
Abstract
Triticeae γ-type seed prolamins have undergone many rounds of gene duplications during evolution. However, the population genetics of total Triticeae γ-prolamin genes has not been studied in an evolutionary perspective so far. Here, evolutionary pattern of γ-prolamin multigene family containing 557 sequences from wheat and closely related species (γ-gliadins), rye (γ-secalin) and barley (γ-hordeins) was investigated. Furthermore, Triticeae genomes were searched for low immuno-reactive celiac disease (CD) epitope contents in their γ-prolamins for the possibility of developing less immunogenic cultivars. Large numbers of haplotypes with segregating sites and mutations in γ-type seed prolamin multigene family were evident. Polymorphism analyses of both promoters and coding regions of species-specific γ-prolamins demonstrated no deviation from neutral theory. The sliding window revealed that most of the singleton polymorphic sites are resided in the variable R1 domain. Furthermore, the significant negative Tajima’s D and H tests’ values of each groups 1, 2, 3, and 8 showed a selective sweep for R and D genome duplication processes. The dN/dS ratio analysis revealed strong purifying selection acting upon duplication of the two ancestral branches, while the neutral selection among overall groups was observed. Our results support previously reported findings that the groups of 2, 3, 8 (D genome) and 10 (A genome) can potentially be recommended for selection approaches or genetic engineering of less immunogenic cereal grains. Furthermore, the results provide new insights on the evolutionary dynamics of γ-prolamin multigene family, polyploidy speciation and phylogeny of Triticeae species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cereals are the most widely grown and consumed staple. The use of cereals for most food products is mainly due to the characteristics provided by gluten that accounts for 80% of total grain protein content (Rosell et al. 2014). Gluten is a complex mixture of proteins comprising of prolamins with gliadins and glutenins in wheat, with equivalents in barley and rye (Shewry et al. 1999). The gliadins are traditionally divided based on their mobility in acidic polyacrylamide gel electrophoresis at low pH into three groups: S-rich α-type gliadins (with 3 inter-chain disulphide bonds (ICDB), γ- gliadins (with 4 ICDB) and S-poor ω-gliadins with no ICDB. Similarly, glutenin subunits are separated into low and high molecular weight (L/HMW) subunits based on SDS-PAGE analysis (Payne et al. 1980). Wheat gliadins are encoded by medium to large multigene families (Payne et al. 1984a), including α-gliadins encoded by the Gli-2 loci on the short arms of group 6 chromosomes, γ-gliadins and ω-gliadins encoded by the Gli-1 loci (Gli-A1, Gli-B1 and Gli-D1) on the short arms of homologous chromosome 1, and are tightly linked to the Glu-3 loci coding for LMW-glutenin (Anderson et al. 2012; Payne et al. 1984a; Tatham and Shewry 1995), while HMW-glutenin encoding genes are on long arms of group 1 chromosomes (Glu-1 loci) (Payne et al. 1984b).
The γ-types are the most ancient Triticeae gluten prolamins that have been studied extensively in wheat (γ-gliadins), rye (γ-secalins) and barley (γ-hordeins) (Qi et al. 2009; Stenman 2011). Studies of the amino acid composition, and molecular mass analysis indicated that rye γ-secalins and barley γ-hordeins are closely related to wheat γ-gliadins. The γ-gliadins that contribute to the visco-elastic properties of the dough are mainly heterogeneous collection of 30–78 kDa monomeric proteins with poor solubility in dilute salt solutions and good solubility in 70% ethanol (Bietz and Wall 1972; Gellrich et al. 2003, 2005; Guo et al. 2012; Qi et al. 2009, 2013; Singh et al. 1990; Shan et al. 2002; Shewry and Tatham 1990; Wieser 2007). The number of γ-gliadins was preliminary estimated to be 15–40 (Anderson et al. 2001; Shewry et al. 2003) and in contrast to alpha-gliadins, only ~ 14% of which are pseudogenes in hexaploid bread wheat (Ohno 2013). From clinical point of view, γ-gliadins show a strong association with celiac disease (CD), a chronic inflammatory condition of small intestine triggered by the ingestion of gluten derived from wheat, barley and rye in up to 10% of the populations (Colomba and Gregorini 2012; Ferretti et al. 2012; Van den Broeck et al. 2009). The disease has high heritability, and shows a strong association with the human leukocyte antigen (HLA) class II DQ2/DQ8 molecules as a major genetic risk factor. The formation of disease lesion in small intestine involves in the activation of gluten-reactive CD4 (cluster of differentiation 4) T-cells. These T-cells recognize particular proline and glutamine-rich gluten peptides (CD-epitope cores) presented by the predisposing HLA-DQ2/8 molecules of antigen presenting cells (APCs), where transglutaminase2 (tTG2) enzyme converts certain glutamine (Q) residues to negatively charged glutamate (E) residues (Van den Broeck et al. 2009; Vaccino et al. 2009; Hausch et al. 2002; Sollid 2002). The resulting intestinal inflammation often causes symptoms related to malabsorption, but in many patients, extra-intestinal symptoms dominate, and in some others, the disease is clinically silent (Sollid and Khosla 2005; Bethune and Khosla 2012). However, the only available and effective treatment for CD patients is a lifelong gluten-exclusion diet. With regard to an immunogenicity response, several sets of CD-epitope cores (9-mer peptides) located on the first variable domain R1 (domain II) region of γ-prolamins were identified (Aggarwal et al. 2012; Altenbach et al. 2010; Goryunova et al. 2012; Gu et al. 2004; Kim et al. 2004; Lionetti and Catassi 2011; Meresse et al. 2012; Qi et al. 2009; Qiao et al. 2005; Salentijn et al. 2012; Shewry et al. 1992; Shewry and Tatham 2016; Sjoström et al. 1998; Stenman et al. 2010; Stepniak et al. 2005).
Search for either natural variants of cereals with different core length or structure in one hand and/or breeding cultivars through crosses with monosomic/nullisomic lines via conventional crossing and selection on the other hand may promise development of wheat varieties being non/less immunogenic (Anderson et al. 2001). Furthermore, evolutionary analysis of gluten seed proteins, L/HMW-GS with great protein structure and low evolutionary variations may have greater potential for improving wheat quality (Li et al. 2007; Wang et al. 2011). In addition, from the phylogeny and heterogeneity analysis of gliadins, it can be demonstrated that γ-prolamin multigene family is highly diverse and shows multiple sets of CD-epitope cores with high variability compared to the conserved dodecamer repeat, common to all α/β- and ω-gliadins in wheat and its closely related species (Goryunova et al. 2012; Noma et al. 2016; Qi et al. 2009).
Here the molecular population genetics and evolutionary pattern of γ-prolamin multigene family from wheat and its closely related species (γ-gliadins), rye (γ-secalin) and barley (γ-hordeins) were established, which have not been reported so far. The roles of selective forces that have driven the polymorphism of these duplicated genes at nucleotides and amino acids levels were assessed. Furthermore, we have introduced Triticeae genomes with low CD-epitopes content to develop the deficient cultivars in immunogenic γ-prolamins using conventional breeding and genetic engineering approaches.
Materials and methods
Sequence retrieval and alignment analysis of γ-prolamin multigenes
Orthologous of Triticeae γ-prolamin genes, wheat γ-gliadins, barley γ-hordeins and rye γ-secalins, were identified that contained 557 DNA sequences from 16 population sets (Table 1). CLUSTALW program (accurate) was used to carry out the multiple-sequence alignments at nucleotide level separately among the total DNA sequences of complete genes and pseudogenes from wheat γ-gliadin and their closely relates species; barley γ-hordeins and rye γ-secalins from GenBank at National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov) (McWilliam et al. 2013). Certain insertions were manually removed after sequence alignments prior to calculating the polymorphism parameters.
In-silico identification of γ-prolamin celiac-immunogenic peptides
The total 16 DNA sequences of γ-prolamin genes, with about 99% identity, from sixteen different species of Triticeae were selected as the query sequences for further bioinformatics analysis. The DNA sequence data of the pseudogenes were excluded from in silico CD-epitope analysis. Accession numbers of the query sequences of DNA/protein sequences were JN849093/AFC98439 (Triticum aestivum L.), JQ269810/AFQ20244 (Triticum monococcum L.), FJ006563/ACJ03424 (Triticum turgidum Desf.), FJ006634/ACJ03495 (Triticum urartu Thumanjan ex Gandilyan), FJ0065715/ACJ03544 (Aegilops sharonensis), JQ269769/AFQ20207 (Aegilops speltoides(Tausch) A`Löve), FJ006712/ACJ03541 (Aegilops bicornis Forsk.), FJ006686/ACJ03515 (Aegilops searsii), FJ006648/ACJ03509 (Aegilops longissima Schweinf. et Muschl.), KF880536/AHJ60680 (Aegilops tauschii Coss.), JQ269731/AFQ20178 (Aegilops umbellulata Zhuk.), HQ875873/AEW46778 (Aegilops comosa), JQ269744/AFQ20184 (Aegilops uniaristata), JQ269704/AFQ20157 (Aegilopes markgrafii (Greuter) K.Hammer), HQ266703/ADP95511 (Secale cereale L.), JQ867079/AFM77738 (Hordeum vulgare L., respectively. CLUSTAL Omega program was used to carry out amino acid sequence alignment among total of 16 γ-prolamin query sequences (McWilliam et al. 2013). Based on a prior report on amino acids composition of the common γ-type CD-epitopes, the corresponding epitope position were identified in the amino acid sequences using MEGA 6.0 program (Tamura et al. 2013) and only perfect matches were considered.
Phylogenetic analysis
For the phylogenetic analysis, the sequence data of diploid, tetraploid and hexaploid Triticum sp., Aegilops sp., S. cereale and H. vulgare with their pseudogenes were retrieved from EMBL/Genbank (August 2011). A total of 461 γ-gliadins, 68 γ-hordein and 89 γ-secalins sequences were included in the phylogenetic analysis (Table 1). The sequence data of Oryza sativa prolamin with accession number of X60979 was considered as the outgroup.
Both the nucleotide and the deduced amino acid sequences of γ-gliadin data set were aligned using MEGA 6.0 (Tamura et al. 2013). The γ-prolamin phylogenetic tree for complete DNA sequence (CDS) was constructed by maximum likelihood method (MLE) under the Tamura and Nei 1993 (TN93) model (Saitou and Nei 1987; Tamura and Nei 1993) using a discrete Gamma distribution (+G = 3.08) in MEGA version 6.0 (Tamura et al. 2013). The 1000 bootstrap replicates were performed, and values greater than 50% frequencies were shown. All positions containing alignment gaps and missing data were eliminated only in pairwise sequence comparisons (Pairwise deletion option).
Evolution and selection pressure analysis
The nucleotide diversity, Tajima’s D test (Tajima 1989), Fay and Wu’s H (Fay and Wu 2000), and a sliding window analysis were carried out using DNASP4.10 (Rozas et al. 2003). The window size for the promoter and transcriptional units (TU) analyses was 50 bp with a step size of 10 bp. The number of base differences per site, number of synonymous differences per synonymous site and number of non-synonymous differences per non-synonymous site from averaging overall sequence pairs within each group and overall sequences were calculated using Nei and Gojobori (Nei 1987) method in MEGA 6.0 (Tamura et al. 2013). Support for individual nodes was assessed through random resampling of sequences with 1000 bootstraps. All positions containing alignment gaps and missing data were eliminated only in pairwise sequence comparisons (Pairwise deletion option). The ratio between synonymous substitutions per site (dS) and non-synonymous substitutions per site (dN), and (dN/dS ratio) were calculated. Sequence divergences between orthologs and paralogs were estimated using Mega version 6.0 (Tamura et al. 2013). The evolutionary distance, k, among γ-prolamin endosperm-specific promoter sequences (500 bp upstream of the coding region) was computed using the kimura 2-parameter substitution model and gaps were treated as missing data. All standard errors of divergence distances were determined using 500 bootstrap replicates. The heterogeneity of polymorphisms to fixed differences, Gmean and DKS statistics were computed by DNA Slider 1.13 (McDonald 1996, 1998). For Gmean and DKS statistics, recombination parameter (R) of 2, 4, 8, 16 and 32 were simulated with 1000 replicates. The highest p value of each statistic was reported.
Results
Sequence polymorphisms of γ-prolamin genes in Triticeae
Sequence polymorphic analysis was revealed that the ORF lengths of the sequences derived from Ae. sharonensis were the most variable (678–1020 bp), and for T. monococcum were the most conserved in length as previously reported by Qi et al. (2009). In total 126 sequences from Triticeae excluding H. vulgare were pseudogenes. All of which contained one or more internal stop codons or frameshift mutations caused by single nucleotide indels (insertions/deletions). The remaining 431 sequences were putatively functional, with no internal stop codons.
Comparative sequence polymorphic analyses between coding DNA sequences (CDS) and promoter sequences of γ-prolamins were revealed substantial differences. Nucleotide sequence analysis of 20 γ-prolamin endosperm-specific promoters revealed 99% identity. γ-prolamins’ promoter regions showed a high nucleotide diversity (π) (Watterson 1975) of 1.25-fold and 1.36-fold greater than that of γ-secalins and γ-gliadins coding regions, respectively. In contrast, the coding region of γ-hordein showed the nucleotide diversity of 0.17673, which was 1.56-fold higher than that of γ-prolamin endosperm specific promoter region. A similar trend was observed when another nucleotide diversity estimate (θw) (Tamura and Nei 1993) was examined. The nucleotide diversity (π) for γ-gliadin coding region was 0.08263; T. tugidum and T. urartu showed the highest (0.14005) and the lowest (0.01966) nucleotide diversity values, respectively (For more detail see Table 1).
Tajima’s D test was not significantly negative in the endosperm-specific promoter region of γ-prolamins (D = − 0.584, p > 0.1, Table 1). In the coding region of γ-gliadins, the D value was not significant excepte for T. urartu with D value of -1.9353 (p < 0.05, Table 1), which was marginally negative among γ-gliadin genes. This indicates that the entire coding region of γ-gliadins from T. urartu was favored by positive selection, and beneficial alleles have been fixed. However, a significant negative D value can also be the result of deriving low-frequency detrimental alleles or a bottleneck effect as described by Huang et al. (2002). Similar to γ-gliadin, neither the promoter nor coding region had significant D values for γ-secalin and γ-hordein genes.
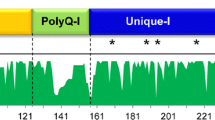
Different regions of γ-prolamin promoter and coding regions were scanned using sliding windows of Tajima’s D test to identify regions that deviated from neutral expectations. In the promoter region (500 bp upstream of the γ-prolamin coding region), D values after about position of 220 were insignificantly positive. In the entire region of promoter, the proportions of positive and negative regions were compatible (Fig. 1A). Furthermore, the CDS sliding window analysis showed the maximum number of singleton polymorphic sites lying in between 1095 and 1490 from the first variable R1 domain (domain II) of the CDS (Fig. 1B). Therefore, both the promoter and the coding region of γ-prolamin multigenes did not deviate from neutrality at nucleotide level.

Sliding windows of Tajima’s D tests along endosperm specific promoter (A) and coding region (B) of γ-prolamins. The scale of the sliding window plot was adjusted for the promoter regions. The last (3′-end) sites of the promoter sequences of γ-prolamin were placed in the same position. The window size is 50 bp, and the step size is 10 bp
In-silico identification of the conserved CD-epitope sites
The amino acid sequence alignment was carried out among the deduced amino acid sequences of the sixteen templates using CLUSTAL Omega program at EMBL (Fig. 2). The seven common CD-epitopes including DQ2.5-glia-γ1 (PQQSFPQQQ), DQ2.5-glia-γ2a (FPQQPQQPF), DQ2.5-glia-γ3 (QQPQQPYPQ), DQ2.5-glia-γ4a (SQPQQQFPQ), DQ2.5-glia-γ4b (PQPQQQFPQ), DQ2.5-glia-γ4c (QQPQQPFPQ) and DQ8-glia-γ1a (QQPQQPFPQ) were identified (the targets for tTG deamidation are bold and underlined) as the most conserved motifs in the domain R1 region of γ-prolamins (Arentz-Hansen et al. 2002; Sjöström et al. 1998; Sollid et al. 2012; Stepniak et al. 2005). Furthermore, DQ2.5-glia-γ4b/c and DQ2.5-glia-γ1 CD-epitopes showed the highest homology (identity = 98.7%) in Triticeae γ-prolamins.

Schematic diagram of the alignment among the 16 deduced amino acid sequences of the γ-prolamins templates from Triticeae. The amino acid sequences of the CD-epitope cores (9-mer peptides) are shown in red letters. In the epitope names, the short terms denote the type of proteins that the epitope derived from: 'glia-γ' denotes γ-gliadin and 'sec' denotes secalin
Clustering and phylogeny relationship analysis
The maximum likelihood (ML) tree of DNA sequences from CDS produced separate clusters of γ-secalins and two well-separated large groups of γ-gliadins with unequal size: 145 consensus sequences belonged to the first large group and 275 consensus sequences belonged to the second large group (Fig. 3). In total ten significant (bootstrap support value of 95% or higher) groups from CDS region were observed. The accession number of HQ875932 from Secale cereale as ancestral sequence of γ-gliadin and γ-secalin was considered as a root of the γ-secalin and γ-gliadin branches. Total DNA sequence data of γ-secalin genes from T. aestivum (AABBDD) and S. cereale (RR) and H. vulgare (VV) were restricted to group 1 in the first ancestral branch. Sequences originating from Triticum species with an A genome (T. monococcum (Am), T. urartu (Au), T. aestivum (AABBDD) and Ae. uniaristata (NN) were restricted to the third branch. Within this branch, all γ-gliadin sequences from T. monococcum (Am), T. urartu (Au) and T. aestivum clustered in group 10. However, the sequences originating from Triticum and Aegilops species with B genome (T. aestivum (AABBDD), T. turgidum (AABB) and Ae. speltoides (BB)) and D genome (T. aestivum (AABBDD) and Ae. tauschii (DD)) were clustered into groups 2, 8 and 7, respectively. All groups except 1, 7, 8 and 10 contained a mixture of sequences of three to six species of Aegilops sp. and Triticum sp.

Molecular Phylogenetic analysis of γ-prolamin from Triticeae by Maximum Likelihood method. The tree is drawn to scale, with branch lengths measured in the 0.1 substitutions per site. The analysis involved 558 nucleotide sequences including an outgroup sequence from Oryza sativa (Accession No. X60979). All positions containing gaps and missing data were eliminated. There were a total of 122 positions in the final dataset. The evolutionary history was inferred by using the Maximum Likelihood method based on the Tamura-Nei model (Tamura and Nei 1993). The tree with the highest log likelihood (− 2899.9916) is shown. Initial tree(s) for the heuristic search were obtained by applying the neighbor-joining method to a matrix of pairwise distances estimated using the maximum composite likelihood (MCL) approach. A discrete Gamma distribution was used to model evolutionary rate differences among sites (3 categories [+G, parameter = 2.3866)]. Evolutionary analyses were conducted in MEGA6 (Tamura et al. 2013)
Genetic variation analysis within and among the groups
In addition to genetic variation analysis within Triticeae species, a comparative evolutionary pattern analysis within and among the groups were carried out using Tajima’s D test in order to determine any deviation from neutral theory. Tajima’s D was significantly negative in groups 3 (D = − 2.312) and 8 (D = − 2.451) (p < 0.01). D values for groups 1 (− 1.560) and 2 (− 1.848) were also marginally negative (p < 0.05, Table 1). Since, most members of groups 2, 3 and 8 were from Ae. tauschii (DD genome) and T. aestivum (AABBDD), the significant negative D values of the groups might be due to the D genome. Thus, it is probable that the γ-gliadins present on the D genome were favored by either positive selection or bottleneck effect that led to the fixation of these beneficial alleles during evolution. With the same argument, it can be stated that positive selection probably has been occurred in γ-secalin from S. cereale (RR genome) according to negative significant D value for group 1. Additionally, the signature of positive selection of the groups was further confirmed by Fay and Wu’s H test, Gmean and DKS statistics (Table 1). In contrast, significant D value within the groups, among two large groups and overall groups were not seen (Table 2).
Fay and Wu’s H test was used to demonstrate if selective sweep, reduction in polymorphism via fixation of an advantageous mutation, has been in process through calculation of differences between high-frequency mutations and intermediate-frequency mutations (Lin et al. 2008). For this a homologous sequence from Oryza sativa was used as an outgroup. In our study, the most significant H values were − 21.78983 (p < 0.01; group 1 with R genome), − 26.16909 (p = 0.0012; group 2 with D genome), − 15.79710 (p = 0.0015; group 3 with D genome) and − 16.90587 (p < 0.02; group 8 with D genome). These negative significant H values are illustrative of a recent selective sweep in the species of S. cereale, T. aestivum and Ae. tauschii.
Furthermore, Gmean, statistics were used to determine number of polymorphic sites. DKS was used as an indication of the heterogeneity of polymorphism to divergence ratios. These statistics were used in the coding DNA sequences of species within groups 1, 2, 3, and 8. Gmean is most sensitive for detecting one or two peaks, and DKS is good at detecting a single low to high change in polymorphism (McDonald 1998). The Gmean and DKS tests were significant for all four groups; indicative of heterogeneity in polymorphism-to-divergence ratios in their coding region (Table 3). In other words, localized selective sweeps were detected. Thus, the selective sweep event in R and D genomes (especially in the γ-prolamin coding region) was supported by four neutrality tests.
Selection pressure in γ-prolamin evolution
The ratios of ω = dN/dS were computed within and among the defined groups (Table 2). The ratio of dN/dS indicates protein evolution. Where the values of these ratios are greater, equal, and smaller than 1, it means positive, neutral and purifying selection, respectively (Yang 2007). Here, group 9 with R genome in S. cereale and Ae. markgrafii (Greuter) K. Hammer (C genome) showed the highest ω = 1.5463, while group 8 with T. aestivum (ABD genome) and Ae. tauschii (D genome) showed the lowest ratio of ω = 0.7198. Thus, it can be stated that strong positive and purifying selection are acting upon C and D genomes, respectively. Furthermore, the high dN/dS ratios of group 1 (ω = 1.3346) and group 7 (ω = 1.2022) indicated the positive selection for S. cereale (R genome), Ae. speltoides (B genome), Ae. comosa (M genome), and T. turgidum (AB genome). Within groups of 2, 3, 8 (D genome), group 5 (AB genome), and 10 (A genome), the marginally purifying selection were observed, while group 4 and 6 both with Ae. searsii, Ae. sharonensis, Ae. bicornis and Ae. longissima (S genome) and Ae. umbellulata (U genome) were favored for neutral hypothesis.
Since, the total 10 groups of γ-prolamins lie in two large branches, we have determined the dN/dS ratio of these large ancestral branches. The results showed that a purifying selection acts on duplication of the total 2 large ancestral groups, while the dN/dS ratio of 1.0470 revealed the neutral selection among overall groups.
Discussion
Gene duplication and subsequent functional divergence are among the contributing factors in evolution of multigene families such as prolamines (Anderson et al. 2001; Nei 1969; Nei and Roychoudhury 1973; Ohno 2013; Shewry et al. 2003; Shewry and Tatham 1990; Stephens 1951; Ohno 1967). Analysis of the γ-gliadin multigene family fits with the birth-and-death evolutionary model with multiple gene duplication and divergence events, which was previously reported by Goryunova et al. (2012). In all Aegilops/Triticum genomes, the signatures of pseudogene formation and gene loss are apparent. All of which have split within a short evolutionary period (2.5–4.5 MYA) for Aegilops/Triticum group (A, S/B, C, D, M, N, U), S. cereale (R genome) and H. vulgare (V genome) (Van Slageren, 1994) and the multigenes were expanded within this period (Goryunova et al. 2012). Here, γ-type seed prolamins, with no prior record of evolutionary studies, were considered for the analysis of genetic diversity in their promoters and TU regions.
A comparative and extensive analysis was carried out on 557 γ-prolamin sequences, including 461 γ-gliadin from Triticum sp. and Aegilops sp. (A, B, D, S, U, M, N, and C genomes), 89 γ-secalin from S. cereale (R genome) and 7 γ-hordein from H. vulgare (V genome). The sequences were retrieved from GenBank and further considered to check on the sequence diversity, gene duplication and the effect of natural selection forces that mirrored on the evolution of the γ-prolamin multigene family. Furthermore, Triticeae genomes were examined to determine the genotypes with low CD-epitope contents. These genotypes might be useful in the development of future cultivars with reduced level of immunogenicity/allergenicity.
DNA sequence polymorphism and genetic diversity
Nucleotide diversity (θw and π) of the γ-prolamin endosperm-specific promoter was greater than that of γ-secalins and γ-gliadins transcriptional units (TUs), except for γ-hordeins. Similar to many genes, γ-prolamin TUs are more conserved than promoter sequences. Accordingly, these two sequence units have been subjected to distinct selection mechanisms and/or demographic histories. In contrast in γ-hordeins, duplication events have been occurred in blocks of “promoter + TU”, resulting in relatively higher sequence conservation within the promoter. Furthermore, high haplotype diversity was evident for γ-prolamins in general.
According to the several reports, ORF lengths of γ-prolamin sequences range from 678 to 1089 bp, while Ae. sharonensis (678 bp) and Ae. speltoides (1089 bp) are respectively the shortest and the longest so far reported by Qi et al. 2009 as well as our alignment results. None of which does contain intron, interrupting the coding DNA sequence (Qi et al. 2009). The protein structure of γ-prolamins comprised of a 20-residue signal peptide, followed by a short N-terminus non-repetitive domain (I), a highly variable repetitive domain (II), a non-repetitive domain containing most of the cysteine residues (III), a glutamine-rich region (IV), and the C-terminal non-repetitive domain containing the final two conserved cysteine residues (V) (Goryunova et al. 2012; Qi et al. 2009). The sliding window analysis revealed that the maximum number of singleton polymorphisms lie in the first variable domain R1 (domain II) region. This domain bears immunogenic epitopes that leads to CD (Anderson et al. 2001; Salentijn et al. 2012). According to the previous report, the long size of the repetitive domain (encompassing about 45% of total γ-gliadins length) contains regular short repeats caused by SNPs (single nucleotide polymorphisms) (Qi et al. 2009). Here, our data was affirmative on the role of SNPs and frequent amino acid sequence variations specifically within the first variable R1 domain with ~ 138–537 bp long, which is mainly responsible for the size of the γ-gliadins heterogeneity.
Different selection forces working on different γ-prolamin genomes
The evolutionary analyses revealed that selection plays a key role in the maintenance of γ-prolamin promoter and coding regions within and among Triticeae species in the form of positive selection, which are presented by negative values of the neutrality test statistics (Tajima’s D, Fu and Li’s D) as previously described by Goryunova et al. (2012) and Qi et al. (2009). Overall, Gli-1 loci are diverse, although γ-type sub-fractions are supposed to be the most ancient family among prolamins (Shewry and Tatham 1990; Sabelli and Shewry 1991). Furthermore, Tajima’s D test was not significantly negative in the γ-prolamins endosperm-specific promoter region and their transcriptional units within Triticeae species. The only exception was T. urartu, which seems that positive selection is in action and beneficial alleles has been fixed. Thus, neutral hypothesis stands for both the promoter and TU sequences of γ-prolamins. Following duplication events, an equivalence mutation and genetic drift most likely have happened among the nucleotide sequences of γ-prolamins from Triticeae species.
The phylogeny analysis grouped Triticeae γ-prolamins into ten clades of two ancestral branches: first including groups of one to five, and second including groups of six to ten. Group 1 and 10 comprised of respectively R genome from S. cereale, V genome H. vulgare and A genome from T. monococcum, while groups 2, 3, 8 comprised of D genome from Ae. tauschii and T. aestivum. The sequences of H. vulgare lie in separate cluster, separating γ-hordeins from γ-secalins and γ-gliadins. Furthermore, γ-hordeins demonstrated greater genetic diversity than other γ-prolamins. Additionally, an extensive endo-reduplication and multiple polymorphic indels within group 1 (R and V genomes), group 10 (A genome), groups 2, 3 and 8 (D genome) was evident. In groups containing varieties of species, the action of horizontal gene transfer through introgression followed by duplication may describe the genetic diversity.
The significant negative Tajima’s D value of each groups 1, 2, 3, 8 revealed the positive selection pressure acting on R and D genome duplication process. Furthermore, the other statistical parameters, Fay and Wu’s H test, Gmean and DKS, on these 4 groups revealed a strong selective sweep within each group. Although within the two ancestral branches and groups inside, neutral theory holds, dN/dS ratio suggests a strong purifying selection for the ancestral groups. The ω < 1 values of individual groups of 2, 3, 5, 8, 10 and separately two ancestral groups indicate that γ-prolamins are an evolutionary older family than was proposed by Goryunova et al. (2012).
Perspective for cereal breeding programs
So far, the only efficient proposed therapy for CD is a life-long gluten-free diet. However, modified/shortened T-cell stimulatory gluten peptides may reduce or even abolish the immunogenicity of gluten consumption in the patients. Moreover, consumption of foods prepared with these kinds of flour may bring the progression of the disease into halt (Salentijn et al. 2012; Shewry and Tatham 1990). Recently, a number of new potential therapeutic alternatives to the gluten-free diet are under development, including enzymatic detoxification of gluten, tissue transglutaminase inhibitors, blocking of HLA-DQ peptide presentation, silencing of gluten-reactive T cells, cytokine therapy and selective adhesion molecule inhibition, and selection or genetic engineering of less toxic grains (Sollid and Khosla 2005; Bethune and Khosla 2012). Several reports have suggested that the genetic differences in gliadins and γ-prolamin genes with short repetitive R1 domain lead to design strategies for making non-toxic and more nutritious cereal varieties (Molberg et al. 2005; Qi et al. 2009; Spaenij-Dekking et al. 2005). We sought for the genomes with indels or endo-reduplication of the R1 domain. Moreover, we have also mainly introduced the conservative CD epitopes in the γ-prolamin TU region using bioinformatics approach. With overlapping between the conservative CD epitopes and the regions with the least polymorphism in the A and D genomes, we have demonstrated that groups 2, 3, 8 (D genome), and 10 (A genome) showed the lowest polymorphism with fewer CD epitopes than other genomes. This suggests that these genomes might be valuable progenitors in breeding programs to possibly generate less-immunogenic lines as previously reported by Wang et al. (2012). Therefore, we believe that the progenitors of these genomes, A and D, have to be used in future breeding programs to develop cereal flours that are less immunogenic in CD patients or gluten sensitive individuals. Additionally, attempts have been made to use RNAi technology to reduce the corresponding transcripts and therefore lowering down the prolamins of seeds to possibly make the flour less immunogenic (Altenbach and Allen 2011; Gil-Humanes et al. 2008, 2010, 2014; Kohnehrouz and Nayeri 2016; Wen et al. 2012). Advent of genome editing technologies (Boettcher and McManus 2015; Lozano-Juste and Cutler 2014) may promise developing of varieties with totally modified R1 domain. Taking such venues to change the genetics of current cereal cultivars may help to overcome the current gluten free diet (GFD) approach for CD patients and gluten sensitive individuals that accounts for nearly 10% of the worldwide population (Sollid and Khosla 2011; Pérez et al. 2012).
References
Aggarwal S, Lebwohl B, Green PH (2012) Screening for celiac disease in average-risk and high-risk populations. Ther Adv Gastroenterol 5(1):37–47
Altenbach SB, Allen PV (2011) Transformation of the US bread wheat ‘Butte 86’and silencing of omega-5 gliadin genes. GM Crops 2(1):66–73
Altenbach SB, Vensel WH, DuPont FM (2010) Analysis of expressed sequence tags from a single wheat cultivar facilitates interpretation of tandem mass spectrometry data and discrimination of gamma gliadin proteins that may play different functional roles in flour. BMC Plant Biol 10(1):7
Anderson OD, Hsia CC, Torres V (2001) The wheat γ-gliadin genes: characterization of ten new sequences and further understanding of γ-gliadin gene family structure. Theor Appl Genet 103:323–330
Anderson OD, Dong L, Huo N, Gu YQ (2012) A new class of wheat gliadin genes and proteins. PLoS One 7(12):e52139
Arentz-Hansen H, McAdam SN, Molberg O et al (2002) Celiac lesion T-cells recognize epitopes that cluster in regions of gliadins rich in proline residues. Gastroenterology 123:803–809
Bethune TM, Khosla C (2012) Oral enzyme therapy for celiac sprue. Methods Enzymol 502:241–271
Bietz JA, Wall JS (1972) Wheat gluten subunits: molecular weights determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Cereal Chem 49(4):416–430
Boettcher M, McManus MT (2015) Choosing the right tool for the job: RNAi, TALEN, or CRISPR. Mol Cell 58(4):575–585
Colomba MS, Gregorini A (2012) Are ancient durum wheats less toxic to celiac patients? a study of α-gliadin from Graziella Ra and Kamut. Sci World J 2012:837416
Fay JC, Wu CI (2000) Hitchhiking under positive Darwinian selection. Genetics 155(3):1405–1413
Ferretti G, Bacchetti T, Masciangelo S, Saturni L (2012) Celiac disease, inflammation and oxidative damage: a nutrigenetic approach. Nutrients 4:243–257
Gellrich C, Schieberle P, Wieser H (2003) Biochemical characterization and quantification of the storage protein (secalin) types in rye flour. Cereal Chem 80(1):102
Gellrich C, Schieberle P, Wieser H (2005) Studies of partial amino acid sequences of γ-40 k secalins of rye. Cereal Chem 82(5):541–545
Gil-Humanes J, Pistón F, Hernando A et al (2008) Silencing of γ-gliadins by RNA interference (RNAi) in bread wheat. J Cereal Sci 48(3):565–568
Gil-Humanes J, Pistón F, Tollefsen S et al (2010) Effective shutdown in the expression of celiac disease-related wheat gliadin T-cell epitopes by RNA interference. Proc Natl Acad Sci USA 107(39):17023–17028
Gil-Humanes J, Pistón F, Altamirano-Fortoul R et al (2014) Reduced-gliadin wheat bread: an alternative to the gluten-free diet for consumers suffering gluten-related pathologies. PLoS One 9(3):e90898
Goryunova SV, Salentijn EM, Chikida NN et al (2012) Expansion of the gamma-gliadin gene family in Aegilops and Triticum. BMC Evol Biol 12(1):215
Gu YQ, Crossman C, Kong X et al (2004) Genomic organization of the complex α-gliadin gene loci in wheat. Theor Appl Genet 109(3):648–657
Guo G, Lv D, Yan X, Subburaj S et al (2012) Proteome characterization of developing grains in bread wheat cultivars (Triticum aestivum L.). BMC Plant Biol 12(1):147
Hausch F, Shan L, Santiago NA, Gray GM, Khosla C (2002) Intestinal digestive resistance of immunodominant gliadin peptides. Am J Physiol Gastrointest Liver Physiol 283:G996–G1003
Huang S, Sirikhachornkit A, Su X et al (2002) Genes encoding plastid acetyl-CoA carboxylase and 3-phosphoglycerate kinase of the Triticum/Aegilops complex and the evolutionary history of polyploid wheat. Proc Natl Acad Sci 99(12):8133–8138
Kim CY, Quarsten H, Bergseng E, Khosla C, Sollid LM (2004) Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci 101(12):4175–4179
Kohnehrouz BB, Nayeri S (2016) Design, cloning and in silico analysis of efficient siRNA-inducing cassette for silencing wheat γ-gliadins. Jordan J Biol Sci 9(1):35–40
Li X, Zhang Y, Gao L, Wang A, Ji K, He Z, Appels R, Ma W, Yan Y (2007) Molecular cloning, heterologous expression, and phylogenetic analysis of a novel y-type HMW glutenin subunit gene from the G genome of Triticum timopheevi. Genome 50(12):1130–1140
Lin YH, Hwang SY, Hsu PY et al (2008) Molecular population genetics and gene expression analysis of duplicated CBF genes of Arabidopsis thaliana. BMC Plant Biol 8(1):111
Lionetti E, Catassi C (2011) New clues in celiac disease epidemiology, pathogenesis, clinical manifestations, and treatment. Int Rev Immunol 30(4):219–231
Lozano-Juste J, Cutler SR (2014) Plant genome engineering in full bloom. Trends Plant Sci 19(5):284–287
McDonald JH (1996) Detecting non-neutral heterogeneity across a region of DNA sequence in the ratio of polymorphism to divergence. Mol Biol Evol 13(1):253–260
McDonald JH (1998) Improved tests for heterogeneity across a region of DNA sequence in the ratio of polymorphism to divergence. Mol Biol Evol 15(4):377–384
McWilliam H, Li W, Uludag M, Squizzato S et al (2013) Analysis tool web services from the EMBL-EBI. Nucleic Acids Res 41(W1):W597–W600
Meresse B, Malamut G, Cerf-Bensussan N (2012) Celiac disease: an immunological jigsaw. Immunity 36(6):907–919
Molberg O, Uhlen AK, Jensen T et al (2005) Mapping of gluten T-cell epitopes in the bread wheat ancestors: implications for celiac disease. Gastroenterology 128(2):393–401
Nei M (1969) Gene duplication and nucleotide substitution in evolution. Nature 221(5175):40–42
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nei M, Roychoudhury AK (1973) Probability of fixation of nonfunctional genes at duplicate loci. Am Nat 107(955):362–372
Noma S, Kawaura K, Hayakawa K et al (2016) Comprehensive molecular characterization of the α/β-gliadin multigene family in hexaploid wheat. Mol Genet Genomics 291:65
Ohno S (1967) Sex chromosome and sex-linked genes. Chromosoma 23(1):1–9
Ohno S (2013) Evolution by gene duplication. Springer Science and Business Media, Berlin
Payne PI, Law CN, Mudd EE (1980) Control by homoeologous group 1 chromosomes of the high-molecular-weight subunits of glutenin, a major protein of wheat endosperm. Theor Appl Genet 58(3–4):113–120
Payne PI, Holt LM, Jackson EA, Law CN, Damania AB (1984a) Wheat storage proteins: their genetics and their potential for manipulation by plant breeding. Philos Trans R Soc Lond B Biol Sci 304(1120):359–371
Payne PI, Jackson EA, Holt LM, Law CN (1984b) Genetic linkage between endosperm storage protein genes on each of the short arms of chromosomes 1A and 1B in wheat. Theor Appl Genet 67:235–243
Pérez LC, de Villasante GC, Ruiz AC, León F (2012) Non-dietary therapeutic clinical trials in coeliac disease. Eur J Intern Med 23(1):9–14
Qi PF, Wei YM, Ouellet T, Chen Q, Tan X, Zheng YL (2009) The γ-gliadin multigene family in common wheat (Triticum aestivum) and its closely related species. BMC Genomics 10(1):168
Qi PF, Chen Q, Ouellet T et al (2013) Identification of 40k γ-secalin genes. J Genet 92(1):151–154
Qiao SW, Bergseng E, Molberg O, Jung G, Fleckenstein B, Sollid LM (2005) Refining the rules of gliadin T cell epitope binding to the disease-associated DQ2 molecule in celiac disease: importance of proline spacing and glutamine deamidation. J Immunol 175(1):254–261
Rosell CM, Barro F, Sousa C, Mena MC (2014) Cereals for developing gluten-free products and analytical tools for gluten detection. J Cereal Sci 59(3):354–364
Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19(18):2496–2497
Sabelli PA, Shewry PR (1991) Characterization and organization of gene families at the Gli-1 loci of bread and durum wheats by restriction fragment analysis. Theor Appl Genet 83(2):209–216
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406–425
Salentijn MJE, Mitea DC, Goryunova VS et al (2012) Celiac disease T-cell epitopes from gamma-gliadins: immune-reactivity depends on the genome of origin, transcript frequency, and flanking protein variation. Bio Med Cent Genomics 13:277
Shan L, Molberg O, Parrot I et al (2002) Structural basis for gluten intolerance in celiac sprue. Science 297(5590):2275–2279
Shewry PR, Tatham AS (1990) The prolamin storage proteins of cereal seeds: structure and evolution. Biochem J 267(1):1
Shewry PR, Tatham AS (2016) Improving wheat to remove coeliac epitopes but retain functionality. J Cereal Sci 67:12–21
Shewry PR, Tatham AS, Kasarda DD (1992) Cereal proteins and coeliac disease. In: Marsh MN (ed) Coeliac disease. Blackwell Scientific Publications, Oxford, pp 305–348
Shewry PR, Tatham AS, Halford NG (1999) The prolamins of the Triticeae. In: Marsh MN (ed) Coeliac Disease. Seed proteins. Springer, Dordrecht, pp 35–78
Shewry PR, Halford NG, Lafiandra D (2003) Genetics of wheat gluten proteins. Adv Genet 49:111–184
Singh NK, Donovan GR, Batey IL, MacRitchie F (1990) Use of sonication and size-exclusion high-performance liquid chromatography in the study of wheat flour proteins. I. Dissolution of total proteins in the absence of reducing agents. Cereal Chem 67(2):150–161
Sjoström H, Lundin KEA, Molberg O et al (1998) Identification of a gliadin T-cell epitope in coeliac disease: general importance of gliadin deamidation for intestinal T-cell recognition. Scand J Immunol 48:111–115
Sollid LM (2002) Coeliac disease: dissecting a complex on inflammatory disorder. Nat Rev Immunol 2:647–655
Sollid LM, Khosla C (2005) Future therapeutic options for celiac disease. Nat Clin Pract Gastroenterol Hepatol 2(3):140–147
Sollid LM, Khosla C (2011) Novel therapies for coeliac disease. J Intern Med 269(6):604–613
Sollid LM, Qiao SW, Anderson RP, Gianfrani C, Koning F (2012) Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules. Immunogenetics 64(6):455–460
Spaenij-Dekking L, Kooy-Winkelaar Y, van Veelen P et al (2005) Natural variation in toxicity of wheat: potential for selection of nontoxic varieties for celiac disease patients. Gastroenterology 129(3):797–806
Stenman S (2011) Coeliac disease-inducing gluten: in vitro harmfulness and detoxification by germinating cereal enzymes. Tampere University Press, Tampere
Stenman SM, Lindfors K, Venalainen JI et al (2010) Degradation of coeliac disease-inducing rye secalin by germinating cereal enzymes: diminishing toxic effects in intestinal epithelial cells. Clin Exp Immunol 161:242–249
Stephens SG (1951) Possible significance of duplication in evolution. Adv Genet 4:247–265
Stepniak D, Vader LW, Kooy Y et al (2005) T-cell recognition of HLA-DQ2-bound gluten peptides can be influenced by an N-terminal proline at p-1. Immunogenetics 57(1–2):8–15
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10(3):512–526
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729
Tatham AS, Shewry PR (1995) The S-poor prolamins of wheat, barley and rye. J Cereal Sci 22(1):1–16
Vaccino P, Becker H, Brandolini A, Salamini F, Kilian B (2009) A catalogue of Triticum monococcum genes encoding toxic and immunogenic peptides for celiac disease patients. Mol Genet Genomics 281:289–300
Van den Broeck HC, Herpen WJMT, Schuit C, Salentijn MJE, Dekking L, Bosch D, Hamer JR, Smulders JMM, Gilissen JWJL, Van der Meer MI (2009) Removing celiac disease-related gluten proteins from bread wheat while retaining technological properties: a study with Chinese Spring deletion lines. BMC Plant Biol 9:41
Van Slageren MW (1994) Wild wheats: a monograph of Aegilops L. and Amblyopyrum (Jaub. & Spach) Eig (Poaceae). Wageningen Agricultural University, Wageningen, p 530
Wang S, Li X, Wang K, Wang X, Li S, Zhang Y, Guo G, Zeller FJ, Hsam SL, Yan Y (2011) Phylogenetic analysis of C, M, N, and U genomes and their relationships with Triticum and other related genomes as revealed by LMW-GS genes at Glu-3 loci. Genome 54(4):273–284
Wang S, Shen X, Ge P, Li J, Subburaj S, Li X, Zeller FJ, Hsam SLK, Yan Y (2012) Molecular characterization and dynamic expression patterns of two types of γ-gliadin genes from Aegilops and Triticum species. Theor Appl Genetics 125(7):1371–1384
Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7(2):256–276
Wen S, Wen N, Pang J et al (2012) Structural genes of wheat and barley 5-methylcytosine DNA glycosylases and their potential applications for human health. Proc Natl Acad Sci 109(50):20543–20548
Wieser H (2007) Chemistry of gluten proteins. Food Microbiol 24(2):115–119
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Acknowledgements
This work is supported by Dean of Research to Shahid Beheshti University.
Author information
Authors and Affiliations
Contributions
S. Nayeri suggested the idea based on her prior work on prolamins and performed the in silico analysis of the data. Both S. Nayeri and N. Farrokhi discussed the results and contributed to the final version of the manuscript. N. Farrokhi supervised the project.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Nayeri, S., Farrokhi, N. The less genetic diversity of Triticeae A and D genomes, the smaller diversity in γ-prolamins CD-epitopes. Genet Resour Crop Evol 65, 2013–2026 (2018). https://doi.org/10.1007/s10722-018-0675-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-018-0675-9