic method) tree based on trnL region of Allium species
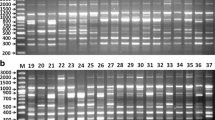
A total of 115 scorable bands were generated among the cultivars using seven ISSR primers. The amplified products exhibited sizes ranging from approximately 100 to 1300 bp (Figs. 11 and 12). The scorable bands produced by each primer varied, ranging from 13 to 24 (Table 2). The overall count of polymorphic alleles amounted to 29.00%, and the percentage of polymorphism was recorded at 25.21%.

ISSR banding pattern of three onion accessions generated by ISSR primers Lanes from left to right; lane 1–1 Kb ladder. Lane 2, 3, 4 – ISSR8US banding pattern for BDUT 1453, 1454 and 1455 respectively. Lane 5, 6, 7 – ISSR 9 banding pattern for BDUT 1453, 1454 and 1455 respectively. Lane 8, 9, 10 – ISSR a banding pattern for BDUT 1453, 1454 and 1455 respectively.

ISSR banding pattern of three onion accessions generated by ISSR primers Lanes from left to right; lane 1–1 Kb ladder. Lane 2, 3, 4 – ISHY 1b banding pattern for BDUT 1453, 1454 and 1455 respectively. Lane 5, 6, 7 – ISHY 2 banding pattern for BDUT 1453, 1454 and 1455 respectively. Lane 8, 9, 10 – ISHY 3 banding pattern for BDUT 1453, 1454 and 1455 respectively. Lane 11, 12, 13 – ISHY 4 banding pattern for BDUT 1453, 1454 and 1455 respectively.
The observed number of alleles (na) was 2, with an effective number of alleles (ne) calculated as 1.6274. Nei’s genetic diversity (h) was determined to be 0.3718, while Shannon’s information index (I) was calculated as 0.5544. The values for Ht, Hs, Gst, and Nm were computed as 0.3718, 0.3314, 0.1085, and 4.1065, respectively. The average number of alleles per locus was found to be 2.3. The estimated In Probability of data was − 20.3, with a mean value of In-likelihood at -19.8 and a variance of In-likelihood as 1.1. The ISSR analysis showed that the mean values of Fst-1, Fst-2, and Fst-3 were calculated as 0.0415, 0.0166, and 0.0076, respectively. Additionally, the average distances of clusters 1, 2, and 3 were determined to be 0.6159, 0.6142, and 0.6136, respectively, as depicted in the bar plot (Fig. 13). Notably, the L (K) achieved by Structure demonstrated a clear distinction among the different cultivars.

Bayesian proportion of individual plants for a K = 3 population model. The population identified by the Structure Software was indicated in different colours
Discussion
To elucidate the phylogenetic relationship between A. cepa and other Allium species, a comparison was conducted using ITS and barcoding sequences. Both the phenetic and cladistic methods of phylogenetic analysis yielded distinct tree topologies for the ITS sequences. The ITS region has been extensively employed for phylogenetic investigations in A. cepa by various researchers (Dubouzet and Shinoda 1998, 1999; Mes et al. 1999; Friesen et al. 2000; Fritsch and Friesen 2002). In particular, Dubouzet and Shinoda (1999) proposed that DNA sequence analysis, specifically utilizing the ITS sequence, serves as a valuable tool for understanding the intragenic organization within the Allium genus.
The initial phase of this study aimed to establish the universality of the five candidate DNA markers. To achieve this, we assessed the DNA markers that were consistently amplified and sequenced in the largest number of analyzed samples. To enhance the clarity of interpreting the outcomes, only the most universally effective primer combination for each candidate DNA marker was examined. The ITS phylogeny results were showing that the three distinct Allium cultivars were failed to merge into a single clade. While studying the phylogenetic clustering patterns, both the NJ and MP methods adopted for ITS region analysis reveals a significant genetic divergence among three Allium clusters studied.
In a study conducted by Ipek et al. (2008), the ITS sequences of diverse Allium species were examined to elucidate the phylogenetic connection between A. tuncelianum and other Allium species. The investigation revealed that both the NJ dendrogram and the consensus tree resulting from parsimony analysis yielded same tree topologies. Intriguingly, both the analyses placed A. tuncelianum within the clade of subgenus Allium, along with Garlic. Previously, the monophyly of section Allium was confirmed by Hirschegger et al. (2010). Four main clades were identified on all ITS analyses. However, the interconnections among these clades and the remaining species within section Allium remained unresolved. The employment of cpDNA-based phylogenetic trees led to the identification of two major clades, though the resultant topology only partially correlated with that of the ITS tree. To trace the presumed parent species of polyploid taxa, a method involving intra-individual polymorphism of the ITS region was utilized. The phylogenetic relationships of the barcoding locus, reported by Da-Cruz (2012), introduced a degree of confusion due to the presence of similarities across different species.
Subsequently, Nguyen et al. (2008) developed a phylogenetic tree employing ITS alone and in conjunction with ETS. This collective approach facilitated a comprehensive assessment of evolutionary relationships between Allium species. Notably, the ITS region autonomously offered substantial insights and determine the broader relationships among species. The incorporation of the second marker (ETS) not only reinforced the phylogenetic positions of the species but also contributed to enhancing resolution within the subgenus.
One striking characteristic of the ITS data is the strangely large intrageneric genetic distances within Allium. Distances exceeding 40% based on Kimura calculations were identified in research conducted by Friesen et al. (2000) and also in the work by Dubouzet and Shinoda (1999). Such distances often typify the most remotely related genera within subfamilies or even families (Baldwin et al. 1995; Blattner and Kadereit 1999; Hsiao et al. 1999; Noyes and Rieseberg 1998). In stark contrast, intrageneric distances within other plant families predominantly remain below the 10% threshold (Baldwin et al. 1995).
These findings position the Allium as either a remarkably rapid-evolving taxon or one of ancient origins, as molecular evolution hasn’t led to the emergence of comparable numbers of taxonomic categories. Moreover, the outcomes of a phylogenetic analysis showcased that all three cultivars belonging to the same species formed distinct clusters, indicative of their genetic divergence.
The CBOL plant working group (2009) proposed rbcL and matK as the standard barcodes for land plants. This combination embodies a pragmatic resolution to the intricate balance between universality, sequence quality, discrimination, and cost. In their study, utilizing rbcL and matK within the examined sample set yielded a species discrimination success rate of 72%, with the remaining species effectively matched to groups of congeneric species with a 100% success rate. Selecting a plant barcode from the available candidate loci posed a challenging task. Each locus (matK, rbcL, trnH-psbA, and trnL) possessed highly desirable attributes for an effective plant DNA barcoding system. However, none of these four loci completely met all the criteria perfectly.
The phylogenetic analysis of matK region of the present study showing a distinct, comprehensive clustering of BDUT1455 from that of other Allium species. Although, the NJ and MP methods of matk region showing a slightly different patterns of genetic divergence and distinct topologies, relationship among three tested cultivars remained consistent with ITS region analytical results.
Among the realm of plastid genes, rbcL emerges as the most regarded. Enhanced accessibility across various land plants has been achieved through advancements in primer design (Fazekas et al. 2008), rendering it well-suited for generating good quality bidirectional sequences. Regarded as an exemplary multi-locus candidate, rbcL demonstrates excellent performance among the most variable regions, facilitating species discrimination.
Both NJ an MP methods of rbcL region analysis resulted a unified clustering pattern of close relationship among three tested cultivars. Despite research reporting very low divergence for rbcL, especially among closely related species (Newmaster et al. 2008; Liu et al. 2010), Liu et al. (2010) highlighted its potential suitability for Bryophyta barcoding.
Insights from Kress et al. (2005) indicated trnH-psbA spacer ranged from 119 to over 100 bp across the studied Angiosperms. This variability in length could potentially lead to alignment difficulties, a concern that could impede the effectiveness of DNA barcoding due to the substantial number of insertions and deletions within trnH-psbA. For instance, a 94% match between two trnH-psbA sequences in Trigonella foenum-graecum L. might result in intraspecific variation or the misidentification of a single voucher. Consequently, utilizing multiple voucher sequences becomes essential, particularly for similar barcoding regions known to be highly similar (Schori and Schowalter 2011).
As reported by Kress and Erickson (2007), trnH-psbA demonstrated robust amplification across a range of land plants using a single pair of primers, while achieving high levels of species discrimination. Nonetheless, the major obstacle for this locus remains the difficulty in obtaining high-quality bidirectional sequences. The conclusions drawn by Friesen et al. (2000) suggest that Allium is either an exceptionally fast-evolving taxon or one with ancient origins. This implies that molecular evolution within Allium does not necessarily coincide with the emergence of a proportionate increase in taxonomic categories. However, the NJ and MP methods adopted for the trnH-psbA sequence analysis reveals a genetic divergence among three cultivars tested.
Research conducted by Cowan et al. (2006) involving 96 species of Sinningia (Gesneriaceae) showcased a remarkable 95% probability of correct identification through trnS-trnG, trnT-trnL, rpl16, trnL-trnF, arpB-rbcL, and ncpGS markers. The NJ and MP methods of trnL region analysis unveils the genetic divergence among three tested Allium cultivars. It is similar to those of other phylogenetic analyses conducted in this present study.
ISSR markers stand out for effectively studying intraspecific variations, particularly due to their proficiency in detecting even low levels of genetic polymorphism in plants (Zietkiewciz et al. 1994). In comparison, other methods like RAPDs and microsatellite-primed PCR markers tend to yield lower levels of polymorphism, while ISSR markers offer enhanced reliability and reproducibility of bands (Sonnante and Pignone 2001). Consequently, RAPD and various other molecular markers have been employed for studying intraspecific polymorphism, whereas ISSR has shown its proficiency in unravelling interspecific diversity (Nagaoka and Ogihara 1997; Hao et al. 2002; Goldman 2008; Bianco et al. 2011; Poczai et al. 2011; Mukherjee et al. 2013). ISSR analysis using the Structure package exposes clear distinction in the banding pattern. Thus, unveiled a significant level of intraspecific diversity among the various cultivars within A. cepa. This is consistent with the findings of Mukherjee et al. (2013), who reported high levels of polymorphism within A. sativum through ISSR analysis.
Conclusion
In conclusion, the molecular evidence presented in this study emphasizes the presence of significant interspecific diversity and intraspecific divergence within A. cepa. This phenomenon could potentially arise from the distinct characteristics of various cultivation sites, leading to random genetic drift. The employed barcode locus in this research yields substantial insights into A. cepa. To enhance the accuracy of identifying this specific species, it is recommended that future research incorporates a broader range of samples from diverse cultivation sites.
Data accessibility
Genebank Accession numbers: KF769488, KF769489, KF769490, KF769494, KF769495, KF769496, KF769500, KF769501, KF769502, KF769506, KF769507, KF769508, KF779156, KF779157, KF779156.
References
Anvarkhah S, Khajeh MH, Hassan Rashed MM, Ali Davari EP, Hashemi H (2013) Identification of three species of genus Allium using DNA barcoding. Intl J Agri Crop Sci 5:1195–1203
Azizi A, Wagner C, Honermeier B, Wolfgang F (2009) Intraspecific diversity and relationship between subspecies of Origanum vulgare revealed by comparative AFLP and SAMPL marker analysis. Plant Syst Evol 281:151–160. https://doi.org/10.1007/s00606-009-0197-1
Bafeel SO, Arif IA, Bakir MA, Khan HA, Farhan AHA, Homaidan AAA, Ahamed A, Thomas J (2011) Comparative evaluation of PCR success with universal primers of maturase K (matK) and ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit (rbcL) for barcoding of some arid plants. Plant Omics 4:195–198
Baldwin S, Pither-Joyce M, Wright K, Chen L, McCallum J (2012) Development of robust genomic simple sequence repeat markers for estimation of genetic diversity within and among bulb onion (Allium Cepa L.) populations. Molecular Breeding 30:1401–1411. https://doi.org/10.1007/s11032-012-9727-6
Baldwin BG, Sanderson MJ, Porter JM, Wojciechowski MF, Campbell CS, Donoghue MJ (1995) The ITS region of nuclear ribosomal DNA: a valuable source of evidence on angiosperm phylogeny. Ann Missouri Bot Gar 82:247–277. https://doi.org/10.2307/2399880
Becker J, Heun M (1994) Microsatellites: allele variation and mapping. Plant Molr Biol 27:835–845
Bhasi A, Senalik D, Simon PW, Kumar B, Manikandan V, Philip P, Senapathy P (2010) RoBust: an integrated genomics resource for the root and bulb crop families Apiaceae and Alliaceae. BMC Plant Biol 10:161. https://doi.org/10.1186/1471-2229-10-161
Bianco CL, Fernandez JA, Migliaro D, Crino P, Egea-Gilabert C (2011) Identification of F1 hybrids of artichoke by ISSR markers and morphological analysis. Mol Breed 27:157–170. https://doi.org/10.1007/s11032-010-9418-0
Blattner FR, Kadereit JW (1999) Morphological evolution and ecological diversification of the forest dwelling poppies (papaveracea: Chelidonioideae) as deduced from a molecular phylogeny of the ITS region. Plant Syst Evol 219:181–197. https://doi.org/10.1007/BF00985578
Brooks HJ, Vest G (1985) Public programs on genetics and breeding of horticultural crops in the United States. Hort Sci 20:826–830. https://doi.org/10.21273/HORTSCI.20.5.826
CBOL Plant Working Group (2009) A DNA barcode for land plants. Proc Natl Acad Sci USA 106:12794–12797. https://doi.org/10.1073/pnas.090584510
Chalbi A, Chikh-Rouhou H, Mezghani N, Slim A, Fayos O, Bel-Kadhi MS, Garcés-Claver A (2023) Genetic diversity analysis of onion (Allium cepa L.) from the Arid region of Tunisia using phenotypic traits and SSR markers. Horticulturae 9(10):1098. https://doi.org/10.3390/horticulturae9101098
Chase MW, Cowan RS, Hollingsworth PM, van den Berg C, Madrinan S et al (2007) A proposal for a standardised protocol to barcode all land plants. Taxon 56:295–299. https://doi.org/10.1002/tax.562004
Chase MW, Fay MF (2009) Barcoding of plants and Fungi. Science 325:682–683
Chen S, Yao H, Han J, Liu C, Song J et al (2010) Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS ONE. https://doi.org/10.1371/journal.pone.0008613
Costion C, Ford A, Cross H, Crayn D, Harrington M, Lowe A (2011) Plant DNA barcodes can accurately estimate species richness in poorly known Floras. PLoS ONE. https://doi.org/10.1371/journal.pone.0026841
Cowan RS, Chase MW, Kress WJ, Savolainen V (2006) 300,000 species to identify: problems, progress, and prospects in DNA barcoding of land plants. Taxon 55:611–616. https://doi.org/10.2307/25065638
Da-Cruz F (2012) Sistemática e filogenética molecular do gênero Hexachlamys (Myrtaceae) através do uso de marcadores plastidiais e nucleares. Programa de Pos-Graduacaoem genetic e Biologia Molecular department de Genetica, Instituto de Bicietica, Universidade Federal do Rio Grande do Sul. http://hdl.handle.net/10183/49275
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Dubouzet JG, Shinoda K (1998) Phylogeny of Allium L. Subgenus Melanocrommyum (Webb Et Berth.) Rouy based on DNA sequence analysis of the internal transcribed spacer region of rDNA. Theor Appl Genet 97:541–549. https://doi.org/10.1007/s001220050929
Dubouzet JG, Shinoda K (1999) Relationships among old and new world Alliums according to ITS DNA sequence analysis. Theor Appl Genet 98:422–433. https://doi.org/10.1007/s001220051088
Fazekas AJ, Burgess KS, Kesanakurti PR et al (2008) Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well. PLoS ONE. https://doi.org/10.1371/journal.pone.0002802
Frey KJ (1999) National plant breeding study-I. Special report 98. Iowa State University, Ames
Friesen N, Fritsch RM, Pollner S, Blattner FR (2000) Molecular and morphological evidence for an origin of the aberrant genus milula within himalayan species of Allium (Alliaceae). Mol Phylogenet Evol 17:209–218. https://doi.org/10.1006/mpev.2000.0844
Friesen N, Klaas M (1998) Origin of some minor vegetatively propagated Allium crops studied with RAPD and GISH. Genet Resour Crop Evol 45:511–523. https://doi.org/10.1023/A:1008647700251
Fritsch RM, Friesen N (2002) Evolution, domestication and taxonomy. In: Rabinowitch HD, Currah L (eds) Allium crop science: recenttt advances. CABI Publishing, New York, pp 5–30
Fu YM, Jiang WM, Fu CX (2011) Identification of species within Tetrastigma (Miq.) Planch, (Vitaceae) based on DNA barcoding techniques. J Syst Evol 49:237–245. https://doi.org/10.1111/j.1759-6831.2011.00126.x
Fujito S, Akyol TY, Mukae T, Wako T, Yamashita KI, Tsukazaki H, Hirakawa H, Tanaka K, Mine Y, Sato S, Shigyo M (2021) Construction of a high-density linkage map and graphical representation of the arrangement of transcriptome-based unigene markers on the chromosomes of onion, Allium cepa L. BMC Genomics 22(1):1–8. https://doi.org/10.1186/s12864-021-07803-y
Gao T, Yao H, Song J, Liu C, Zhu Y et al (2010) Identification of medicinal plants in the family Fabaceae using a potential DNA barcode ITS2. J Ethno Pharmacol 130:116–121. https://doi.org/10.1016/j.jep.2010.04.026
Goldman JJ (2008) The use of ISSR markers to identify Texas bluegrass inter specific hybrids. Plant Breed 127:644–646. https://doi.org/10.1111/j.1439-0523.2008.01526.x
Gostimsky SA, Kokaeva ZG, Konovlac FA (2005) Studying plant genome variation using molecular markers. Russ Genet 41:378–388. https://doi.org/10.1007/s11177-005-0101-1
Gounaris Y, Skoula M, Fournaraki C, Drakakaki G, Makris A (2002) Comparison of essential oils and genetic relationship of Origanumx intercedents to its parental taxa in the island of Crete. Biochem Syst Ecol 30:249–258. https://doi.org/10.1016/S0305-1978(01)00079-5
Gurcharan SS (2004) Plant systematics: an integrated approach. Science publishers, Enfield, NH
Hajibabaei M, Singer GAC, Hebert PDN, Hickey DA (2007) DNA barcoding: how it complements taxonomy, molecular phylogenetics and population genetics. Trends Genet 23:167–172. https://doi.org/10.1016/j.tig.2007.02.001
Hao G, Lee DH, Lee JS, Lee NS (2002) A study of taxonomical relationships among species of Korean Allium sect. Sacculiferum (Alliaceae) and related species using inter-simple sequence repeat (ISSR) markers. Bot Bull Acad Sin 43:63–68
Hayden MJ, Tabone TL, Nguyen TMS, Keiper FJ, Fox RL, Chalmers KJ, Eglinton JA (2010) An informative set of SNP markers for molecular characterization of Australian barley germplasm. Crop past Sci 61:70–83. https://doi.org/10.1071/CP09140
Hebert PDN, Cywinska A, Ball SL, DeWaard JR (2003) Biological identification through DNA barcodes. Proc R Soc Lond Series B 270:313–321. https://doi.org/10.1098/rspb.2002.2218
Herlina L, Reflinur R, Sobir S, Maharijaya A, Wiyono S (2019) The genetic diversity and population structure of shallots (Allium cepa var. aggregatum) in Indonesia based on R gene-derived markers. Biodiversitas J Biol Divers 20(3):696–703. https://doi.org/10.13057/biodiv/d200312
Hirschegger P, Jakse J, Tronteli P, Bohanec B (2010) Origins of Allium ampeloprasum horticultural groups and a molecular phylogeny of the section Allium (Allium: Alliaceae). Mol Phylogenet Evol 54:488–497. https://doi.org/10.1016/j.ympev.2009.08.030
Hollingsworth PM, Forrest LL, Spouge JL, Hajibabaei M, Ratnasingham S et al (2009) Refining the DNA barcode for land plants. Proc Natl Acad Sci USA 106:12794–12797. https://doi.org/10.1073/pnas.111681210
Hsiao C, Jacobs SWL, Chatterton NJ, Asay KH (1999) A molecular phylogeny of the grass family (Poaceae) based on the sequence of nuclear ribosomal DNA (ITS). Aust Syst Bot 11:667–688. https://doi.org/10.1071/SB97012
Ipek M, Ipek A, Simon PW (2008) Genetic characterization of Allium tuncelianum: an endemic edible Allium species with garlic odor. Sci Hortic 115:409–415. https://doi.org/10.1016/j.scienta.2007.11.002
Jabbes N, Geoffriau E, Clerc VL, Dridi B, Hannechi C (2011) Inter simple sequence repeat fingerprints for assess genetic diversity of Tunisian garlic populations. J Agri Sci 3:77–85
Jayaswall K, Sagar R, Jayaswal D, Kumar A, Singh SP, Seth R, Sharma H, Kumar D, Mahajan V, Kumar S, Singh M (2024) Development of Allium cepa potential intron polymorphism markers for molecular breeding of alliums. South Afr J Bot 164:209–220. https://doi.org/10.1016/j.sajb.2023.11.050
Jayaswall K, Sharma H, Bhandawat A, Sagar R, Jayaswal D, Kumar A, Chaturvedi P, Mahajan V, Kumar S, Singh M (2022) Chloroplast derived SSRs reveals genetic relationships in domesticated alliums and wild relatives. Genet Resour Crop Evol 69:363–372. https://doi.org/10.1007/s10722-021-01235-z
Jayaswall K, Sharma H, Bhandawat A, Sagar R, Yadav VK, Sharma V, Mahajan V, Roy J, Singh M (2019) Development of intron length polymorphic (ILP) markers in onion (Allium cepa L.), and their cross-species transferability in garlic (A. sativum L.) and wild relatives. Genet Resour Crop Evol 66:1379–1388. https://doi.org/10.1007/s10722-019-00808-3
Khade YP, Salunkhe SR, Manjunathagowda DC, Sinhasane SR, Mahidar Gowd TY, Mahajan V, Singh M (2022) Molecular characterization of short-day onion genotypes by intron length polymorphic (ILP) markers. Genet Resour Crop Evol 69:2077–2086. https://doi.org/10.1007/s10722-022-01398-3
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. https://doi.org/10.1007/BF01731581
Kress WJ, Erickson DL (2007) A two locus global DNA barcode for land plants: the coding rbcLgene complements the non-coding trnh-psba spacer region. PLoS ONE. https://doi.org/10.1371/journal.pone.0000508
Kress WJ, Wurdack KJ, Zimmer EA, Weigt LA, Janzen DH (2005) Use of DNA barcodes to identify flowering plants. Proc Natl Acad Sci USA 102:8369–8374. https://doi.org/10.1073/pnas.050312310
Labra M, Miele M, Ledda B, Grassi F, Mazzei M, Sala F (2004) Morphological characterization, essential oil composition and DNA genotyping of Ocimum basilicum L. cultivars. Plant Sci 167:725–731. https://doi.org/10.1016/j.plantsci.2004.04.026
Lahaye R, van der Bank M, Bogarin D, Warner J, Pupulin F, Gigot G, Maurin O, Duthoit S, Barraclough TG, Savolainen V (2008) DNA barcoding the floras of biodiversity hotspots. Proc Natl Acad Sci USA 105:2923–2928. https://doi.org/10.1073/pnas.0709936105
Lewontin RC (1972) Testing the theory of natural selection. Nature 236:181–182
Liu Y, Yan HF, Cao T, Ge XJ (2010) Evaluation of 10 plant barcodes in Bryophyta (Mosses). J Syst Evol 48:38–46. https://doi.org/10.1111/j.1759-6831.2009.00063.x
Mallor C, Arnedo-Andrés MS, Garcés-Claver A (2014) Assessing the genetic diversity of Spanish Allium cepa landraces for onion breeding using microsatellite markers. Sci Hort 170:24–31. https://doi.org/10.1016/j.scienta.2014.02.040
McCallum J, Leite D, Pither-Joyce M, Havey MJ (2001) Expressed sequence markers for genetic analysis of bulb onion (Allium cepa L.). Theoretical and Applied Genetics 103:979–991. https://doi.org/10.1007/s001220100630.103
Mes THM, Fritsch RM, Pollner S, Bachmann K (1999) Evolution of the chloroplast genome and polymorphic ITS regions in Allium subg. Melanocrommyum. Genome 42:237–247. https://doi.org/10.1139/g98-123
Moon JS, Kim B, Kim S (2023) Evaluation of homozygosity levels of onion (Allium cepa L.) inbred lines using molecular markers evenly distributed in a high-resolution linkage map. Euphytica 219(9):96. https://doi.org/10.1007/s10681-023-03226-w
Mukherjee A, Sikhar B, Ghosh B, Banerjee A, Ghosh E, Bhattacharya M, Roy SC (2013) RAPD and ISSR analysis of some economically important species, varieties, and cultivars of the genus Allium (Alliaceae). Turk J Bot 37:605–618. https://doi.org/10.3906/bot-1208-18
Nagaoka T, Ogihara Y (1997) Applicabillity of inter-simple sequence repeat polymorphisms in wheat for use as DNA markers in comparison to RFLP and RAPD markers. Theor Appl Genet 94:597–602. https://doi.org/10.1007/s001220050456
Nei M (1972) Genetic distance between populations. Amer Naturalist 106:283–292. https://doi.org/10.1086/282771
Newmaster SG, Fazekas AJ, Ragupathy S (2006) DNA barcoding in land plants: evaluation of rbcL in a multigene tiered approach. Can J Bot 84:335–341. https://doi.org/10.1139/b06-047
Newmaster SG, Fazekas AJ, Steeves RAD, Janovec J (2008) Testing candidate plant barcode regions in the Myrisiticaceae. Mol Ecol Notes 8:480–490. https://doi.org/10.1111/j.1471-8286.2007.02002.x
Nguyen NH, Driscoll HE, Specht CD (2008) A molecular phylogeny of the wild onions (Allium; Alliaceae) with a focus on the western north American center of diversity. Mol Phylogenet Evol 47:1157–1172. https://doi.org/10.1016/j.ympev.2007.12.006
Novak J, Lukas B, Bolzer K, Grausgruber-Groger S, Dengenhardt J (2008) Identification and characterization of simple sequence repeat markers from a glandular Origanum vulgare expressed sequence tag. Mol Ecol Notes 8:599–601. https://doi.org/10.1111/j.1471-8286.2007.02059.x
Noyes RD, Rieseberg LH (1998) ITS sequence data support a single origin for north American Astereae (Asteraceae) and reflect deep geographic divisions asters. S.l. Am J Bot 86:398–412. https://doi.org/10.2307/2656761
Pathirana R, Carimi F (2022) Management and utilization of plant genetic resources for a sustainable agriculture. Plants. https://doi.org/10.3390/plants11152038
Peng X, Liu JJ, Xiang Y, Huang S (2006) A practical handbook of plant molecular biotechnology. Chemical Industry Press, Beijing
Poczai P, Matyas KK, Szabo I, Varga I, Hyvonen J, Cernak I, Gorji AM, Decsi K, Taller J (2011) Genetic variability of Thermal Nymphaea (Nymphaeaceae) population based on ISSR markers: implications on relationships, hybridization, and conservation. Plant Mol Biol Rep 29:906–918. https://doi.org/10.1007/s11105-011-0302-9
Raj AC, Sharangi AB, Das A, Pramanik K, Upadhyay TK, Almutairi M, Khan MI, Ahmad I, Kausar MA, Saeed M (2022) Assessing the genetic divergence of onion (Allium Cepa L.) through morpho-physiological and molecular markers. Sustainability 14(3):1131. https://doi.org/10.3390/su14031131
Reddy PM, Sarla N, Siddiq EA (2002) Inter simple sequence repear (SSR) polymorphism and its application in plant breeding. Euphytica 128:9–17. https://doi.org/10.1023/A:1020691618797
Rizkalla AA, Attia SAA, El-Hady AAA, Hanna NS, Nasseef JE (2012) Genetic diversity based on ISSR and protein markers associated with Earliness Trait in Wheat. World Appl Sci J 20:23–33
Schori M, Schowalter AM (2011) DNA barcoding as a means for identifying Medicinal plants of Pakistan. Pak J Bot 43:1–4
Segarra-Moragues JG, Gleiser G (2009) Isolation and characterisation of di and tri nucleotide microsatellite loci in Rosmarinus officinalis (Lamiaceae), using enriched genomic libraries. Conserv Genet 10:571–575. https://doi.org/10.1007/s10592-008-9572-7
Singh PR, Mahajan V, Verma A, Sagar R, Jayaswall K, Shukla N, Gedam P, Singh M (2021) Identification and characterization of white onion (Allium cepa L.) genotypes for high total soluble solid content through molecular markers. Genet Resour Crop Evol 68:957–967. https://doi.org/10.1007/s10722-020-01037-9
Sonnante G, Pignone D (2001) Assessment of genetic variation in a collection of Lentil using molecular tools. Euphytica 120:301–307. https://doi.org/10.1023/A:1017568824786
Taberlet P, Coissac E, Pompanon F, Gielly L, Miquel C, Valentini A (2007) Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res. https://doi.org/10.1093/nar/gkl938
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526. https://doi.org/10.1093/oxfordjournals.molbev.a040023
Tautz D (1989) Hypervariability of simple sequence as a general source: a polymorphic DNA markers. Nucleic Acids Res 17:6463–6471
Tibayrenc M (2005) Bridging the gap between molecular epidemiologists and evolutionists. Trends Microbiol 13:575–580. https://doi.org/10.1016/j.tim.2005.09.004
Trindade H (2007) Molecular biology of aromatic plants and spices. Flavour Frag J 25:272–281. https://doi.org/10.1002/ffj.1974
Vijayan K, Tsou CH (2010) DNA barcoding in plants: taxonomy in a new perspective. Curr Sci 11:1530–1541
Vos P, Hogers R, Bleeker M, Reijans M, Van Der T, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414. https://doi.org/10.1093/nar/23.21.4407
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetids. In: Innis MA, Gelfand D, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, New York, pp 315–322
Williams JG, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18:6531–6535. https://doi.org/10.1093/nar/18.22.6531
Zarei H, Fakheri BA, Naghavi MR, Mahdinezhad N (2020) Phylogenetic relationships of Iranian Allium species using the matK (cpDNA gene) region. J Plant Biotechnol 31:15–25. https://doi.org/10.5010/JPB.2020.47.1.015
Zietkiewciz E, Rafalksi A, Labuda D (1994) Genome fingerprinting by simple sequence repeat (SSR)-Anchored polymerase chain reaction amplification. Genomics 20:176–183. https://doi.org/10.1006/geno.1994.1151
Acknowledgements
The first author gratefully acknowledges Dr. M.B. Viswanathan, former Professor and Head, Department of Botany, Bharathidasan University, for given the lab facility for DNA and PCR analyses and all the authors are gratefully acknowledged Mr. R. Anand for his assistance in sample collection.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
All the authors were equally contributed towards the conception and design of the project. N.C.J.P.L and R.R produced, assembled and analysed the data. The manuscript was written by N.C.J.P.L and D.M with valuable contributions from R.R. and J.R.B. All authors were reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
No approval is required for the study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Packia Lekshmi, N.C.J., Brindha, J.R., Mahamuni, D. et al. Genomic profiling reveals intraspecific divergence and interspecific diversity in Allium cepa L.. Genet Resour Crop Evol (2024). https://doi.org/10.1007/s10722-024-01870-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10722-024-01870-2