
Abstract
The blueberry (Vaccinium L. section Cyanococcus Rydb.) is a health promoting and economically important small fruit crop. Structured genetic diversity and relatedness were studied in 63 blueberry wild clones and cultivars , using eleven EST-PCR and nine microsatellite markers. Markers were found to be polymorphic and detected 249 alleles with mean polymorphic information content of 0.80 for EST-PCR; and 164 alleles with mean polymorphic information content of 0.77 for microsatellite markers. The average resolving power was 4.6 for EST-PCR and 2.6 for microsatellite markers. The average values of expected and observed heterozygosity, inbreeding coefficient and Shannon’s index were higher for the EST-PCR markers than those of microsatellites. Multivariate clustering analyses using neighbor joining and principal coordinate analyses formed five groups and clustered the genotypes according to their place of origin that were also confirmed by STRUCTURE analysis and analysis of molecular variance. While 33% of variation was found among the geographic groups, the variation among the communities within the groups was 23% and among genotypes within the communities was 44%, in combined analysis. The availability of high genetic diversity among the wild clones will contribute significantly in germplasm management and their utilization in the current blueberry improvement program.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Blueberries belong to the genus Vaccinium L., which contains between 400 and 500 species of shrubs or small trees worldwide (Vander Kloet 1988; Vander Kloet and Dickinson 2009). Research strongly suggests the association of blueberry antioxidants with numerous health benefits (Halliwell 2007; Joseph et al. 2003; Willis et al. 2005). Lowbush blueberries are native to eastern Canada and the north-east USA, and are predominantly the tetraploid “sweet lowbush blueberry” V. angustifolium Ait., but also include V. myrtilloides Michaux.
Genetic diversity has an impact on higher levels of biodiversity such as morphological and biochemical (Templeton 1996). Generally the wild species are more diverse than the cultivated varieties. The development of new cultivars requires selection of superior wild growing plants and crossing them with half-high or highbush blueberries.
Microsatellites or simple sequence repeats (SSRs) are among the available molecular markers that gained huge importance for studying genetic diversity, mapping, marker-assisted breeding and population genetic analyses for their highly codominant and polymorphic nature (Chapman et al. 2009). As a large number of ESTs are available in public domain, their use is cost-effective and efficient in a breeding program. The present study was conducted with 56 wild blueberry clones, six cultivars and one selection to identify EST-PCR and microsatellite primers suitable for fingerprinting the present set of blueberry genotypes, to assess the properties of markers and their importance in assessing the diversity in these blueberries and to evaluate genetic diversity and population structure among these genotypes. Eleven EST-PCR and nine microsatellite primer pairs were used.
Materials and methods
Plant material
The wild lowbush blueberry clones belonging to V. angustifolium Ait. (Table 1) were collected from four Canadian provinces: Newfoundland and Labrador (37, designated as NL1 to NL37), Prince Edward Island (11, designated as PE1-PE11), Quebec (six, designated as QC1, QC2, QC4, QC5, QC7 and QC9) and from New Brunswick (two, designated as NB1 and NB2). The other genotypes included were three half-high blueberry cultivars: ‘Patriot’ (PT), ‘Chippewa’ (CH) and ‘St. Cloud’ (SC); one highbush blueberry cultivars ‘Polaris’ (PO); two lowbush blueberry cultivars ‘Fundy’ (FU) and ‘Brunswick’ (BR); and one lowbush blueberry selection (designated as FO) obtained from the open-pollinated seedlings of lowbush blueberry cultivar FU. Half-high blueberries are the cultivars derived from the crossing of lowbush and highbush blueberries (V. corymbosum L.) (Galletta and Ballington 1996). The clones were collected in August 2001 from the wild, based on optimal berry colour, plant vigour, berry size, berry yield and apparently free from insect and disease. During the collection process, it was taken into consideration that the distance between the two selected plants within a same community was more than 10 m (Debnath 8,9,). All genotypes were grown and maintained in a greenhouse of the St. John’s Research and Development Centre, Agriculture and Agri-Food Canada, St. John’s, Newfoundland and Labrador, Canada.
DNA extraction
A modified DNA extraction method was used based on Kim et al. (1997). Young leaves were sampled from plants maintained in the greenhouse. Two hundred mg of leaf samples were transferred to the vials containing 300 μL lysis buffer [100 mM EDTA (pH 8), 200 mM Tris-HCl (pH 8), 1 M NaCl and 2% PVP-40; Sigma Chemical Co, Oakville, ON, Canada], and two big (6.35 mm) and few small beads (1.6–2.0 mm) (Glemnills Inc., Clifton, NJ, USA). Homogenization was done with FastPrep®-24, MP Biomedicals, and homogenizer was allowed to run for 3 times at the speed of 5400 rpm for 45 s with the 1-min interval in between. Then, 18 μL of 1 M Dithiothreitol (DTT) (Sigma-Aldrich, St. Louis, MO, USA) and 100 μL of 20% Sodium dodecyl sulphate (SDS) (Sigma-Aldrich) were added and the vials were kept at 65 °C for 60 min followed by centrifugation at 5000 rpm for 1 min. Five hundred μL of 7.5 M ammonium acetate (Sigma-Aldrich) solution was added to the lysates in new vials to precipitate the proteins, and vials were kept on ice for 60 min and then centrifuged at 9000 rpm for 9 min at 4 °C. Supernatants were transferred to new vials and equal volume of 99% isopropanol was added to precipitate the DNA. DNA pellets were dissolved in 100 μL of TE buffer and stored at 4 °C. DNA concentration and purity was measured using JENWAY GenovaNano spectrophotometer (Bibby Scientific Ltd., Stone, Staffordshire, UK).
Gel electrophoresis
Eleven EST-PCR (CA16, CA54, CA175, CA287, CA1029, CA1105, CA1423, CA1590, CA1785, NA27 and NA1068; Bel et al. 2008) and nine microsatellite (seven EST-SSR: CA236, CA421, CA483, NA741, NA800, NA961 and NA1040, and two genomic SSR: VCC_I2 and VCC_S10; Boches et al. 2005) primer pairs that worked well with the present material were used for the study. The primers were procured form Integrated DNA Technologies, Inc. (IDT) (Coralville, Iowa, USA). PCR reactions were performed using TopTaq DNA polymerase kit from QIAGEN (Qiagen, 181 Bay Street, Suite 4400, Toronto, ON, Canada). For PCR, the reaction mixture volume of 25 μL was used (Debnath 2014), which contained 2.5 μL of TopTaq 10× PCR buffer [Qiagen TopTaq contains Tris.Cl, KCl, (NH4)2SO4, 1.5 mM MgCl2, stabilizers; pH 8.7 at 20 °C], 0.2 μM of dNTP mix (Sigma Chemical Co.), 200 μM of each forward and reverse primer, 25 ng of DNA, 0.7 units of TopTaq DNA polymerase (Sigma-Aldrich) and PCR grade distilled water (dH2O) (Sigma-Aldrich). Reaction mixtures were amplified in PTC-100 Programmable Thermal Controller (MJ Research Inc., Watertown, MA, USA) using an initial “hot start” of 94 °C for 10 min denaturation step, followed by 40 cycles of 40 s denaturation step at 94 °C, 70 s annealing step at the approximate annealing temperature (ranged from 44 and 62 °C), and 120 s extension step at 72 °C. The reaction was terminated with the final extension of 10 min at 72 °C before holding the samples at 4 °C for analysis. Amplified fragments along with low ranger (100 bp) and mid ranger (1 kb) DNA Ladders (Norgen Biotek, Thorold, ON, Canada), were separated using electrophoresis on 1.4% agarose gel. The gels were stained with the GelRed (Biotium, 3159 Corporate Place Hayward, CA, USA) dye solution following the manufacturer instruction. Images were captured using UV gel imaging system INGENIUS-3 by Syngene (Beacon House, Cambridge, UK) by using the settings for GelRed dye (Biotium, Inc., Hayward, CA, USA). The sizes of the fragments were calculated by GeneTools software (Syngene) based on comparison with the standard size marker. The test with each marker pair was done three times for each sample. The only loci amplified in each instance (i.e. all three trials) with reproducibility were scored and included in the analysis. Loci and the gels showing monomorphic band pattern were not included in the analysis. Bands of similar molecular weight and migration distance across individuals were presumed to be homologous (Adams and Rieseberg 1998; Debnath 2014).
Data analysis
Due to the polyploid nature of the blueberries, co-dominant scoring for EST-PCR and SSR markers was complicated for the heterozygous samples (Esselink et al. 2003). In polyploid species, multiple genomes can amplify the same product, which makes it difficult to differentiate the genotypes based on the intensity of the band. Therefore, it is advisable to score polyploidy banding pattern as presence (1) or absence (0) of the band in a particular genotype (Esselink et al. 2003; Horvath et al. 2011). Presence (1) or absence (0) of each allele which was treated as a separate locus and the matrix was created for EST-PCR and SSR markers, since wild lowbush blueberry clones and blueberry cultivars are tetraploid species (2n = 4x = 48), and because homozygous and heterozygous loci could not be differentiated. Genotypic diversity was calculated based on allele phenotype where alleles at individual loci were scored for presence or absence (Esselink et al. 2004; Bian et al. 2014). PowerMarker v3.25 (Liu and Muse 2005) software was used to calculate the diversity measurements such as the number of alleles per locus (NA), expected heterozygosity (HE), observed heterozygosity (HO) and polymorphic information content (PIC). For each locus, genetic diversity was computed using Shannon’s index (I) (Shannon and Weaver 1949): \( I = \sum Pi log_{2} Pi \), where, Pi is the frequency of the presence or absence of the ith allele. The bootstrap option of PowerMarker was used to create 1000 dendrograms using neighbor joining (NJ). These 1000 dendrograms were used to create a strict consensus tree, which gives an idea about common pattern. In these analyses, dendrograms were rooted at midpoint. The binary data was also subjected to principle coordinate (PCo) analysis to partition the variance using GenAlEx 6.501 (Peakall and Smouse 2012) and the first two components were plotted into two-dimensional plots. PCo and NJ analyses were done using marker data of EST-PCR, SSR and combined data for both marker types.
Structure analysis
In order to infer the population structure of the entire set of genotypes without considering the pre-existing subspecies classification or geographical information, the model-based program, STRUCTURE version 2.3.4 (Pritchard et al. 2000) was used. The software provides Bayesian approach to infer population structure by using marker datasets to identify the number of clusters (K) to which the program then assigns each individual genotype (Pritchard et al. 2000). For STRUCTURE analysis, all loci were assumed to be independent and each K population is assumed to follow Hardy–Weinberg equilibrium. The runs were completed with 100,000 burns in iterations and 100,000 subsequent Monte Carlo Markov Chain (MCMC) runs with K ranging from 1 to 9. To check the reliability of the results between the runs with the same K, five replicates were run for each assumed K value. The results obtained from the STUCTURE software package were evaluated in a website based STRUCTURE HARVESTER program (http://taylor0.biology.ucla.edu/structureHarvester/#) (Earl and Vonholdt 2012). Optimal value of K was identified using two methods, one as described by Pritchard (Pritchard et al. 2000) using L(K) and the other developed to determine delta K (∆K) (Evanno et al. 2005). In case of first method, when posterior probabilities of K is approaching to plateaus (or continues increasing slightly) and has high variance between runs (Rosenberg et al. 2001) then that K is considered true value of K. In second method, data calculated based on the second order rate of change of the likelihood the ∆K (Evanno et al. 2005) shows a clear peak at the true value of K in graph.
Analysis of molecular variance (AMOVA)
Arlequin version 3.5.1.2 (Excoffier et al. 2005) was used to calculate AMOVA among and within populations and subpopulations. Pairwise difference method was used to calculate the difference with 1000 permutations. The p-value was set at 0.05. AMOVA analysis was performed among five groups based on geographic locations/place of origin of the genotypes; where group 1 included wild clones from Newfoundland and Labrador, group 2 from Prince Edward Island and group 3 from Quebec. Group 4 presented two New Brunswick (NB) clones, two lowbush cultivars ‘Fundy’ and ‘Brunswick’ and a selection from ‘Fundy’ open pollinated seedlings. All these genotypes originated from NB. Group 5 presented all half-high and highbush blueberry cultivars. Pairwise difference method was used to calculate the genetic distance and 1000 permutations were performed. The p-value was set at 0.05 significance level.
Results
Diversity parameters
For 11 EST-PCR primers, the total number of alleles (NA) was 249, with an average of 23, ranging from 4 for primer pairs CA1029, CA1590 and NA27 to 52 for CA16 (Table 2). The expected heterozygosity (HE) ranged from 0.511 for NA27 to 0.977 for CA1785, while observed heterozygosity (Ho) ranged from 0.171 for CA1029 to 0.536 for CA175. The inbreeding coefficient (F) value for the EST-PCR primers ranged from 0.329 for CA175 to 0.774 for CA16, with the average of F = 0.599. While Shannon index (I), ranged from 0.879 for NA27 to 4.099 for CA16 and NA1068, with the average 2.619; the polymorphic information content (PIC) varied from 0.441 for NA27 to 0.976 for CA16 and CA1785, with the average PIC value of 0.801.
Microsatellites produced 164 alleles. The allele number was lowest for CA236 and highest for NA1040 followed by VCC_10 (Table 3). The average allele number was 18. The primer pair NA1040 also exhibited highest values for HE, Ho, I and PIC while CA236 was the poorest for HE, F, I and PIC values. The Ho values ranged from 0.144 for CA483 to 0.444 for NA1040 and the F values, from 0.076 to 0.839 for CA438 with an average of 0.549. While the I values ranged from 0.580 to 3.972 with the average of 2.03, PIC values which are essential for detecting primer’s polymorphism, ranged from 0.282 to 0.977 with the average 0.765 (Table 3).
NJ analysis
In the NJ analysis for EST-PCR, there were five major clusters, three of which (clusers I, II and V) were comprised of NL clones only. Although the biggest cluster (cluster IV) contained blueberry clones from all other provinces and included lowbush blueberry cultivars and a selection (FO), there were four distinct subclasses where most of the genotypes collected or originated from each province were well separated (supplementary Fig. 1). The SSR markers also divided the blueberry genotypes into five groups but the grouping was not exactly the same as was observed with EST-PCR markers. In SSR analysis, clusters I was divided into two subclusters where the first subcluster conatined NB clones (NB1 and NB2), lowbush blueberry cultivars FU and BR and the selection FO. The same grouping of these genotypes was also observed in EST-PCR analysis where they were in one sub-sub-cluster of cluster IVb. The second SSR-derived subcluster of cluster I contained six NL clones. Cluster II contained five PE and 12 NL clones and cluster III had 15 NL clones. Cluster IV was divided into two subclusters, one with highbush cultivars PO, PT, CH and SC; and the other with six QC clones. Cluster V contained six PE clones (supplementary Fig. 2).
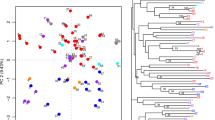
Combined analysis for both EST-PCR and microsatellite markers divided 63 genotypes into five distinct clusters in NJ dendrogram (Fig. 1). Cluster I contained five NL clones, NL23, NL27, NL28, NL32 and NL33 and were divided into two subclusters. Cluster II was divided into two subclusters, IIa and IIb where IIa contained all PE clones and IIb contained all QC clones. Cluster III contained 17 (NL7, NL9, NL12, NL15, NL16, NL18–20, NL22, NL24, NL29–31, NL34–37) NL clones and cluster IV had 15 NL clones (NL1–6, NL8, NL10, NL11, NL13, NL14, NL17, NL21, NL25 and NL26). Cluster V was divided into two subclusters Va and Vb. Subcluster Va contained all the genotypes related to NB province, which included two NB clones NB1 and NB2; two lowbush blueberry cultivas FU and BR and one selection FO. Subcluster Vb contained one highbush (PO) and three half-high blueberry cultivars (PT, CH, SC) (Fig. 1).

Neighbor joining (NJ) dendrogram of 63 blueberry genotypes based on the proportion of shared allele distance for EST-PCR and SSR markers. Numbers refer to branch lengths
PCo analysis
In PCo analysis with EST-PCR markers, five major clusters were formed in which the genotypes were distributed according to their geographic collection sites. The first three axes together represented 34% of total genetic variation (axis 1 = 15%, axis 2 = 10% and axis 3 = 9%). Cluster I contained highbush blueberry cultivar PO and half-high blueberry cultivars PT, CH and SC. The lowbush cultivars FU and BR, clones NB1 and NB2 and the selection FO were grouped in cluster II while cluster III contained all PE clones; cluster IV, all QC clones and Cluster V contained all NL clones (supplementary Fig. 3).
The PCo analysis with SSR markers also produced five major clusters and distributed the genotypes according to their geographic collection sites, but combined QC and PE clones in one cluster (cluster V) and divided NL clones into two separate groups (clusters III and IV). Two NL clones (NL2 and NL6) were not included in any cluster. Cluster I contained the half-high and highbush blueberry cultivars PO, PT, CH and SC and cluster II, NB1, NB2, FU, BR and FO (supplementary Fig. 4). The first three axes together represent 36% of total genetic variation (axis 1 = 14%, axis 2 = 12% and axis 3 = 10%).
Combined PCo analysis revealed that the first three axes together represent 29% of total genetic variation (axis 1 = 12%, axis 2 = 9% and axis 3 = 8%). Five major clusters were created where the genotypes were distributed according to their geographic collection sites. Cluster I contained four blueberry cultivars (PO, PT, CH and SC). Cluster II included two NB clones (NB1 and NB2), two lowbush blueberry cultivars (BR and FU) and the selection FO. These five genotypes originated from NB province. While cluster III contained all PE clones, cluster IV had all 37 NL clones. Cluster V contained had six QC clones (QC1, QC2, QC4, QC5, QC7 and QC9) (Fig. 2).

Principle co-ordinate analysis of 63 blueberry genotypes using EST-PCR and microsatellite markers
Structure analysis
In individual and combined analyses, plots of probability of data (Ln) for K and ∆K determined that the genotypes were divided into six (K = 6) clusters (Pritchard et al. 2000; Evanno et al. 2005). However, in ∆K graph, there were two peaks, one at K = 2 and the other at K = 6 (data not shown). It can be considered that K = 2 shows broader structure and K = 6 finer. Although ΔK helps in identifying the correct number of clusters in most situations, it should not be used exclusively and should be used together with the other information provided by structure, such as Ln (K) itself (Evanno et al. 2005). In addition, when bar plot was created using K = 6, it showed clustering of genotypes according to their geographic collection site as seen in Fig. 3. Thus, clustering at K = 6 is also considered for this data even though it did not give highest peak in ΔK graph, but Pritchard et al. (2000) gives rationale to select K = 6 for Ln (K) graph.

Q-plot showing Bayesian clustering of 63 genotypes, for K = 6, based on analysis of combined genotypic data of EST-PCR and microsatellite using STRUCTURE software. Each genotype is represented by a vertical bar. The colored subsections within each vertical bar indicate membership coefficient (Q) of the accession to different clusters. Identified clusters are I, II, III, IV, V and VI. (Color figure online)
In EST-PCR marker data, cluster I was comprised of six QC clones and NL8. NL8 showed high admixture of ~40% from cluster VI. Cluster II contained half-high and highbush blueberry cultivars PO, PT, CH and SC and Cluster III had 11 PE clones and NL 24. NL 24 had around 40% admixture from cluster IV. While cluster IV contained 16 NL clones, cluster V had two NB wild clones, two lowbush blueberry cultivars (FU and BR), selection FO and NL28. NL28 contained ~40–45% admixture from multiple clusters. Cluster VI included 18 NL clones: NL1–6, NL10, NL26, NL27 and NL29–37 (supplementary Fig. 5).
In microsatellite marker analysis, cluster I contained 14 NL clones and cluster II included all 11 PE clones. While cluster III was composed of six QC clones, cluster IV contained two NB clones, two lowbush blueberry cultivars and the selection FO. Cluster V included the half-high and highbush blueberry cultivars PO, PT, CH and SC. Cluster VI had 23 NL clones (supplementary Fig. 6).
In combined analysis, STRUCTURE plot divided 63 genotypes into clusters that resembled the genotypic distribution and grouping seen in NJ and PCO analyses. Cluster I contained 23 NL clones many of which showed admixture of ~5–70% from other STRUCTURE groups (Fig. 3). Cluster II was comprised of 11 PE clones and cluster III had blueberry cultivars PO, PT, CH and SC. Cluster IV included 14 NL clones, some of which showed admixture of ~18–70%. Cluster V was comprised of six QC clones and cluster VI contained two NB wild clones (NB1 and NB2), two lowbush blueberry cultivars (FU and BR) and the selection FO (Fig. 3).
AMOVA
The results for AMOVA analysis with eleven EST-PCR markers found the variation of 37% among the groups, 17% among the communities within the groups and 46% among genotypes within communities (Table 4). These values were 27% among the groups, 32% among the communities within the groups and 41% among genotypes within communities with nine SSR markers. Analysis with the combined data of EST-PCR and microsatellite markers indicated that the variation among the groups was 33% was, among the communities within the groups, 23% and among genotypes within the communities was 44% (Table 4).
Discussion
Although EST-PCR (Bell et al. 2008; Rowland et al. 2010; Debnath 2014), EST-SSR (Boches et al. 2005; Debnath 2014) and genomic SSRs (Boches et al. 2006) have been used in distinguishing blueberry, report on a wide range of wild lowbush blueberry germplasm of diverse origin is scarce. EST-PCR, EST-SSR and genomic SSR markers are co-dominant, multi-allelic and reproducible, but are expensive and laborious to develop. Primers developed for highbush blueberry cultivar Bluecorp (Rowland et al. 2003) were utilized in the present study. This mitigates the cost of cDNA preparation.
EST-PCR and SSR primers showed different discriminating capacities in genetic diversity analysis. The capacity of a marker’s potential in detecting genetic polymorphism and variation was shown with the parameters such as allele number (NA), polymorphic information content (PIC), expected (HE) and observed heterozygosity (HO), inbreeding coefficient (F) and Shannon’s index (I). Generally, the higher the value of any parameter for a marker system, the greater its capacity in detecting polymorphism and variation between genotypes. The higher value also suggests that the marker is more informative (Ojango et al. 2011).
In our study, we found that EST-PCR analyses yielded a high mean allele number of 23 per locus (primer pair) while SSR produced 18 in 63 blueberry genotypes. These results are comparable with those of Boches et al. (2006) but exhibit higher values than those of Bian et al. (2014) and Liu et al. (2014) in Vaccinium spp. The availability of more alleles per locus in the present study indicates that the wild lowbush blueberries are more diverse and are expected to contribute more in developing new cultivars. In this study, the average PIC value was higher for EST-PCR markers (0.80) than for SSR markers (0.77). In our study, we did not find much difference between the mean PIC values of EST-PCR and microsatellite markers, which suggests that both markers are highly polymorphic and able to find genetic diversity among blueberry genotypes. Marker heterozygosity that gives an idea on marker’s ability to provide heterozygous information, was very high for expected heterozygosity indicating that the current blueberry genotypes are very heterozygous in nature. HE (0.82) and HO (0.34) values for EST-PCR were higher than those of SSR values (HE = 0.78; HO = 0.32) indicating that EST-PCR markers are more powerful in showing heterozygosity among genotypes. EST-PCR markers were more powerful than those of SSRs in showing inbreeding behavior among genotypes. As blueberries grow in wild, there are greater chances of inbreeding. In contrast to other parameters such as expected heterozygosity, Shannon’s index does not require knowledge of allele frequencies. Therefore, it is an accurate measure of diversity in polyploidy species where allele frequencies cannot be determined with certainty due to the difficulty in distinguishing the copy number of individual alleles (Boches et al. 2006). In present studies, the average Shannon’s index of 2.61 for 11 EST-PCR, and 2.03 for nine SSR markers, were higher than those reported by Debnath (2014), and Bian et al. (2014) in blueberries confirming that the present material consists of more diverse genotypes than those reported earlier.
Patterns of clustering based on NJ and PCo analyses were similar for most of the genotypes. A combined analysis of EST-PCR and microsatellite markers revealed five clusters for the NJ dendrogram. PCo analysis formed five groups in combination of both markers and separated half-high and highbush blueberry cultivars from wild clones. This kind of association and grouping was also seen in NJ dendrograms. Model based Bayesian cluster analysis divided NL clones into two separate clusters although NL clones were seen together in one cluster by Debnath (2014). This can be due to the fact that more number of genotype and markers were included in the current study.
As it is seen from the graphs of STRUCTURE, NJ and PCo analyses, few clones classified by the STRUCTURE analysis, fall into different groups of NJ or PCo analyses. In STRUCTURE software, the loci within a population are assumed to be in Hardy Weinberg equilibrium (HWE) and linkage disequilibrium although non-random mating, random genetic drift, mutations, gene flow, selection and meiotic drive may also disturbed HWE (Hardy 1908; Debnath 2014). This might be the reason for the same. In all cluster analyses, it was largely found that separate clusters were made according to the respective geographic origin of the genotypes.
The AMOVA analysis detected abundant variation among genotypes within communities, among communities with groups (provinces) and among groups (provinces). These results on variation among genotypes within communities were confirmed with the previous studies (Debnath 2009, 2014). In present study, high level of variation was also observed among communities within groups and among groups, which was less than reported by Debnath (2014). This can be explained by existence of diverse gene pool from province to province.
The results indicate that the current blueberry wild clones and cultivars are very diverse and variations among genotypes can be also observed among genotypes collected from the same communities. There was no report on variability study with EST-PCR and SSR markers with so many genotypes as used in this study. Eleven EST-PCR, seven EST-SSR and two genomic SSR markers were helpful and sufficient to differentiate 63 blueberry genotypes. These analyses will be helpful in DNA fingerprinting, for selecting useful clones as parents in a breeding program, for management of blueberry germplasm and for conservation of intellectual property rights.
References
Aalders LE, Hall IV, Jackson LP (1977) Brunswick lowbush blueberry. Can J Plant Sci 57:301
Adams RP, Rieseberg LH (1998) The effects of non-homology in RAPD bands on similarity and multivariate statistical ordination in Brassica and Helianthus. Theor Appl Genet 97:323–326
Bell DJ, Rowland LJ, Polashock JJ, Drummond FA (2008) Suitability of EST-PCR markers developed in highbush blueberry for genetic fingerprinting and relationship studies in lowbush blueberry and related species. J Am Soc Hortic Sci 133:701–707
Bian Y, Ballington J, Raja A, Brouwer C, Reid R, Burke M, Wang X, Rowland LJ, Bassil N, Brown A (2014) Patterns of simple sequence repeats in cultivated blueberries (Vaccinium section Cyanococcus spp.) and their use in revealing genetic diversity and population structure. Mol Breed 34:675–689
Boches PS, Bassil NV, Rowland LJ (2005) Microsatellite markers for Vaccinium from EST and genomic libraries. Mol Ecol Notes 5:657–660
Boches P, Bassil NV, Rowland L (2006) Genetic diversity in the highbush blueberry evaluated with microsatellite markers. J Am Soc Hortic Sci 131:674–686
Chapman MA, Hvala J, Strever J, Matvienko M, Kozik A, Michelmore RW, Tang S, Knapp SJ, Burke JM (2009) Development, polymorphism, and cross-taxon utility of EST-SSR markers from safflower (Carthamus tinctorius L.). Theor Appl Genet 120:85–91
Debnath SC (2009) Development of ISSR markers for genetic diversity studies in Vaccinium angustifolium. Nord J Bot 27:141–148
Debnath SC (2014) Structured diversity using EST-PCR and EST-SSR markers in a set of wild blueberry clones and cultivars. Biochem Syst Ecol 54:337–347
Earl DA, Vonholdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Esselink GD, Smulders MJM, Vosman B (2003) Identification of cut rose (Rosa hybrida) and rootstock varieties using robust sequence tagged microsatellite site markers. Theor Appl Genet 106:277–286
Esselink GD, Nybom H, Vosman B (2004) Assignment of allelic configuration in polyploids using the MAC-PR (microsatellite DNA allele counting-peak ratios) method. Theor Appl Genet 109:402–408
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 1:47–50
Finn C, Luny J, Wildung D (1990) Half-high blueberry cultivars. Fruit Var J 44:63–68
Galletta GJ, Ballington JR (1996) Blueberries, cranberries and lingonberries. In: Janick J, Moore JN (eds) Fruit breeding, volume II: vine and small fruit crops. Wiley, Prentice Hall, New York, pp 1–107
Halliwell B (2007) Dietary polyphenols: good, bad, or indifferent for your health? Cardiovasc Res 73:341–347
Hardy GH (1908) Mendelian proportions in a mixed population. Science 28:49–50
Hepler P, Draper A (1976) Patriot blueberry. HortScience 11:272
Horvath A, Sanchez-Sevilla JF, Punelli F, Richard L, Sesmero-Carrasco R, Leone A, Hoefer M, Chartier P, Balsemin E, Barreneche T, Denoyes B (2011) Structured diversity in octoploid strawberry cultivars: importance of the old European germplasm. Ann Appl Biol 159:358–371
Joseph JA, Denisova NA, Arendash G, Gordon M, Diamond D, Shukitt-Hale B, Morgan D (2003) Blueberry supplementation enhances signaling and prevents behavioral deficits in an Alzheimer disease model. Nutr Neurosci 6:153–162
Kim CS, Lee CH, Shin JS, Chung YS, Hyung NI (1997) A simple and rapid method for isolation of high quality genomic DNA from fruit trees and conifers using PVP. Nucl Acids Res 25:1085–1086
Liu KJ, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Liu Y, Liu S, Liu D, Wei Y, Liu C, Yang Y, Tao C, Liu W (2014) Exploiting EST databases for the development and characterization of EST-SSR markers in blueberry (Vaccinium) and their cross-species transferability in Vaccinium spp. Sci Hort 176:319–329
Lyrene P (2002) Blueberry. HortScience 37:252
Ojango JM, Malmfors B, Okeyo AM (2011) Module 4: quantitative methods to improve the understanding and utilisation of animal genetic resources. [Animal genetic training resources (AGTR). International Livestock Research Institute (ILRI)]. http://agtr.ilri.cgiar.org/index.php?option=com_content&view=article&id=28&Itemid=268
Okie W (1997) Register of new fruit and nut varieties. Brooks and Olmo list 38. HortScience 32:785–805
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rosenberg NA, Burke T, Elo K, Feldmann MW, Freidlin PJ, Groenen MAM, Hillel J, Maki-Tanila A, Tixier-Boichard M, Vignal A, Wimmers K, Weigend S (2001) Empirical evaluation of genetic clustering methods using multilocus genotypes from 20 chicken breeds. Genetics 159:699–713
Rowland LJ, Mehra S, Dhanaraj AL, Ogden EL, Slovin JP, Ehlenfeldt MK (2003) Development of EST-PCR markers for DNA fingerprinting and genetic relationship studies in blueberry (Vaccinium, section Cyanococcus). J Am Soc Hortic Sci 128:682–690
Rowland LJ, Ogden EL, Ehlenfeldt MK (2010) EST-PCR markers developed for highbush blueberry are also useful for genetic fingerprinting and relationship studies in rabbiteye blueberry. Sci Hortic 125:779–784
Shannon CE, Weaver W (1949) The mathematical theory of communication. University of Illinois Press, Urbana
Templeton AR (1996) Translocation as conservation tool. In: Szaro RC, Johnston DW (eds) Biodiversity in managed landscapes: theory and practice. Oxford University Press, Oxford, pp 315–325
Vander Kloet SP (1988) The genus Vaccinium in North America. Agriculture Canada Research Branch, Ottawa
Vander Kloet SP, Dickinson TA (2009) A subgeneric classification of the genus Vaccinium and the metamorphosis of V. section Bracteata Nakai: more terrestrial and less epiphyticin habit, more continental and less insular in distribution. J Plant Res 122:253–268
Willis L, Bickford P, Zaman V, Moore A, Granholm AC (2005) Blueberry extract enhances survival of intraocular hippocampal transplants. Cell Transpl 14:213–223
Acknowledgements
The authors like to thank Darryl Martin, Glenn Chubbs, Sarah Leonard, Juran Goyali and Amrita Ghosh for their excellent technical help.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Neighbor joining (NJ) dendrogram of 63 blueberry genotypes based on the proportion of shared alleles distance for EST-PCR markers. Numbers refer to branch lengths (TIFF 379 kb)
Supplementary Fig 2
Neighbor joining (NJ) dendrogram of 63 blueberry genotypes based on the proportion of shared allele distance for microsatellite markers. Numbers refer to branch lengths (TIFF 404 kb)
Supplementary Fig. 3
Principle co-ordinate analysis of 63 blueberry genotypes using EST-PCR marker (TIFF 140 kb)
Supplementary Fig. 4
Principle co-ordinate analysis of 63 blueberry genotypes using microsatellite marker (TIFF 148 kb)
Supplementary Fig. 5
Q-plot showing Bayesian clustering of 63 genotypes, for K=6, based on analysis of EST-PCR using STRUCTURE software. Each genotype is represented by a vertical bar. The colored subsections within each vertical bar indicate membership coefficient (Q) of the accession to different clusters. Identified clusters are I, II, III, IV, V and VI (TIFF 409 kb)
Supplementary Fig. 6
Q-plot showing Bayesian clustering of 63 genotypes, for K=6, based on analysis of microsatellite using STRUCTURE software. Each genotype is represented by a vertical bar. The colored subsections within each vertical bar indicate membership coefficient (Q) of the accession to different clusters. Identified clusters are I, II, III, IV, V and VI (TIFF 325 kb)
Rights and permissions
About this article

Cite this article
Tailor, S., Bykova, N.V., Igamberdiev, A.U. et al. Structural pattern and genetic diversity in blueberry (Vaccinium) clones and cultivars using EST-PCR and microsatellite markers. Genet Resour Crop Evol 64, 2071–2082 (2017). https://doi.org/10.1007/s10722-017-0497-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-017-0497-1