
Abstract
The present study evaluated the population structure and genetic diversity using inter simple sequence repeat (ISSR) markers in 18 natural populations belonging to three species of Eplingiella (E. cuniloides, E. fruticosa and E. brightoniae), found growing naturally in the semiarid region of Northeast Brazil. Samples of 265 plants were analyzed using nine primer combinations, which generated 131 informative bands. Eplingiella spp. populations showed moderate genetic diversity (percentage of polymorphic bands, PPB = 75.6–96.9%, Nei's genetic diversity He = 0.31–0.39, Shannon's information index I = 0.33–0.48). Molecular variance analysis revealed that within populations, variations contributed more (74%) to the genetic diversity than between population variations (26%), with percentage of the genetic differentiation coefficient (GST = 0.29). The mean value of FST was 0.175, demonstrating good differentiation between populations. The analysis of the structure by the Bayesian method revealed the formation of two groups (K = 2), with many migrant individuals and a high level of miscegenation. The hierarchical cluster dendrogram grouped the 18 populations into two major clusters, with good support for the main clades (100%). According to principal component analysis, the two main principal components explained 21.06% of the total variation. The ISSR markers used were effective in identifying the variability of natural populations of Eplingiella spp., and population structure demonstrated recent diversification of species. The results shed more light on the genetic variation and evolutionary dynamics of Eplingiella, helping to formulate effective breeding strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lamiaceae is one of the most important families in terms of aromatic properties, comprising about 250 genera and more than 7000 species, which are common to the Mediterranean and subtropical regions (Bridi et al. 2021; Assaf et al. 2022; Fusani et al. 2022). Within this family, the genus Hyptis is considered to have great economic importance, and comprises about 280 species, which after a new circumscription based on molecular phylogenies have been split into 11 genera, including the genus Eplingiella Harley & J.F.B. Pastore (Harley and Pastore 2012).
Eplingiella comprises important aromatic species which are a relevant source of essential oils and secondary metabolites. They are used in food as well as in traditional and modern medicine, including the production of pharmaceuticals. Among these are Eplingiella fruticosa (Salzm. ex Benth) Harley & J.F.B. Pastore, Eplingiella cuniloides (Epling) Harley & J.F.B. Pastore, and Eplingiella brightoniae Harley (Harley 2014). These species occur in the semiarid region of the Brazilian Northeast, in impoverished sandy soils and are characterized by their shrub size, with small xeromorphic leaves and flowers with short pedicels (Harley and Pastore 2012; Harley 2014).
Eplingiella fruticosa (syn. Hyptis fruticosa Salzm. ex Benth.), known popularly as ‘alecrim de vaqueiro’, is native to the coastal region of the Brazilian Northeast (Silva et al. 2019). Pharmacological research has validated its analgesic (Melo et al. 2020), antinociceptive (Lima et al. 2013), anti-inflammatory (Andrade et al. 2010), antioxidant (Pinto et al. 2021) and vasorelaxant (Moreira et al. 2010) properties, as well as effects against neurodegenerative diseases (Beserra-Filho et al. 2019). E. cuniloides is a perennial shrub, endemic to dry and sandy areas in plateaus, found in the municipality of Morro do Chapéu, Bahia, Brazil. This species is also popularly known as ‘alecrim de vaqueiro’, but its potential use has not yet been scientifically investigated. E. brightoniae is a bushy shrub, up to 1–1.5 m tall, strongly aromatic, native to Serra do Curral Frio (Curral Frio Mountains), straddling the border between the municipalities of Umburanas and Sento Sé, in Bahia, Brazil. Its occurrence is so restricted that it is described as an endangered species, since its natural habitat is constantly subjected to fires and deforestation (Harley 2014).
The concern with the loss of variability and the extinction of species with medicinal potential has increased the demand for works involving collection, preservation and genetic conservation potential of these valuable species (Hashemifar and Rahimmalek 2018). Thus, plant breeders have been concentrating their efforts on maintaining variation in natural germplasm in an attempt to improve their breeding programs. Genetic variability studies represent an important tool for understanding the distribution relationships of species (Munda et al. 2022). For this purpose, the analysis of molecular markers is considered to be a robust tools for the evaluation of genetic diversity of several species of Lamiaceae (Triguero-Piñero et al. 2021; Koohdar and Sheidai 2022; Talebi et al. 2022). Among these markers, the ISSR (Inter Simple Sequence Repeat) markers have been widely used for the evaluation of genetic relationships of medicinal plants (Gupta et al. 2021; Feijó et al. 2022; Koohdar and Sheidai 2022; Peng et al. 2023). They are dominant markers, and have the advantages of being simple, quick, highly reproducible and highly polymorphic, besides requiring small DNA amounts and being highly informative (Gupta et al. 2021; Ghanbari et al. 2022).
However, the few molecular marker-based studies of Eplingiella species are mainly restricted to characterization of genotypes (Silva et al. 2017). To our knowledge, use of molecular markers for assessment of the structure and genetic diversity of natural populations of E. fruticosa, E. cuniloides and E. brightoniae, especially from the Northeast region of Brazil, has not been reported. This limits the implementation of reasonable conservation strategies and effective management practices of natural populations.
Based on these concerns, we conducted the present study with the following objectives: i) to evaluate for the first time the genetic variation within and among populations of selected species of Eplingiella (E. fruticosa, E. cuniloides and E. brightoniae) using inter simple sequence repeat (ISSR) markers, ii) to determine the level of population differentiation and relationships between molecular and geographical distribution patterns of this species, and iii) to evaluate the genetic structure of the Eplingiella spp. populations and species studied. The information obtained provides new insights into the population genetic structure of these species to guide future actions for conservation and new collections for breeding.
Material and methods
Plant material
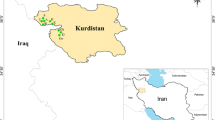
Two hundred and sixty-five samples from 18 natural populations of Eplingiella spp. (14 E. fruticosa, 3 E. cuniloides and 1 E. brightoniae) were collected in the states of Bahia and Sergipe, as listed in Table 1 and Fig. 1. These species were registered with platform of the National System of Genetic Heritage Management and Associated Traditional Knowledge (SISGEN) under reference number A1C1DEB.

Geographic locations of Eplingiella fruticosa (Salzm. ex Benth) Harley & J.F.B. Pastore (circles) Eplingiella cuniloides (Epling) Harley & J.F.B. Pastore (triangles) and Eplingiella brightoniae Harley (squares) populations sampled from the semiarid regions of Northeast Brazil
Sampling was carried out considering the heterogeneous distribution of individuals in the habitats, with an average of 15 individuals per population. These were named according to the continuous numbering recorded on the extraction form. Extractions, quantifications and data capture were carried out at the Laboratory of Molecular Plant Systematics (LAMOL) of Universidade Estadual de Feira de Santana (UEFS). Geographic distances between populations ranged from 21 km (between EF2088 and EF2089) to 634 km (between EF1850 and EF1893).
The species were identified by Dr. José Floriano Barêa Pastore at UEFS and the voucher of each population were deposited in the Herbarium da Universidade Estadual de Feira de Santana (HUEFS), listed in Table 1.
It is interesting to note that E. brightoniae is a species restricted to two municipalities in Bahia (Umburana and Sento Sé), but in the present study we collected samples only in Umburana, since we were unable to locate this species in Santo Sé due to indiscriminate burning in that region at the time. Therefore, we only represent a single population of E. brightoniae.
DNA extraction
Aliquots of total DNA from each sample were used in analyses, with the remaining material incorporated in the DNA Bank belonging to the Laboratory of Molecular Systems of Plants/UEFS. Fresh leaf samples (500 mg) were collected in CTAB gel (35% NaCl2 and 3% CTAB) for DNA extraction using the 2X CTAB protocol (Doyle and Doyle 1987). The extracted DNA was quantified by electrophoresis in 1% agarose gel in comparison with a 100pb ladder marker (Invitrogen®).
PCR amplification
A total of 16 ISSR primers were tested. The polymerase chain reaction (PCR) was carried out with total reaction volume of 10 μl, containing 4.0 µL of the Top Taq Master Mix Kit (Qiagen Inc., Hilden, Germany), 2.0 µL of high-quality ultrapure H2O (part of the amplification kit), 1.6 µL of TBT (Samarakoon et al. 2013), 2.0 µL of diluted sample DNA (5 mg/mL) and 0.4 µL of the primer (0.5 M). The amplification reactions were performed in an Esco Healthcare Swift® Max Pro thermocycler, starting with a denaturation cycle at 94 °C for 90 s, followed by 40 amplification cycles with denaturation at 94°C for 45 s, annealing at 45–52 °C for 45s, and extension at 72 °C for 90 s, plus two variations, the first at 94 °C and the second at 44 °C, both for 45 s, followed by a final extension at 72 °C for 7 min, for the 265 individuals.
The PC reaction products were separated and visualized in 2.0% agarose gels with SB buffer (Brody and Kern 2004), and compared to a 100-base-pair ladder marker with ethidium bromide staining. To verify the reproducibility of the results, each PCR and gel amplification run was repeated twice, and only the amplified ISSR fragments present in both lanes were considered. Gels were photographed and analyzed using the software GelCompar II (Applied Maths), to establish standardized comparisons between all samples in different gels and establish the homologous bands based on size.
ISSR-PCR data analysis
The ISSR bands were counted using the binary scoring system, recording the presence or absence of bands as 1 or 0, respectively. A binary matrix (1/0) was prepared with GelCompar II.
Genetic variation within and between populations was estimated using the following parameters: number of polymorphic bands (NPb); percentage of polymorphic bands (%PPb); and expected heterozygosity or genetic diversity of Nei (1973) (He). Genetic structure was estimated using the genetic differentiation coefficient (GST) (Culley et al. 2002), Shannon’s information index (I) and the genetic distance between populations (FST). For the purposes of population analysis and conservation (Pearse and Crandall 2004), the latter two metrics are of great importance for assessing population structure and gene flow (Takahata and Nei 1984).
Analyzes were performed using POPGENE v.1.32 (Yeh et al. 1999) and GenAlEx. 6.5 (Peakall and Smouse 2012). Nei's genetic distance (1978) and genetic structuring (FST) were estimated using AFLP-Surv. 1.0 (Vekemans et al. 2002). From these distances (run of 1000 repetitions), cluster analysis was performed based on Nei's genetic distance matrix (1973) and the neighbor-joining algorithm, using PHYLIP v. 3.695 (Felsenstein 1989). One thousand bootstrap repetitions were performed to assess the statistical confidence of each branch.
Principal component analysis (PCA) and analysis of molecular variance (AMOVA) were performed using GenAlEx 6.5 (Peakall and Smouse 2012). AMOVA was carried out in two ways, first by assessing variability at one level, between populations and within populations, and then at two levels, partitioning the variability also between species.
The population structure was estimated using the STRUCTURE 2.2 software based on the Bayesian clustering algorithm (Pritchard et al. 2000), and the result was confirmed by Structurama (Huelsenbeck and Andolfatto 2007; Huelsenbeck et al. 2007; Huelsenbeck et al. 2011). Population structure was analyzed by setting the number of sub-populations (K-values) from 1 to 10, and each run was repeated five times. For each run, a burn-in period of 50,000 and a Markov chain Monte Carlo replication of 100,000 were set. The statistical parameter (ΔK) was used to determine the number of groups. Structure Harvester was used to determine the peak value of ΔK according to the method described by Evanno et al. (2005).
Results and discussion
Genetic diversity
Among the 16 primers tested, only nine produced amplification of polymorphic ISSR patterns (Table 2). A total of 131 polymorphic bands were detected among nine ISSR primers from the 18 populations of Eplingiella obtained, with an average of 14.55 bands (loci) per primer (Table 2), indicating a high level of polymorphism.
Genetic diversity estimates from ISSR are summarized in Table 3. PCR amplification of the 265 individuals with the nine primers generated a total of 131 (100%) polymorphic bands (NPb), corresponding to an average number of 119 (90.8%) polymorphic bands per population. All primers produced high rates of polymorphism for the 18 populations, and the presence of exclusive bands between them was not identified. For the population of E. fruticosa, the proportion of polymorphic bands at the population level (%PPb) varied from 75.3% (EF1989) to 93.9% (EF1888, EF1893), with an average of 89.58% polymorphism. Silva et al. (2017), when evaluating the genetic diversity of E. fruticosa in the Brazilian state of Sergipe using ISSR markers, identified high polymorphism. They selected eight primers with 72 amplified bands, varying from 8 to 11 with a mean of nine bands per primer. The parameters that determine the degree of genetic diversity among individuals (expected heterozygosity –He and Shannon’s information index—I) showed similar trend as the PPB. The He ranged from 0.30 (EF1989) to 0.38 (EF1850, EF1888) and the Shannon’s index (I) tvaried from 0.37 (EF1989) to 0.47 (EF1850). The values found by us for He, I are comparatively higher than the levels reported for E. fruticosa by Silva et al. (2017), although the primers used to determine the genetic diversity of this species are not exactly the same in the present study.
The population represented by E. cuniloides presented %PPb, ranging from 85.5 (EC1842) to 92.4 (EC1851), He values between 0.32 (EC1842) and 0.38 (EC1851), and Shannon index (I) ranging from 0.39 (EC1842) to 0.46 (EC1851). E. brightoniae presented %PPb (96.9), He (0.39) and I (0.48). The different levels of genetic diversity were probably due to the locations of the sampled populations. The results obtained in our study are in line with those reported by other authors who have worked with natural populations of different species of Lamiaceae. Gupta et al. (2021) identified 90% polymorphic bands among Ocimum populations, and Jedrzejczyk and Rewers (2018) found 100% polymorphic bands for natural populations of Mentha L. On the other hand, Kameli et al. (2013) observed low levels of diversity in populations of Satureja spp., with percentage of polymorphic bands varying between 52.63% and 73.68%, although with high averages for Shannon's (0.46) and Nei's (0.42) indices. These species, however, have a restricted and fragmented distribution, which may have led to a narrowing of their genetic bases (Nybom 2004; Rodrigues et al. 2013).
Genetic structure
Analysis of molecular variance (AMOVA) revealed significant variations (P < 0.01), and the majority of the variance observed occurred within populations (71%), while the remaining 29% was variation between populations, indicating a strong genetic structure (Table 4). Generally, the presence of high variability within the population detected in the present study meant the divergence of the individual within a single population. This is in agreement with reports on other Lamiaceae plants that also indicated greater within-population variation, such as Mentha cervina L. (Rodrigues et al. 2013), Perovskia abrotanoides Karel and P. atriplicifolia (Hashemifar and Rahimmalek 2018), Salvia rosemarinus Schleid. (Zigeneet al. 2020), and Melissa officinalis L. (Koohdar and Sheidai 2022).
Considering two levels of partitioning, when populations were grouped by species, the proportion of within-population variance decreased (69%), as well as among-population variance (27%), with 4% of the variation being attributed to the differentiation among species. This can indicate there is very little differentiation at the species level, or that the method used was not sensitive to genetic differences between them, although the result was statistically significant. In addition, these rates reveal the great diversity among the sampled populations, confirming the values of He and I.
The average value of the genetic differentiation coefficient (GST) among populations of Eplingiella spp. was 0.29, indicating a high degree of genetic differentiation between populations, as well as ΦST shown in Table 4, which represents the value of individuals taken at random within each population in relation to individuals taken at random in the entire sample. Nei (1978) classified (GST) into three classes: low < 0.05), moderate (0.05 < GST < 0.15) and high (G GST > 0.15). In this study, the average value of the genetic differentiation coefficient (GST) among populations of Eplingiella spp. was (0.29), in a moderate class (total = 0.29), indicating a high degree of genetic differentiation between populations, as well as ΦST shown in Table 4, which represents the value of individuals taken at random within each population in relation to individuals taken at random in the entire sample.
A high and significant (P < 0.01) genetic differentiation, according to the Φ-statistic (ΦST), was also observed among species, among populations and within populations. The highest value ΦST was found within populations (ΦST = 0.449) and the lowest was observed among species (ΦST = 0.279). Similar patterns of genetic differentiation have been observed for other plant species (Flihi et al. 2022).
The AMOVA also revealed a moderate wright genetic differentiation between populations (FST = 0.175). If the FST value is between 0 and 0.05, genetic differentiation between populations is low; if it is between 0.05 and 0.15, differentiation is moderate; if it is between 0.15 and 0.25, it is high; and if it is above 0.25, differentiation is very high (Wright 1978, Hartl and Clark 1997). A recent study carried out by Duan et al. (2022) stated that the genetic differentiation coefficient FST is a vital parameter to reflect the degree of genetic structure. The results obtained here are in line with those reported by Saidi et al. (2013) and Silva et al. (2017), who also found a moderate genetic diversity in Satureja L. (Lamiaceae) and E. fruticosa (Lamiaceae) species, respectively.
The genetic structure of the 18 populations of Eplingiella spp. was determined by employing the Bayesian clustering method based on STRUCTURE (version 2.2) analysis, resulted in the formation of three peaks (K = 2, K = 6 and k = 8), indicating three possibilities of assigning individuals to real populations (Figs. 2, 3). Applications of this method include demonstrating the population structure present, identifying genetically distinct populations, assigning individuals to populations, and identifying migrant or mixed populations and individuals (Pritchard et al. 2010). The peak K = 2, indicating the formation of two groups for the 18 populations of Eplingiella spp., was the largest, albeit with many migrant individuals, demonstrating a high level of miscegenation among them. This result was further confirmed by the STRUCTURAMA analysis (Huelsenbeck et al. 2011), strengthening the possibility of the best fit being formation of two large genetic groups among the sampled populations.

Estimated ∆K of the 18 natural populations of Eplingiella Harley & J.F.B. Pastore during 2 runs of each K value

Population structure inferred by the STRUCTURE software based on ISSR data at K = 2; K = 6; K = 8. Populations: E. fruticosa (EF1850, EF1855, EF1864, EF1888, EF1893, EF1921, EF1922, EF1932, EF1988, EF1989, EF2088, EF2089, EF2091, EF2347); E. cuniloides (EC1851, EC1842, EC1851, EC1856) and E. brightoniae (EB2326)
Despite the high level of miscegenation, populations of E. fruticosa (EF1850, EF1855, EF1864, EF1888, EF1893, EF1921), E. cuniloides (EC1842, EC1851, EC1856) and E. brightoniae (EB2326) have predominantly the same gene pool, therefore being allocated in the same group (Fig. 3), even though having a strong interspecific genetic mixture. This mixture involved populations of E. fruticosa (EF1850–EF1921), E. cuniloides (EC1842–EC1856) and E. brightoniae (EB2326). Archibald et al. (2006a) reported that it is possible for co-migrant bands to represent different loci, which appear homologous because they are approximately the same size, or loci that are identical due to convergences. In addition, allogamous species, with recent diversification, tend to present greater allelic variation within populations, enabling conserving much of the ancestral genetic load (Maia 2010).
The E. fruticosa populations (EF1988, EF1989, EF2088, EF2089, EF2091 and EF2347) predominantly shared the same gene pool, being gathered in the opposite group (Fig. 3). On the other hand, in populations EF1922 and EF1932, the gene pool was shared between characteristics of the two groups formed, making it impossible to analyze their relationship within each group individually. This was because in this method the sampled individuals are assigned (probabilistically) to populations, or together to two or more populations if their genotypes indicate they are mixed (Pritchard et al. 2010). It is also important to highlight that the studied species have very similar morphological characteristics, and also E. cuniloides and E. brightoniae present very restricted occurrences and share these occurrences with E. fruticosa. Based on this information, there may be genetic exchanges between them at some level. In addition, the pattern presented reveals plesiomorphism of traits close to the three species.
The second peak (K = 6) showed the possible formation of six populations, although with strong miscegenation (Fig. 3). The groups were arranged as follows: first, individuals from populations of E. fruticosa (EF1850, EF1855), E. cuniloides (EC1851 and EC1856), forming the group represented mostly in red; then individuals of E. fruticosa (EF1864, EF1888 and EF1893), represented in blue. In relation to the remaining populations (EF1921, EF1922, EF1932 and EF1988), they formed the predominantly cyan group, but with the first two showing a higher level of genetic mixing. Populations of E. fruticosa (EF1989, EF2089 and EF2091), with representation in lilac; EF2088, EF2347 (E. fruticosa) and EB2326 (E. brightonia), represented in yellow, and the isolated population EC1842 (E. cuniloides), in green. In this situation, the strong mixture identified may be related to the low intensity of the peak along with the distribution in a larger number of groups, which may have forced the grouping of weakly related individuals in the same population. However, there are cases where the grouping is justified by the ease of biological explanation, at a certain level disregarding the percentages of miscegenation.
Similar behavior was observed for the individuals grouped in the third possibility, where the peak K = 8 was also of low intensity and formed a greater number of groups. The individuals sampled were assigned to eight populations, also with a strong genetic mix, grouping individuals from population EF1850 (E. fruticosa), and EC1851 (E. cuniloides), marked with predominant blue color; EF1855 (E. fruticosa) and EC1856 (E. cuniloides), in purple; EF1864 isolated in cyan; EF1888, EF1893 and EF1921(E. fruticosa) in orange; and EF1922, EF1932, EF1988 and EF1989 (E. fruticosa) predominantly in yellow.
Cluster analysis based on neighbor joining methods classified the 18 populations into two main clades (Fig. 4) with good support (100%), which were analogous to the ones identified by STRUCTURE. Clade I consisted of five (27.8%) populations (EF1850, EF1855, EC1842, EC1851 and EC1856) belonging to two different species (E. fruticosa and E. cuniloides). Therefore, it is a mixed formation, and most of the populations (80%) allocated in this clade had the same geographic origin (Morro do Chapéu, BA). Within this clade, subgroups were formed, composed by EF1850 and EC1851 populations, which despite belonging to different species, apparently retain a very close genetic composition, so there is probably still a genetic exchange between them (both are from Morro do Chapéu). This can change the configuration of the species. Similar behavior was observed for the subgroup formed by the populations EC1842, EC1856 and EF1855. The data presented here suggest the occurrence of gene flow.

Neighbor-joining dendrogram based on Nei's genetic distance matrix for the 18 populations of Eplingiella Harley & J.F.B. Pastore. Populations: E. fruticosa (EF1850, EF1855, EF1864, EF1888, EF1893, EF1921, EF1922, EF1932, EF1988, EF1989, EF2088, EF2089, EF2091, EF2347); E. cuniloides (EC1851, EC1842, EC1851, EC1856) and E. brightoniae (EB2326)
The isolation of the EB2326 population (100%) in clade II can be explained by the fact it belongs to the species E. brightoniae Harley, which is endemic and restricted to the rocky fields of Serra do Curral Frio, straddling the border between the municipalities of Umburanas and Sento Sé (Harley 2014). In clade II, the subgroup formed by EF1864 (from Jambeiro, Bahia), EF1888 (from São Cristóvão. Sergipe) and EF1893 (from Japaratuba, Sergipe) may be related to the places of origin, because despite the distance between these populations (Table 1), the edaphoclimatic characteristics of the three places are similar (data not shown). In addition, the inclusion of populations EF1864 and EF1888 in this clade may be related to the resilience factor, since they coexist in locations that are frequently subject to burning. In the subgroups formed by EF1921; EF1932 and EF1922, EF1989, the relationship between populations also tended to converge due to environmental pressures, since these were collected in places with similar soil and climate characteristics, with sandy-stony soil, full sunlight, and good water supply throughout the year (800 to 1300 mm). Finally, the last subgroup analyzed, with the populations EF1988, EF2088, EF2089, EF2091 and EF2347, converged due to the environmental characteristic of being collected in transitional sites (ecotones) between the Caatinga and Atlantic Forest biomes, with predominance of the second biome.
In general, the stratification of populations and subpopulations of the three species of Eplingiella spp. studied reinforce the idea of a common ancestor, of recent diversification in a short evolutionary interval. In addition to the moderate genetic variability present, these species are possibly still in the process of genetic differentiation.
The results of the principal component analysis (PCA) generated by using genetic distance-based analysis were very consistent with the result obtained by NJ clustering, where first two principal coordinates explained only 21.06% of the total variation (Fig. 5), demonstrating the intermixed but integrated genetic relationship of the populations studied. In this case, it was not possible to observe, through the scatter plot (Fig. 5), the separation of populations into groups. The pattern demonstrated in this analysis suggests the putative existence of three species corresponding to a single group (one species), which is widely diverse.

Principal component analysis of the genetic distance matrix of 265 individuals from 18 populations of Eplingiella Harley & J.F.B. Pastore, based on 131 ISSR loci. Populations: E. fruticosa (EF1850, EF1855, EF1864, EF1888, EF1893, EF1921, EF1922, EF1932, EF1988, EF1989, EF2088, EF2089, EF2091, EF2347); E. cuniloides (EC1851, EC1842, EC1851, EC1856) and E. brightoniae (EB2326)
Genetic variation based on molecular markers is an effective and functional method used in breeding programs (Zigene et al. 2020). It is interesting to note that the number of polymorphic bands identified (131 bands) in our study was sufficient to delimit the genetic structure of the groups formed. Furthermore, the recent diversification of this group may explain the level of homology identified here. The allogamy factor related to the recent radiation of taxa can generate a series of problems in defining the genetic structure of populations, a situation commonly reported in studies of native and endemic species of recent taxonomic evolution (Archibald et al. 2006a; 2006b).
Conclusions
The use of ISSR-like dominant markers proved to be an efficient method to identify the variability existing between natural populations of Eplingiella spp. It was possible to identify important traits of the genetic relationship between the populations studied, to support measures for the conservation and rational use of these genetic resources. Populations EB2326, EF1888 and EF1893 displayed the greatest diversity, indicating that protective measures and greater collection efforts can be directed to areas of their natural occurrence in order to safeguard the highest percentage of the species' gene pool. Furthermore, it was possible to question the population structure of the three species of the genus, and also their small genetic differences, supporting the hypothesis of recent diversification and persistent genetic link between them.
Abbreviations
- ISSR:
-
Inter simple sequence repeat
- PPB:
-
Percentage of polymorphic bands
- He:
-
Nei's genetic diversity
- I:
-
Shannon's information index
- NPb:
-
Number of polymorphic bands
- G ST :
-
Genetic differentiation coefficient
- F ST :
-
Wright’s genetic differentiation coefficient between populations
- PCA:
-
Principal component analysis
References
Andrade AM, Oliveira JPR, Santos ALLM, Franco CRP, Antoniolli AR, Estevam CS, Thomazzi SM (2010) Preliminary study on the anti-inflammatory and antioxidant activities of the leave extract of Hyptis fruticosa Salzm. ex Benth. Lamiaceae Braz J Pharmacog 20:962–968
Archibald JK, Crawford DJ, Santos-Guerra A, Mort ME (2006a) The utility of automated analysis of intersimple sequence repeat (ISSR) loci for resolving relationships in the Canary Island species of Tolpis (Asteraceae). Am J Bot 93:1154–1162
Archibald JK, Mort ME, Crawford DJ, Santos-Guerra A (2006b) Evolutionary relationships within recently radiated taxa: comments on methodology and analysis of inter-simple sequence repeat data and other hypervariable, dominant markers. Taxon 55:747–756
Assaf M, Korkmaz A, Karaman S, Kulak M (2022) Effect of plant growth regulators and salt stress on secondary metabolite composition in Lamiaceae species. S Afr J Bot 144:480–493
Beserra-Filho JIA, Macêdo AM, Leão AHFF, Bispo JMM, Santos JR, Oliveira-Melo AJ, Menezes PP, Duarte MC, Araújo AAS, Silva RH, Quintans-Júnior LJ, Ribeiro AM (2019) Eplingiella fruticosa leaf essential oil complexed with β-cyclodextrin produces a superior neuroprotective and behavioral profile in a mice model of Parkinson’s disease. Food ChemToxicol 124:17–29
Bridi H, Meirelles GC, von Poser GL (2021) Subtribe hyptidinae (Lamiaceae): a promising source of bioactive metabolites. J Ethnopharmacol 264:1–30
Brody JR, Kern SE (2004) Sodium boric acid: atris-free, cooler conductive medium for DNA electrophoresis. Biotechniques 36:2–4
Culley TM, Wellen SG, Sakai AK (2002) The evolution of wind pollination in angiosperms. Trends Ecol Evol 17:361–369
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Duan B, Li W, Yu Y, Guan Y, Mu S, Li Z, Li X, Kang X (2022) Microsatellite analysis of genetic diversity in wild and cultivated Portunus trituberculatus in Bohai Bay. Mol Biol Rep 49:2543–2551
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Feijó EVRS, Barbosa BL, van den Berg C, Oliveira LM (2022) Genetic diversity of Lippia origanoides Kunth. in natural populations using ISSR markers. Ciênc Agrotec 46:1–8
Felsenstein J (1989) PHYLIP phylogeny inference package (Version 3.2). Cladistics 5:164–166
Flihi J, Rhimi A, Yangui I, Flihi J, Messaoud C, Ali IBE (2022) Genetic diversity and population structure of Tunisian wild Kermes oak (Quercus coccifera L.): assessment by ISSR molecular markers and implication for conservation. Mol Biol Rep 49:6215–6224
Fusani P, Ronga D, Carminati D, Mandrioli M, Manicardi GC, Giannì S, Tava A (2022) Composition and biological activity of essential oils from Artemisia roxburghiana Besser and Elsholtzia fruticosa Rehder cultivated in Italy. Ind Crop Prod 187:1–9
Ghanbari MA, Salehi H, Moghadam A (2022) Genetic diversity assessment of Iranian kentucky bluegrass accessions: I. ISSR markers and their association with habitat suitability within and between different ecoregions. Mol Biotechnol 64:1244–1258
Gupta P, Mishra A, Lal RK, Dhawan SS (2021) DNA fingerprinting and genetic relationships similarities among the accessions/species of Ocimum using SCoT and ISSR markers system. Mol Biol 63:446–457
Harley RM (2014) Eplingiella brightoniae, a new species of hyptidinae (Lamiaceae: Ocimeae) from Northerbn Bahia. Brazil Kew Bulletin 69:1–5
Harley RM, Pastore JFB (2012) A generic revision and new combinations in the Hyptidinae (Lamiaceae), based on molecular and morphological evidence. Phytotaxa 58:1–55
Hartl DL, Jones EW (2005). DNA structure and DNA manipulation. – In: Genetics: analysis of genes and genomes. 5th edn, Sudbury:Jones and Bartlett pub. pp 36– 85
Hashemifar Z, Rahimmalek M (2018) Genetic structure and variation in Perovskia abrotanoides Karel and P. atriplicifolia as revealed by molecular and morphological markers. Sci Hortic 230:169–177
Huelsenbeck JP, Andolfatto P (2007) Inference of population structure under a Dirichlet process model. Genetics 175:1787–1802
Huelsenbeck JP, Andolfatto P, Huelsenbeck ET (2011) Structurama: bayesian inference of population structure. Evol Bioinform Online 7:55–59
Jedrzejczyk I, Rewers M (2018) Genome size and ISSR markers for Mentha L. (Lamiaceae) genetic diversity assessment and species identification. Ind Crops Prod 120:171–179
Kameli M, Hesamzadeh Hejazi SM, Ebadi M (2013) Assessment of genetic diversity on populations of three Satureja species in Iran using ISSR markers. Ann Biol Res 4:64–72
Koohdar F, Sheidai M (2022) Genetic diversity and population structure in medicinal plant Melissa officinalis L. (Lamiaceae). Genet Resour Crop Evol 69:1753–1758
Lima ACB, Paixão MS, Melo M, Santana MT, Damascena NP, Dias AS, Porto YCBS, Fernandes XA, Santos CCS, Lima CA, Quintans Junior LJ, Estevam CS, Araujo BS (2013) Orofacial antinociceptive effect and antioxidant properties of the hydroethanol extract of Hyptis fruticosa Salmz ex Benth. J Ethnopharmacol 146:192–197
Maia MCC (2010) Sistema reprodutivo de populações alogamas e autógamas: modelo básico e equilíbrio. Rev Agro@mbiental Online 4:53–54
Melo AJO, Heimarth L, Carvalho AMS, Quintans JSS, Serafini MRS, Araújo AAS, Alves PB, Ribeiro AM, Saravanan S, Quintans-Júnior LJ, Duarte MC (2020) Eplingiella fruticosa (Lamiaceae) essential oil complexed with β-cyclodextrin improves its anti-hyperalgesic effect in a chronic widespread non-inflammatory muscle pain animal model. Food Chem Toxicol 135:3–9
Moreira IJA, Moreno MON, Fernandes MFG, Fernandes JB, Moreira FV, Antoniolli AR, Santos MRV (2010) Vasorelaxant effect of Hyptis fruticosa Salzm. Ex Benth. Lamiaceae, dichloromethane extract on rat mesenteric artery. Braz J Pharmacog 20:762–766
Munda S, Saikia RJ, Begum T, Bhandari S, Gogoi A, Sarma N, Tamang R, Lal M (2022) Evaluation of genetic diversity based on microsatellites and phytochemical markers of core collection of Cymbopogon winterianus Jowitt. germplasm. Plants 11:1–20
Nei M (1973) Analysis of genetic diversity in subdivided populations (population structure/genetic variability/hetrozygosity/gene differentiation). Proc Natl Acad Sci 70:3321–3323
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:3583–3590
Nybom H (2004) Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol 13:1143–1155
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539
Pearse DE, Crandall KA (2004) Beyond F-ST: Analysis of population genetic data for conservation. Conserv Genet 5:585–602
Peng Y, Nafee-Ul A, Liu M, He Q, Liang Z (2023) Assessment of genetic diversity in bupleurum spp. basing agronomic traits, medicinal components and ISSR markers. Genes 14:1–14
Pinto JAO, Oliveira AKS, Santos EWP, Silva AO, Blank AF, Corrêa CB, Nogueira PCL, Arrigoni MF (2021) Essential oils of Eplingiella fruticosa populations: chemical, antioxidant, and cytotoxic analyses. Res Soc Dev 10:e341101623723
Pritchard J, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pritchard J, Wen X, Falush D (2010). Documentation for structure software. [Documentation file] Available with the program at https://www.pritchbsduchicagoedu/structurehtml
Rodrigues L, van den Berg C, Povoa O, Momteiro A (2013) Low genetic diversity and significant structuring in the endangered Mentha cervina populations and its implications for conservation. Biochem Syst Ecol 50:51–61
Saidi M, Movahedi K, Mehrabi AA (2013) Characterization of genetic diversity in Satureja bachtiarica germplasm in Ilamproviance (Iran) using ISSR and RAPD markers. Int J Agric Sci 5:1934–1940
Samarakoon T, Wang SY, Alford MH (2013) Enhancing PCR amplification of DNA from recalcitrant plant specimens using a trehalose-based additive. Appl Plant Sci 1:1–3
Silva DC, Diniz LEC, Blank AF, Nizio DAC, Pinto JAO, Pereira KLG, Arrigoni-Blank MF (2017) Assessment of genetic diversity of a native population of Eplingiella fruticosa: a plant with therapeutic potential. Genet Mol Res 16:1–10
Silva DC, Arrigoni-Blank MF, Bacci L, Blank AF, Faro RRN, Pinto JAO, Pereira KLG (2019) Toxicity and behavioral alterations of essential oils of Eplingiella fruticosa genotypes and their major compounds to Acromyrmex balzani. Crop Prot 116:181–187
Takahata N, Nei M (1984) Fst and Gst statistics in the finite island model. Genetics 107:501–504
Talebi SM, Tabaripour R, Matsyura A (2022) Genetic diversity and population structure of diverse Iranian Nepeta L. taxa. Genet Resour Crop Evol 69:285–296
Triguero-Piñero P, Vega C, Aparicio A, Albaladejo RG (2021) Isolation of microsatellite markers for the endemic Phlomislychnitis (Lamiaceae). Mol Biol Rep 48:8233–8238
Vekemans X, Beauwens T, Lemaire M, Roldán-Ruiz I (2002) Data from amplified fragment length polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Mol Ecol 11:139–151
Wright S (1978) Evolution and the genetics of populations, vol IV. University of Chicago Press, Chicago, Variability within and among natural populations
Yang L, Hisorievd H, Kurbonovad P, Boboeve M, Bobokalonovd K, Feng Y, Lia W (2021) High genetic diversity and low differentiation of endangered Ferula tadshikorum Pimenov in Tajikistan. Glob Ecol Conserv 28:1–8
Yeh FC, Yang R, Boyle T (1999). POPGENE 1.32: Population genetic analysis. Programa para computador livre distribuído pelos autores. 1999. Disponível em: <ftp://ftp.microsoft.com/Softlib/MSLFILES/HPGL.EXE>. Acessado em: 20/01/2022
Zigene ZD, Asfaw BT, Bitima TD (2020) Analysis of genetic diversity in rosemary (Salvia rosemarinus Schleid.) using SSR molecular marker for its management and sustainable use in Ethiopian genebank. Genet Resour Crop Evol 68:279–329
Acknowledgements
The authors thank the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for financial support (001) and for the postdoctoral research grant given to the fifth (PNPD/UEFS 15950830814). We also thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for financial support.
Funding
This research was supported by Grant from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (PNPD/UEFS 15950830814).
Author information
Authors and Affiliations
Contributions
AS: Conceptualization, Methodology, Investigation, Data curation, Writing-Original draft preparation; LO: Conceptualization, Investigation, Methodology, Resources, Data curation, Writing—original draft, Writing—review & editing, Supervision, Funding acquisition; JFBP: Conceptualization, Formal analysis, Data curation, Writing—original draft, Supervision; CvdB: Conceptualization, Resources, Methodology, Data curation, Formal analysis, Writing—original draft, Supervision; TS: Conceptualization, Visualization, Formal analysis, Writing—review & editing; ES: Visualization, Writing—review & editing.
Corresponding author
Ethics declarations
Confict of interest
The authors declare that they have no confict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Silva, A.d., de Oliveira, L.M., Pastore, J.F.B. et al. Genetic diversity and population structure of Eplingiella species (Lamiaceae) using ISSR markers. Genet Resour Crop Evol 70, 2801–2813 (2023). https://doi.org/10.1007/s10722-023-01607-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-023-01607-7