
Abstract
Galectin-3 is associated with the development and malignancy of several types of tumor, mediating important tumor-related functions, such as tumorigenesis, neoplastic transformation, tumor cell survival, angiogenesis, tumor metastasis and regulation of apoptosis. Therefore, synthetic galectin-3 inhibitors are of utmost importance for development of new antitumor therapeutic strategies. In this review we present an updated selection of synthetic glycoconjugates inhibitors of tumor-related galectin-3, properly addressed as monosaccharide- and disaccharide-based inhibitors, and multivalent-based inhibitors, disclosuring relevant methods for their synthesis along with their inhibitory activities towards galectin-3. In general, Cu(I)-assisted 1,3-dipolar azide-alkyne cycloaddition (CuAAC) reactions were predominantly applied for the synthesis of the described inhibitors, which had their inhibitory activities against galectin-3 evaluated by fluorescence polarization, surface plasmon resonance (SPR), hemagglutination, ELISA and cell imaging assays. Overall, the presented synthetic glycoconjugates represent frontline galectin-3 inhibitors, finding important biomedical applications in cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Carbohydrate-protein interactions have been in evidence since they are involved in many biochemical and biological processes, such as downstream cell signaling, cell-cell and matrix- extracellular interactions, cell growth regulation, apoptosis and cancer metastasis [1–3]. During tumor development the alteration in cell survival and growth, as well as cell migration and antitumor immunity can be correlated with a diversity of glycosylation mechanisms on cell surface, leading to wide complex structures to be decoded by glycan-binding proteins (lectins) [3–6]. Thus, for deep understanding of what factors take place during the initial and late stages of neoplastic diseases it is crucial to consider the identification and expression of carbohydrate-binding proteins, such as galectins, as important targets for development of antitumor drugs [7–9]. Members of galectins (around 15), which have been isolated and sequenced in a wide variety of tissues from different species [9], are defined based on their affinity for β-galactosides-containing saccharides and amino acid sequence similarity in their CRDs (Carbohydrate Recognition Domain). Galectin-3, for instance, is the only chimeric galectin found in vertebrates that has a C-terminal CRD, which binds to β-galactosides, and the N-terminal domain that contains a serine phosphorylation site, which is critical for galectin-3 cell signaling, multivalency and cross-linking activity [8, 9].
Galectin-3 has been largely implicated in tumor development, being involved in cell proliferation, apoptosis, cell adhesion, invasion, angiogenesis and metastasis [8, 11, 12]. Galectin-3 has differential expression and location dependent on the type of tumor, being highly expressed in colon, head and neck, gastric, endometrial, thyroid, liver, lung, bladder and breast cancer. Moreover, decreased expression of galectin-3 has been shown in colorectal, prostate, kidney and pituitary cancer, although increased levels in the cytoplasm were observed in colorectal, tongue, and prostate cancer, both correlated with disease progression and metastasis [8, 12, 13]. Cytoplasmic galectin-3 presents anti-apoptotic function related with several but not completely understood mechanisms, such as galectin-3 phosphorylation promoting its nuclear exportation into the cytoplasm and possible interaction with the B-cell lymphoma 2 (Bcl-2) protein resulting in inhibition of cytochrome C release. Although the mechanism is not well known, some relevant papers have described that galectin-3 has an Asp-Trp-Gly-Arg (NWGR) motif in the C-terminal domain that shows similarity with Bcl-2 and therefore seems to participate in interaction between both galectin-3 and this apoptosis regulator. The consequence of this inhibitory effect is the impairment of apoptosis from this pathway [8, 12–14].
Indeed, galectin-3 anti-apoptosis role is assigned by the tumor suppressor p53, which suppresses the transcription of galectin-3, leading to p53 induced apoptosis. Thus, this galectin can be considered a target for apoptosis and downstream events via p53 [15]. In contrast, pro-apoptotic function has been associated with the nucleus and with extracellular activity in activated T cells and in B-cell lymphoma [8, 12–14] (Fig. 1).

Modulation of tumor development by Galectin-3 (Gal-3). Gal-3 participates in cell-cycle progression by promoting downregulation of A and E Cyclins a; upregulation of inhibitors p21 (WAF 1) and p27 (KIP1) a; and also induction of D1 after β-catenin binding b. An extracelullar stimulus favors Gal-3 tumor growth through oncogenic Ras (KRas) interaction with subsequently phosphatidylinositol-3-kinase (PIK-3) signal transduction b. Gal-3 antiapoptotic function is shown by Bcl2 binding that can stabilize the mitochondrial membrane potential inhibiting apoptosis b. Otherwise, Gal-3 transcription can be suppressed by apoptotic molecule p53 b. Gal-3 modulates angiogenesis d and indirectly metastasis c followed by up-regulation of Integrin and Adhesion Molecules expression and also, endothelial cells migration with increased pro-inflammatory cytokines production
In addition, galectin-3 may mediate cell transformations and growth by interacting with oncogenic Ras (KRas), promoting Ras signal transduction through subsequently phosphatidylinositol-3-kinase (PIK-3) activation, and also by controlling cell cycle process upon regulation of cyclins, such as downregulation of A, E; upregulation of the cell-cycle inhibitors p21 (WAF 1) and p27 (KIP1); and induction of D1 followed by β-catenin binding [8, 12]. Lastly, galectin-3 promotes angiogenesis and indirectly metastasis by inducing endothelial cells migration through increased pro-inflammatory cytokines production and, therefore, up-regulation of molecules, such as Integrins, E-selectin, Intercellular Adhesion Molecule 1 (ICAM-1) and Vascular Cell Adhesion Molecule 1 (VCAM-1) [16] (Fig. 1). Thus, considering all these tumor-promoting effects of galectin-3, the development of synthetic galectin-3 inhibitors represents a frontline strategy in the search for new antitumor drugs and accurate cancer diagnosis.
CRDs of galectins contain approximately 135 amino acid (aa) residues, which directly lead to the specificity of galectins for saccharides and are composed by subsites (A-E) which, along their 135 aa, form a groove able to bind up to a tetrasaccharide [17–19].Galactose binds to the most conserved subsite C, whereas the second more important subsite D is occupied by another pyranoside represented by (1 → 4)Glc/GlcNAc or (1 → 3) GlcNAc/GalNAc linked to galactose (subsite C) [17, 19, 20]. Interestingly, the preferential interaction of these subsites of each galectin CRD with different carbohydrates illustrate the diversity in their binding specificity and biological activities [17, 21]. The CRD of galectin-3 is comprised of eight conservative amino acids (Arg144, His158, Asn160, Arg162, Asn174, Trp181, Glu184 and Arg186) responsible for the lectin binding to carbohydrates. In general, the main interactions of galectin-3 to the natural disaccharide ligands Lac/LacNAc are represented by hydrogen bonds between the OH groups of Gal (C-4 and C-6) and Glc/GlcNAc (C-3) with His158, Asn160, Arg162, Glu184 and Asn174, and by Van der Waals contacts of Gal and Glc/GlcNAc residues with Trp181 and Arg186 [22]. Therefore, chemical modifications of natural ligands, such as at C-3 of Gal and at C-1 of Glc/GlcNAc can maintain the cited interactions and increase additional contacts, such as with Arg144 (subsite B) [22–25]. In the search for effective galectin-3 inhibitors, the synthesis of carbohydrate based 1,2,3-triazole analogs by Cu(I)-assisted 1,3-dipolar azide–alkyne cycloaddition (CuAAC) has been largely applied, considering the several advantages of the formed triazole ring, such as its property to act as a rigid link, its stability toward oxidation, reduction and hydrolysis, besides the fact that CuAAC reactions are, generally, easily executed, fast and highly selective [26, 27].
On these bases, this review encompasses an updated selection (2006–2016) of synthetic glycoconjugates inhibitors of tumor-related galectin-3, disclosuring relevant methods for their synthesis along with their inhibitory activities towards galectin-3. It’s noteworthy to mention that important published reviews on galectin-3 inhibitors rely mostly on crystallographic and biological aspects [12, 28–33], lacking deeper description on their synthesis.
Synthetic glycoconjugates as galectin-3 inhibitors
For the sake of clearer disposal and better comprehension, the selected synthetic galectin-3 inhibitors were properly divided in monosaccharide- and disaccharide-based inhibitors, and multivalent-based inhibitors, as follows in the next sections.
Monosaccharide-and disaccharide-based inhibitors
Taking into account the low affinities of naturally occurring carbohydrate ligands for galectins, besides their low physiological stabilities due to acid sensitive glycosidic bonds, and their high polarity, synthesis of anomeric and O-3 triazole analogs of galactosides has been developed to obtain higher affinity inhibitors of galectin-3. Thus, Cu(I)-assisted 1,3-dipolar azide-alkyne cycloaddition (CuAAC) reactions between galactosyl alkynes 1 and 2 with azides 3 or 4, using CuI/DIPEA as catalytic system in THF, provided the triazole-galactosyl derivatives 5–7 (Scheme 1) [34]. The best inhibitory activities in the qualitative hemagglutination inhibition assays were verified for compounds 1 and 5, which showed to be 40 times more active than galactose at 1.25 mM concentration (Table 1) [34].

Synthesis of triazoles 5–7 by CuAAC reaction
The distinct sugars galactose and mannose display common structural features in that the stereochemical relationship of the galactose axial O-4 and equatorial O-3 resembles the axial O-2 and equatorial O-1 of β-mannose. Thus, this similarity suggested the possibility to synthesize β-mannoside-based galectin-3 inhibitors, by exploring the easier access to C-1 in mannose as compared to C-3 in galactose. In this regard, a series of 1H-(1,2,3)-triazol-1-yl β-mannosides was synthesized by copper(I)-catalyzed 1,3-dipolar cycloaddition of azido 8 with methyl propiolate, followed by Zemplén transesterification (NaOMe/MeOH) and treatment with different amines, affording a panel of trizole amides (Scheme 2). The best affinities for galectin-3, as determined by fluorescence polarization assay, were verified for compounds 9 (Kd 1.4 mM) and 10 (Kd 1.5 mM) (Table 1), which, although active, did not represent optimal mimetics of 3-(triazol-1-yl)-galactosides for galectin-3 [35].

Synthesis of β-mannoside-based triazoles 9 and 10
The affinity enhancement for the different subsites (A-D) of galectins by combining structural fragments to galactoside or lactoside derivatives may represent a significant approach to get effective galectin-3 inhibitors [36, 37]. In this perspective, two galactosyl oximes having C3-triazole fragments were prepared by CuAAC reactions between the 3-azido-Gal-indol-carbaldoxime 11 and the corresponding alkynes methyl propiolate 12 and phenyl acetylene 13, in the presence of Cu wire in propanol (Scheme 3). The obtained compounds 14 and 15 showed promising affinities for galectin-3, with Kd values of 11 μM and 17 μM, respectively, in fluorescence polarization assays (Table 1). The improved affinity and selectivity of 14 and 15 for galectin-3 was further justified through their modeling into the binding sites of different galectins [19]. It was then observed that, apart from interacting with subsites B and C present in all galectins, these compounds were able to establish an extra strong interaction, through their indole aldoxime fragments, with the Arg144 residue present in a distinct cavity existent only in Gal-3 [19, 29].

Synthesis of galactosyl oximes 14 and 15 with subsite B-binding 1,2,3-triazoles at C3
Following the strategy of getting ligands able to interact with the different subsites (A-D) of galectins, a set of 3-triazol-galactosides, 3´-triazol-N-acetyllactosamine (LacNAc) and di-triazol-thiodigalactoside derivatives were synthesized, under Cu(I)-catalyzed reactions, as potential galectin-3 inhibitors [38]. Initially, a series of triazoles was obtained at the C3 position of galactose by click reaction (CuI/DIPEA in toluene, rt.-40 °C) of 3-azide-GalSMe 16 with different monosubstituted-aryl alkynes, as exemplified for triazole 17 (Scheme 4) Subsequently, considering that LacNAc is far superior to galactose as natural ligands for galectins, several LacNAc derivatives carrying 4-carbamoyl and 4-aryl triazoles at the galactose C3´ were obtained by click reactions of LacNAc C3´-azido 18 and corresponding alkynes, as shown for compound 19 (Scheme 4). Lastly, a panel of thiodigalactoside triazole amides was obtained by a multi-step synthetic route, starting with the preparation of the thiodigalactoside triazole ester 20 by cycloaddition reaction between the galacto-azide 21 and methyl propiolate 12. The formed triazole was then treated with HBr and to the resulting bromide 22 was added dried sodium sulfide, which led to its dimerisation. As the last step, reactions of 23 with a series of primary amines in methanol afforded the respective amides, as illustrated for the thiodigalactoside triazole butyl-amide 24 (Scheme 4). In general, the obtained compounds proved to be better inhibitors of galectin-3 if compared to other galectins subtypes (7, 8 N and 9 N), in competitive fluorescence polarization assays. The highest inhibitory activities toward galectin-3 were verified for compounds 19 (Kd 0.66 μM) and 24 (Kd 0.029 μM), related to LacNAc- and thiodigalactoside-triazoles series, respectively (Table 1) [38].

Synthesis of naphtyl-3-triazol-galactoside 17, naphtyl-3´-triazol-N-acetyllactosamine 19 and butylamine-di-triazol-thiodigalactoside 24 by Cu(I)-catalyzed reactions
Considering the potential of β-D-Galp-(1–3)-β-D-GlcpN (lacto-N-biose) disaccharide to interact with two essential galectin-3 subsites (C and D), a library of N-functionalized octyl-lacto-N-biose was synthesized aiming stronger binding to galectin-3 via interactions with its additional subsites [39]. The synthesis of this library was initiated by previous preparation of octyl-lacto-N-biose 25 by NIS/AgOTf-promoted glycosylation reaction between the suitably protected building blocks thioethyl galactoside 26 and octyl-N-Troc-glucosamine 27, followed by N-Troc deprotection (Zn/AcOH) and N-acylation with a range of commercially available acetyl chlorides and carboxylic anhydrides in pyridine (Sheme 5) [39]. After removal of the benzylidene and benzyl groups (Pd(OH)2/C), and O-deacetylation (NaOMe/MeOH), the obtained lacto-N-biose library was screened for galectin-3 binding by microscale frontal affinity chromatography coupled to mass spectrometry (FAC/MS), which is a technique that allows fast rank ligands and also gives access to their dissociation constants (Kd). It involves the preparation of a microscale tubing column by immobilizing a biotinylated galectin-3 onto streptavidin controlled-pore glass (CPG) beads, with subsequent elution of a library-containing solution. The affinities of compounds to galectin-3 are thus dictated by their order of elution, with the strongest-binding ligands eluting last. According to the screening performed by FAC/MS, best affinity to galectin-3 was verified for compound 28 (Kd 10.6 μM) (Table 1), which eluted much slower (retention time 11 min) than the sole lacto-N-biose (retention time 1.0 min and Kd 73.3 μM) [39] (Scheme 5).

Synthesis of NH derivatized octyl-lacto-N-biose 28
The sugar talose has emerged as an attractive scaffold for the design of novel galectin-3 inhibitors since the inverted C2 configuration, relative to galactose, offers possibilities for inserting affinity-enhancing talose O2 substituents able to stablish interactions with polar amino acids, not previously accessed by galactose-based galectin-3 inhibitors. Moreover, considering that talosides appear not to be naturally present in mammalians, and hence no endogenous talose-processing enzymes are present, they may represent desired hydrolytically stable inhibitors [40]. Therefore, a series of methyl 3-O-(4-methylbenzoyl)-β-D-talopyranoside O2 derivatives was synthesized and tested as galectin-3 inhibitors. Starting from the known benzylidene-protected methyl β-D-talopyranoside 29, subsequent synthetic steps involving acetylation, sulfation and PCl3-mediated reactions, followed by cleavage of benzylidene using aqueous acetic acid, afforded the inhibitors 30, 31 and 32 (Scheme 6). Evaluation of these talosides by fluorescence polarization assay showed corresponding dissociation constants of 0.55 mM, 0.25 mM and 0.60 mM for compounds 30, 31 and 32 (Table 1), reflecting higher galectin-3 inhibition if compared to methyl β-D-galactopyranoside (Kd 4.4 mM) [40].

Synthesis of O2 and O3-derivatized methyl β-D-talopyranosides 30–32
In continuous work to get β-D-talosides as galectins inhibitors, 3-amido-3-deoxy-talosides were synthesized via formation of idoside intermediates, followed by insertion of equatorial 3-NH2 through a substitution or oxidation/reductive amination sequence, and subsequent conversion to aromatic amides. Thus, a synthetic route starting from benzylidene-protected methyl β-D-galactopyranoside 33 afforded the 3-amino-3-deoxy-β-taloside 34, which was then acylated to form selected aromatic amides, such as compounds 35 and 36 (Scheme 7). In fluorescence polarization assays, these compounds showed to be active against galectin-3, presenting corresponding Kd values of 0.57 mM and 0.72 mM (Table 1), which are higher than methyl β-galactoside (4.4 mM) [41, 42].

Synthesis of 3-amido-D-talopyranosides 35 and 36 as galectin-3 inhibitors
The synthesis of anomeric and O3 modified analogs of natural monosaccharide (Gal) and disaccharides (Lac/LacNAc), especially those containing aromatic groups on the galactose C3 position, has been explored since they are able to establish favorable cation-π interactions with arginine residues, leading to galectin-3 inhibition [42–44]. Thus, aiming to favor these interactions and considering the potential of O-3 triazole-galactose analogs to interact with Gal-3 CRD, a series of 1,2,3-triazole amino acids-derived-3-O-galactosides, such as compounds 37–41, derived from corresponding phenylalanine, tryptophan, lysine, aspartic acid and tyrosine amino acids, was synthesized. The synthesis of protected compounds 42–46 was carried out by click 1,3-dipolar cycloaddition reactions between azido-functionalized amino acids 47–51 and 3-O-propynyl-GalOMe 52, in a microwave reactor utilizing the catalytic system CuSO4/sodium ascorbate and DMF as solvent, followed by hydrogenolysis (10 % Pd-C/H2) and deacetylation (NaOMe) reactions (Scheme 7). The obtained deprotected compounds 37–41 were then submitted to Surface Plasmon Resonance (SPR) assays for evaluation of their binding affinity to galectin-3. All tested 1,2,3-triazole amino acid-derived-3-O-galactosides 37–41 showed high binding affinity for galectin-3, with lower KD values being verified for compounds 37 (7.96 μM) and 39 (4.56 μM), comprising derived-phenylalanine and lysine side chains amino acids, respectively (Table 1) [10] (Scheme 8).

Synthesis of 1,2,3-triazole-linked glycoconjugates 37–41 by click chemistry
Apart from effective cation-π interactions with arginine residues, computer simulations have shown that additional anionic O2 substituents of inhibitors may provide favorable polar interactions around the arginine guanidinium hydrogens [45]. In this context, anionic O2 derivatives of methyl 3-deoxy-3-(4-methylbenzamido)-1-thio-β-D-galactopyranoside, such as compounds 53, 54 and 55 containing sulfate or phosphate, have been synthesized as inhibitors against galectin-3. Starting from the known galactoside 56, the synthesis of 53–55 involved the previous preparation of the key intermediate 57 (Scheme 9a), suitable for further derivatization of O2 position. Thus, sulfation of 57 using sulfur trioxide was straightforward to obtain 58, whereas treatment of 57 with phosphorus trichloride or pivaloyl chloride/benzyl alcohol led to the phosphonate-derived 59 and benzylphosphate 60, respectively (Scheme 9a). Finally, debenzylidenation of 58–60 afforded the final deprotected inhibitors 53–55, which showed corresponding Kd values of 87 μM, 150 μM and 120 μM against galectin-3, as measured by fluorescence polarization assay (Table 1) [45, 46].
In a different approach aiming to compare the effects of different aglycon moieties on galectin-3 binding, a library of aryl thiolactosides was prepared by phase transfer catalysis reaction (PTC) [47]. Starting from acetobromolactose 61, the phase transfer catalyzed nucleophilic displacement of bromide by aryl thiols (TBAHS), followed by oxidation of the formed sulfides (mCPBA) (Scheme 9b), afforded a library of anomeric sulfones, which were submitted to hemagglutination assays for evaluation of galectin-3 inhibition. Among the tested S-lactosides, best activities were verified for those containing aromatic aglycons, such as compound 62 that showed to be 3 times more active than lactose, with an inhibitory property of 0.31 mM (Table 1) [47]. It’s valuable to mention that the same compound showed an increased inhibitory activity against galectin-1 (40 μM), reflecting its distinct binding modes towards galectins-3 and −1, as further evaluated by electron density calculations [47].

a Synthesis of anionic O2 derivatives of methyl 3-deoxy-3-(4-methylbenzamido)-1-thio-β-D-galactopyranoside 53–55. b Synthesis of the aromatic S-lactoside 62
The effect of aglyconic modifications on galectin-3 inhibition was also investigated by means of the preparation of a library of triazolyl lactoside/ N-acetyllactosamine derivatives [48]. Firstly, the CuAAC reactions (CuSO4, ascorbate, THF/H2O) between the lactosyl azide 63 and disctinct alkynyl alcohols, propargyl aryl ethers and naphthyl propargyl ether afforded a panel of seven lactosides, subsequently O-deacetylated using NaOMe/ MeOH (Scheme 10). Regarding LacNAc derivatives, their synthesis started with the preparation of the propargyl β-D-LacNAc disaccharide 64 by a highly regioselective glycosylation between the prop-2-ynyl-6-O-protected GlcNAc 65 and galactosyl trichloroacetimidate 66 (BF3·Et2O, DCM), followed by CuAAC reactions with a family of triazoles, furnishing a series of eight triazolyl LacNAc derivatives, after removal of the TBDPS protecting group (TBAF) and de-O-acetylation (NaOMe/MeOH) (Scheme 10) [48]. Both Lac/LacNAc libraries were then submitted to solid phase assays, based on the adsorption of the glycoprotein asialofetuin on microtiter plate wells, which is a known galectin-3 ligand, and on the use of labeled galectin-3. The tested compounds were then expected to interfere with the glycoprotein-galectin-3 interaction, being verified three- to fourfold potency in relation to Lac for compounds 67 and 68, at galectin-3 concentration of 0.4–0.5 mM (Table 1). Further NMR spectroscopy analysis with 15N-labeled galectins was also performed to assess the impact of structural differences among the tested compounds on selective binding to galectin-3 and the closely related galectin-1 [48].

Synthesis of corresponding triazolyl lactoside/ N-acetyllactosamine derivatives 67 and 68
Thiodigalactoside has also been shown to bind to galectin-3 with a similar affinity to lactose or LacNAc. In fact, X-ray crystallography and molecular modeling showed that the two galactose residues of thiodigalactoside (TDG) were bound identically in the β-galactoside binding site, whereas the second galactose of thiodigalactoside was bound in the same place as the GlcNAc of LacNAc, with identical hydrogen bonding networks between the disaccharides and the protein [24, 49]. Therefore, derivatization of thiodigalactoside at C-3 of galactose, especially with aromatic amides, may lead to effective cation-π interactions with arginine residues of galectin-3, such as Arg144 and Arg186 [44]. In this regard, thiodigalactosides bearing two identical amides at the two 3-positions (i.e. C 2 -symmetrical compounds) were synthesized from galacto azide 69, in a synthetic route involving a sequential reduction to amine, acylation with respective aromatic acyl chlorides to give the amides, bromination (HBr), treatment with dried sodium sulfide and final deprotection (Scheme 11a). Among the obtained diamides, 70 (Kd 0.046 μM), 71 (Kd 0.050 μM) and 72 (Kd 0.049 μM) (Scheme 11a) presented higher inhibitory activities against galectin-3 in fluorescence polarization assays. For the sake of comparison, unsymmetrical thiodigalactosides, such as 73 and 74, having two different amides at C-3 and C-2 positions of galactose residues were synthesized from dimethoxybenzamide-substituted galactose 75, in a sequential 5 steps route (Scheme 11b). Compounds 73 (Kd 0.069 μM) and 74 (Kd 0.052 μM) showed binding affinities to galectin-3 comparable to the symmetrical diamides 70–72 in fluorescence polarization assays (Table 1) [44].

a Synthesis of symmetrical thiodigalactosides 70–72. b Synthesis of unsymmetrical thiodigalactosides 73 and 74
Considering the limited understanding of galectin-3 roles both in and outside the cell, the development of molecular probes able to detect and identify galectin-3 in the organism is of great relevance [50]. Thus, a series of galectin-3 probes bearing acetophenone or benzophenone moieties as photolabels, and based on ester-, amide- or triazole-derived thiodigalactosides, was synthesized [51]. Firstly, the acetophenone-based probe 76 was obtained by coupling of suitably benzylidene-protected thiodigalactoside 77 with 4′-O-succinimide-ester-4-(prop-2-ynyloxy)acetophenone 78 in basic media, followed by deprotection reaction (AcOH/ H2O) (Scheme 12a). Subsequent synthesis of amide-linked probe 79 was carried out by Staudinger reduction of the azido-thiodigalactoside 80 using PPh3, followed by coupling to the photolabel 81 (0.1 M NaHCO3) and Zemplen deprotection (Scheme 12b). Regarding the triazole-linked probe 82, it was obtained by click chemistry reaction between the azido-thiodigalactoside 80 and the photolabel-linked dialkyne 83, using CuSO4/ sodium ascorbate as catalytic system, followed by removal of the acetyl groups (Scheme 12b). According to fluorescence polarization assays, higher binding affinities to galectin-3 were verified for compounds 76 (Kd 2 μM) and 79 (Kd 0.9 μM) (Table 1), which also showed the most pronounced labeling under fluorescence SDS-PAGE gel-based assays [51].

Synthesis of acetophenone-based probe 76 a, amide-linked probe 79 and triazole-linked probe 82 b
Aiming to increase arginine-arene interactions and to improve galectin-3 selectivity, inhibitors for galectin-3 containing bulky aromatic groups were synthesized from thiodigalactoside and lactosamine by derivatization of the galactose C3. The synthesis of thiodigalactoside-based inhibitors, such as 84 and 85, was carried by CuAAC reactions between the common intermediate azido-thiodigalactoside 80 and the aromatic alkynes phenylacetylene 86 and 3-hydroxyphenylacetylene 87, using CuSO4/sodium ascorbate under microwave irradiation, followed by deacetylation reactions (Scheme 13a). In relation to LacNAc derivatives with an ether-linked aromatic moiety, compound 88, bearing the 4-(4-phenoxybenzyl) group, was obtained by glycosylation reaction between derivatized galactoside 89 and glucosamine 90, followed by careful deacetylation without removal of the phthaloyl protecting group (Scheme 13b). For the synthesis of 4-(4-phenoxyphenyl)triazole-linked lactosamine 91, the intermediate 92 was firstly synthesized by glycosylation reaction (TfOH, NIS) between the 3-azido functionalized methylthio donor 93 and the bromide acceptor 94. Subsequently, the obtained 92 was submitted to click reaction (CuSO4, sodium ascorbate) with 1-ethynyl-4-phenoxybenzene, followed by azide introduction (NaN3) and careful deacetylation, affording compound 91 (Scheme 13c) [43]. The binding affinities of compounds 84, 85, 88 and 91 to galectin-3 were evaluated by fluorescence polarization assays, being verified low Kd values of 0.044 μM, 0.013 μM, 1.2 μM and 2.2 μM (Table 1), respectively, reflecting the positive influence of their larger aromatic groups to establish favourable aryl-arginine interactions with galectin-3 [43].

Synthesis of aromatic thiodigalactosides 84 and 85 a, and lactosamines 88 and 91 b and c
In order to investigate the binding properties of selenoglycosides toward galectin-3 [52], symmetrical selenodigalactoside (SeDG) 95 and diselenodigalactoside (DSeDG) 96 were prepared from β-galactopyranosyl isoselenuronium bromide 97, in three-steps synthetic routes (Scheme 14) [53]. These two selenides were submitted to solid phase binding assays, based on their capacity to block binding of labeled galectin to surface-imobilized neoglycoprotein (Lac-BSA), and to fluorescent cell surface assays. SeDG and DSeDG presented corresponding IC50 values of 1.4 mM and 3.8 mM (Table 1), comparable to TDG (IC50 1.1 mM), under fixed concentrations of 15 μg/mL for galectin-3 and 0.25 μg/well of lactosylated BSA. In fluorescent cell assays, involving evaluation of stained colon adenocarcinoma cells (SW480) in the presence of SeDG/ DSeDG or Lac (1 mM) and labeled galectin-3 (5 μg/mL), both selenides presented better galectin-3 inhibition than lactose, displaying fluorescence binding of 96 % [52]. In addition to inhibitory properties towards galectin-3, selenoglycosides opens up the way for development of new bioanalytical approaches directed to human lectins, such as sensor activity involving 77Se -bearing carbohydrate ligands [52].

Synthesis of symmetrical selenodigalactoside (SeDG) 95 and diselenodigalactoside (DSeDG) 96
Multivalent-based inhibitors
Since lectin-carbohydrate interactions are generally weak, special efforts are often required to achieve tighter binding of carbohydrate-based ligands to lectins. One of the established strategies to improve such interactions is based on the concept of the “glycoside cluster effect”, which is related to the number and geometry of carbohydrate residues and also depends on their steric bulk, density and relative distance, as well as on their three-dimensional arrangement [54, 55]. According to this concept, the affinity enhancements observed for multimeric ligands over monomeric species is often greater than predicted from the sum of the constitutive interactions. Within this context, multimeric lactosides based on carbohydrate scaffolds with valences ranging from 1 to 4 and different linker lengths were synthesized by CuAAC reactions as inhibitors of galectin-3 (Scheme 15) [56]. To achieve this purpose, commercially available glucose, maltose and maltotriose were used as core structures to generate multivalency, by means of the preparation of their corresponding azido derivatives, in parallel to the synthesis of lactoside moieties tethered by alkyne-containing oligo(ethylenoglycol) (EG) linkers. Subsequently, click reactions between the obtained azido-sugar cores and alkyne lactosides, in the presence of CuSO4/sodium ascorbate under microwave irradiation, led to a series of mono- and multivalent lactoside triazoles such as compounds 98–103 (Scheme 15) [56]. According to competitive fluorescent polarization assays, the highest affinity for galectin-3 was observed for the tetravalent derivative 102 (Kd 16 μM) (Table 1), which showed four times higher inhibitory potency than the corresponding derivative with one lactose residue.

a General procedure of Cu(I)-catalyzed reactions between azido-sugar cores and alkyne lactosides, exemplified for 98. b Structures of mono- 99 and multivalent lactoside triazoles 100–103
Following the purposes of evaluating the glycoside cluster effect between multivalent ligands and galectin-3, polyfunctional unnatural amino acids like phenyl-bis-alanine (PBA) and phenyl-trisalanine (PTA), bearing two orthogonal functionalities (amine and ester), were employed as scaffolds for the synthesis of a panel of mono-, di- and trivalent lactoside derivatives. Alkyne precursors derived from the PBA and PTA were thus firstly prepared by coupling reactions with propiolic acid and then reacted with 2-azidoethyl-lactose 104, by means of 1,3-dipolar cycloaddition reaction (copper (I) iodide, DIPEA), generating a series of triazole-lactosides (Scheme 16) [57]. The binding affinities of the obtained compounds to galectin-3 were assessed by competitive fluorescent polarization assays, being the highest affinity verified for compounds 105 (Kd 17 μM) and 106 (Kd 27 μM) (Table 1), which were about 10 times more potent than methyl β-lactoside (Kd 220 μM). Further evaluation of the cluster effect of the trivalent-105 and divalent-106 lactoside towards galectin-3 showed cluster effect values around 4.0 and 1.5, when comparing 105 and 106 to the corresponding methyl β-lactoside or to the monomers 107 and 108. Thus, these different values point out the importance to calculate the cluster effect with respect to a reference monomer, such as 107 or 108, instead of lactose when analyzing multivalent galectin-3 inhibitors. It’s interesting to mention that compound 106 presented higher inhibition toward the closely related galectin-1 (Kd 3.2 μM) and also much stronger cluster effects of 30 and 7.7 when compared to methyl β-lactoside and to monomer 108, respectively. This stronger binding of 105 and 106 to galectin-1 may be due to additional interactions of the second lactose on another site on the same CRD, or, in a lesser extent, to their possible cluster effect towards this dimeric galectin [58].

Synthesis of PBA and PTA-derived lactosides 105–108 by CuAAC reactions
In the search for novel galactose-1,2,3-triazole glycoconjugates, the trimeric triazole galactohybrid 109 was synthesized from the sugar 3-azido-3-deoxy-1,2:5,6-di-O-isopropylidene-α-D-galactofuranose 110. Thus, CuAAC reaction between the azidogalactofuranose 110 and the 1,3,5-triethynylbenzene 111 gave the trivalent galacto-cluster 112, which was then treated with an aqueous solution of trifluoroacetic acid for cleavage of isopropylidene protecting groups, affording the final galactohybrid 109 as a mixture of its α- and β-pyranose forms (Scheme 17). As determined by fluorescence anisotropy assay, compound 109 exhibited interesting galectin-3 binding with Kd 50 μM (Table 1) [59].

Synthesis of the trimeric triazole galactohybrid 109 by CuAAC reaction
Envisioning the possibility to interact with two CRDs from distinct galectin-3 monomers, the divalent lactoside represented by 1,2,3-triazole di-lactose-derived glycoconjugate 113 was synthesized as a potential galectin-3 inhibitor. The synthesis of 113 was carried out by click chemistry reaction between the corresponding azide and alkyne precursors 114 and 115, affording the per-acetylated product 116, which was then deacetylated (NaOMe/ MeOH) to give the final divalent lactoside 113 (Scheme 18a) [10]. Compound 113 was submitted to surface plasmon resonance (SPR) assays, which increase the chance of multivalency effects due to immobilization of galectin-3 on the SPR surface, being verified strong interaction with galectin-3 (Kd 0.15 μM) (Table 1). In fact, molecular dynamics simulations revealed the capacity of glycoconjugate 113 to bridge two independent galectin-3 CRDs, creating a non-covalent cross-link between two monomers, which may explain its submicromolar affinity towards galectin-3 [10].
The structure of glycoconjugate 113 resembles described lactulose amines [60, 61], such as N,N´-dilactulose-octamethylenediamine (D-LDO) (Scheme 18b), which satisfactorily inhibited the binding of galectin-3 to microwells coated with 90 K (a highly glycosylated protein previously shown to interact with galectin-3), with IC50 in the order of 20–40 mM [61].

a Synthesis of the 1,2,3-triazole lactose-derived glycoconjugate 113 by click chemistry. b Chemical structure of N,N´-dilactulose-octamethylenediamine (D-LDO)
With the aim to get glycolytically stable lactosides as effective multivalent galectin-3 inhibitors, transition metal-catalyzed cross-coupling reactions have been efficiently used to introduce carbon-carbon bonds [62, 63], generating dimeric thio-lactosides, such as 117. Compound 117 was synthesized by Glaser Cu(I)-catalyzed homo-coupling reaction between two units of the lactoside alkyne 118, followed by deacetylation reaction (NaOMe/MeOH) (Scheme 19a). In addition, the Cu(I) catalyzed 1,3-dipolar cycloaddition reaction also proved to be effective to get the glycolytically stable triazole dimeric lactoside 119, which was obtained after click reaction (CuI/ DIPEA) between the lactoside alkyne 118 and the lactoside azide 63, and final deacetylation reaction (NaOMe/MeOH) (Scheme 19b). Compounds 117 and 119 were tested by hemagglutination assays at a 1 μM concentration of galectin-3, presenting inhibitory IC50 of 160 μM against galectin-3 (Table 1) [64].

Synthesis of multivalent dimeric lactosides 117 a and 119 b
Following the concept of multivalency for gaining improved affinity toward galectin-3, the rigid multivalent lactose ligand 120 was prepared by a synthetic procedure, which mechanism involves the initial formation of thiourea-linked sugar, with subsequent conversion to a rigid 2-aminothiazoline moiety. Therefore, the synthesis of 120 started with the previous preparation of the alkyne-branched tetraamino 121 by conjugation of the building blocks 122 and 123, containing the corresponding free amino and carboxyl groups, in the presence of the coupling reactant BOP (Scheme 20a). Subsequently, compound 121 was reacted with lactose β-isothiocyanate 124 in acetonitrile/iPr2Net and then deacetylated (NaOMe/MeOH), yielding the tetravalent lactose derivative 120 (Scheme 20b). The inhibitory potency of compound 120 toward galectin-3 was determined with the use of a solid phase based assay, according to what the biotin-labeled galectin-3 was allowed to bind to the carbohydrate moieties of asialofetuin (ASF) coated onto the surface of microtiter plate wells (the matrix) in competition with the lactoside 120. It was verified an IC50 of 0.07 μM for compound 120, representing an enhancement, relative to lactose, of almost 4300-fold. However, under a direct binding assay with galectin-3 using Trp fluorescence (fluorescence titration), an apparent Kd value of 14 μM was observed for compound 120, corresponding to much lower potency (44-fold relative to lactose) if compared to the previous solid phase assay (Table 1) [65].

a Synthesis of the alkyne-branched tetraamino 121. b Synthesis of the tetravalent lactose derivative 120
The synthesis of selective multivalent glycoprotein-based glyco-ligands as galectin-3 inhibitors may have a great impact on tumor-related mechanisms mediated by this lectin [66]. In this regard, human serum albumin (HSA) was used as the scaffold and an Asn-linked complex type N-glycan was used as the glycan building block. Sialoglycopeptide (SGP), isolated from chicken egg yolk, was thus used as starting glycan by means of previous treatment with pronase to remove the peptide portion, leaving the asparagine (Asn)-linked N-glycan (SCT-Asn), which was then labeled with an alkyne group by reaction with an activated ester, N-(4-pentynoyloxy)succinimide in an aqueous solution (Scheme 21a). In paralell, HSA, containing free lysine residues on its surface, was tagged with azide functionality by treatment with an excess of the activated azide derivative, NHS-PEG4-azide, in an aqueous NaHCO3 solution resulting in the introduction of multiple azide groups in the protein (Scheme 21b). Subsequently, the SCT-Asn-alkyne 125 and HSA-azide were conjugated via Cu(I)-catalyzed alkyne-azide cycloaddition reaction, utilizing CuSO4, L-ascorbic acid and the copper ligand BTTP, in phosphate buffer (pH .5), affording the HSA-based neoglycoprotein 126, which was treated with neuraminidase for removal of sialic acids and exposure of galactose moieties (Scheme 21b). The HSA-Gal glycoprotein 126, containing about 20 N-glycans, showed high affinity to galectin-3 in SPR assays, with EC50 0.073 μM (Table 1), demonstrating its strong multivalent effect on galectin-3 inhibition. In addition, the inhibition of the attachment of galectin-3 to immobilized cancer cells by HSA-Gal glycoprotein 126 was assessed by ELISA assays, which showed that 126 was able to block the attachment of galectin-3 to both PC3 prostate and A549 alveolar cancer cells in a dose-dependent manner (IC50 for PC3 cells, 0.122 μM; IC50 for A549 cells, 0.338 μM), suggesting that this synthetic HSA based glyco-ligand may find applications for blocking the function of galectin-3 in distinct tumor processes such as cancer cell aggregation, adhesion and metastasis [67].

a Synthesis of alkyne-labeled N-glycan 125. b Synthesis of the HSA-based glycoprotein 126
The involvement of tumor-related galectin-3 is also known to be related to its capacity to bind to TF antigen on MUC1 mucin, a large transmembrane protein that is overexpressed and heavily glycosylated on cancer cells [68]. In fact, clustering formation in response to MUC1-galectin binding exposes adhesion molecules and increases homotypic aggregation with heterotypic adhesion to endothelium, which favors tumor escape in the circulation and eventually metastasis [69]. In this context, galectin-3 inhibitors represented by multivalent lactose-functionalized dendrimers may have a fundamental role on the elucidation of the mechanism by which cancer cellular aggregation occurs. Lactose-functionalized poly(amidoamine) (PAMAM) dendrimers were thus prepared by a synthetic route involving initial catalytic transfer hydrogenation (ammonium formiate/ 10 % Pd-C) of p-nitrophenyl-β-D-lactoside 127, giving the p-aminophenyl derivative 128, which was then treated with thiophosgene in 80 % aqueous ethanol for conversion to the isothiocyanate 129. Subsequently, 129 was submitted to conjugation reaction with dendritic PAMAM, in a mixture of DMF:H2O (1:1) containing DIPEA, to afford glycodendrimers 130–133, displaying 15 to 100 lactose end groups attached to the dendrimer framework (Scheme 22) [70, 71]. The effects of lactose-functionalized dendrimers 130–133 on cellular aggregation in three different cancer cell lines (A549, DU-145, and HT-1080) and in the presence of galectin-3, were then evaluated by cells image analysis, Western blotting and fluorescence microscopy. According to the obtained results, the smaller dendrimer 130 inhibited galectin-3 induced aggregation for all three tested cancer cell lines, whereas the larger dendrimer 133 caused cancer cells to aggregate through a galectin-3 pathway. These distinct observed effects indicate that inhibition by 130 occurred in a competitive mode, diverting galectin-3 binding to TF antigen on MUC1, while dendrimer 133 provided too many sites for galectin-3, increasing cross-linking of the cells and thus enhancing aggregation [71]. Thus, aggregation can be favored or not depending on the number of lactose groups on the dendrimer [71].

Synthesis of lactose-functionalized dendrimers 130–133
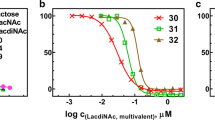
Considering the potential of multivalent glycan ligands to improve binding affinity towards galectin-3, designed neo-glycoproteins constituted by two kinds of tetrasaccharides (LacNAc-LacNAc 134 or LacDiNAc-LacNAc 135) and having bovine serum albumin (BSA) as scaffold for multivalent presentation were prepared by combination of chemo-enzymatic synthesis and chemical conjugation to lysine residues of BSA [72]. Initially, the trisaccharide intermediate 136 was synthesized from the monosaccharide GlcNAc-linker-tBoc 137, in a sequential chemo-enzymatic route involving the glycosyltransferases β4GalT (galactosyltransferase) and β3GlcNAcT (N-acetylglucosaminyltransferase), and the corresponding UDP-Gal and UDP-GlcNAc activated nucleotide sugars as donor substrates (Scheme 23a). Once the intermediate 136 was obtained, it was reacted with the donors UDP-Gal and UDP-GalNAc, respectively, in the presence of the corresponding enzymes β4GalT and β4GalTY284L (N-acetylgalactosaminyltransferase), affording the tetrasaccharides LacNAc-LacNAc 134 and LacDiNAc-LacNAc 135, respectively (Scheme 23a) [72–74]. In order to favor further coupling to BSA, tetrasaccharides 134 and 135 were previously reacted with squaric acid diethyl ester 138 (Et3N/ EtOH) to form the corresponding squaric acid monoamide esters 139 and 140 (Scheme 23b). Subsequently, conjugation of 139 and 140 to lysine groups of BSA was carried out in borate buffer (pH .0), with an incubation period of six days, affording neo-glycoproteins 141a-k and 142a-k with variable numbers of LacNAc-LacNAc and LacDiNAc-LacNAc, reaching numbers for modified lysine residues between 2 and 29 per BSA molecule (Scheme 23b). The binding properties of neo-glycoproteins 141a-k and 142a-k as ligands for human galectin-3 were then evaluated by an ELISA-type assay, based on the incubation of different amounts of galectin-3 (2–12 μM) on immobilized neo-glycoproteins (5 pmol per well). According to the obtained results, lowered Kd values were verified for 142d-f with about 9, 14 and 21 LacDiNAc-LacNAc glycans, respectively, reaching a Kd value for galectin-3 in the nanomolar range (30 nM for 142f) (Table 1). Thus, these data emphasize high selectivity of LacDiNAc-LacNAc glycans toward galectin-3, finding important biomedical applications, especially in cancer related research [72].

a Chemo-enzymatic synthesis of tetrasaccharides 134 and 135 using activated nucleotide sugars as donor substrates and recombinant glycosyltransferases for complete conversions. b Two-step synthesis of neo-glycoproteins 141a–k and 142a–k
Considering the correlation of galectin-3 expression with cancer aggressiveness, metastasis and apoptosis, it can be considered an emerging cancer marker [75]. Therefore, the development of chemical probes is of great relevance for detection of the cancer-linked galectin-3. In fact, they represent alternative tools to the often used antibody-based methods to detect active galectin-3 [76]. In this context, the synthesis of multivalent probe 143 was initiated by linking the lactoside azide 144 to the divalent alkyne 145 using click chemistry reaction (CuSO4/ sodium ascorbate) (Scheme 24a). Subsequently, an alkyne group was introduced into the obtained triazolic compound 146 by means of a BOP coupling of propargyl amine to the scaffold carboxylic acid function, giving compound 147, which was then deacetylated (NaOMe/ MeOH) to afford the probe 143. For probe evaluation, distinct biological protein mixtures containing galectin-3 were sequentially treated with varying concentrations of the probe 143 and the fluorescein-N3 construct 147 (reporter molecule) (Scheme 24b). Based on what was verified in SDS-PAGE, the obtained fluorescent gel images showed a strong and dominant band corresponding to galectin-3 in the tested protein mixtures, being the most favourable concentration of probe 143 determined to be around 5 μM, with a two-fold excess of the N3 reporter 147. In fact, this type of probe in combination with various reporter molecules may represent a valuable tool for deciphering galectin-3 roles, especially in cancer, besides aiding in its diagnosis [76].

a Synthesis of the probe 143 using click chemistry strategy. b Schematic representation of galectin-3 detection by treatment of Gal-3-probe formed complex with the fluorescent reporter 147
Conclusions
In this review we presented relevant synthetic routes for obtaining monosaccharide-, disaccharide- and multivalent-based galectin-3 inhibitors, involving regioselective modifications of distinct carbohydrate scaffolds (galactose, mannose, talose, lactose or thiodigalactose) and glycoproteins. Cu(I)-assisted 1,3-dipolar azide-alkyne cycloaddition (CuAAC) reactions were predominantly applied to the synthesis of the described inhibitors, allowing the insertion of varying substituent groups at different sugar positions via triazole moiety. When comparing the inhibitory activities towards galectin-3, thiodigalactoside derivatives, especially those containing aromatic aglycons, showed to be the most effective galectin-3 inhibitors, highlighting their capacity to interact with extended galectin-3 subsites. In general, multivalent carbohydrate ligands strongly inhibited galectin-3 and it is also worth mentioning that lower Kd values were verified for multivalent inhibitors tested by solid phase assays such as ELISA and SPR, which can be explained by increased chances of multivalency effects when immobilizing galectin-3 on a solid surface.
Taken together, the synthetic glycoconjugates described in this review represent frontline galectin-3 inhibitors, finding important therapeutic and diagnostic applications in galectin-3 mediated tumor processes, and open up the way for the design and synthesis of novel galectin-3 inhibitors candidates.
References
Boligan K.F., Mesa C., Fernandez L.E., von Gunten S.: Cancer intelligence acquired (CIA): tumor glycosylation and sialylation codes dismantling antitumor defense. Cell. Mol. Life Sci. 72, 1231–1248 (2015)
Dall'Olio F., Malagolini N., Trinchera M., Chiricolo M.: Mechanisms of cancer-associated glycosylation changes. Front. Biosci. (Landmark Ed). 17(670–699), (2012)
Stowell S.R., Ju T., Cummings R.D.: Protein glycosylation in cancer. Annu. Rev. Pathol. 10, 473–510 (2015)
Nakahara S., Raz A.: Biological modulation by lectins and their ligands in tumor progression and metastasis. Anti Cancer Agents Med. Chem. 8, 22–36 (2008)
Boscher C., Dennis J.W., Nabi I.R.: Glycosylation, galectins and cellular signaling. Curr. Opin. Cell Biol. 23, 383–392 (2011)
Garner O.B., Baum L.G.: Galectin-glycan lattices regulate cell-surface glycoprotein organization and signalling. Biochem. Soc. Trans. 36, 1472–1477 (2008)
Hockl P.F., Wolosiuk A., Sáez J.M., Bordoni A.V., Croci D.O., Terrones Y.T., Illia G.J., Rabinovich G.A.: Glyco-nano-oncology: Novel therapeutic opportunities by combining small and sweet. Pharmacol. (2016). doi:10.1016/j.phrs.2016.02.005
Ahmed H., AlSadek D.M.: Galectin-3 as a potential target to prevent cancer metastasis. Clin. Med. Insights Oncol. 25, 113–121 (2015)
Arthur, C. M.; Baruffi, M. D.; Cummings, R. D.; Stowell, S. R.: Galectins: methods and protocols. Stowell, S. R.; Cummings, R. D. (eds.); human press, Vol. 1, Chapter 1, pp 1–35 (2015)
Marchiori M.F., Souto D.E., Bortot L.O., Pereira J.F., Kubota L.T., Cummings R.D., Baruffi M.D., Carvalho I., Campo V.L.: Synthetic 1,2,3-triazole-linked glycoconjugates bind with high affinity to human galectin-3. Bioorg. Med. Chem. 23, 3414–3425 (2015)
van den Brüle F., Califice S., Castronovo V.: Expression of galectins in cancer: a critical review. Glycoconj. J. 19, 537–542 (2004)
Liu F.T., Rabinovich G.A.: Galectins as modulators of tumour progression. Nat. Rev. Cancer. 5, 29–41 (2005)
Califice S., Castronovo V., Bracke M., et al.: Dual activities of galectin-3 in human prostate cancer: tumor suppression of nuclear galectin-3 vs tumor promotion of cytoplasmic galectin-3. Oncogene. 23, 7527–7536 (2004)
Sun W., Li L., Yang Q., Shan W., Zhang Z., Huang Y.: G3-C12 peptide reverses galectin-3 from foe to friend for active targeting cancer treatment. Mol. Pharm. 12, 4124–4136 (2015)
Cecchinelli B., Lavra L., Rinaldo C., Iacovelli S., Gurtner A., Gasbarri A., Ulivieri A., Del Prete F., Trovato M., Piaggio G., Bartolazzi A., Soddu S., Sciacchitano S.: Repression of the antiapoptotic molecule galectin-3 by homeodomain-interacting protein kinase 2-activated p53 is required for p53-induced apoptosis. Mol. Cell. Biol. 26, 4746–4757 (2006)
Yang R.Y., Rabinovich G.A., Liu F.T.: Galectins: structure, function and therapeutic potential. Expert. Rev. Mol. Med. (2008). doi:10.1017/S1462399408000719
Leffler H., Carlsson S., Hedlund M., Qian Y., Poirier F.: Introduction to galectins. Glycoconj. J. 19, 433–440 (2004)
Knibbs R.N., Agrwal N., Wang J.L., Goldstein I.J.: Carbohydrate-binding protein 35. II. Analysis of the interaction of the recombinant polypeptide with saccharides. J. Biol. Chem. 268, 14940–14947 (1993)
Tejler J., Salameh B., Lefflerc H., Nilsson U.J.: Fragment-based development of triazole-substituted O-galactosyl aldoximes with fragment-induced affinity and selectivity for galectin-3. J. Org. Biomol. Chem. 7, 3982–3990 (2009)
Bum-Erdene K., Gagarinov I.A., Collins P.M., Winger M., Pearson A.G., Wilson J.C., Leffler H., Nilsson U.J., Grice I.D., Blanchard H.: Investigation into the feasibility of thioditaloside as a novel scaffold for galectin-3-specific inhibitors. Chembiochem. 14, 1331–1339 (2013)
Stowell S.R., Karmakar S., Stowell C.J., Baruffi M.D., McEver R.P., Cummings R.D.: Human galectin-1, −2, and −4 induce surface exposure of phosphatidylserine in activated human neutrophils but not in activated T cells. Blood. 109, 219–227 (2007)
Rapoport E.M., Kurmyshkina O.V., Bovin N.V.: Mammalian galectins: structure, carbohydrate specificity, and functions. Biochem. Mosc. 73, 393–405 (2008)
Giguere D., Andre S., Bonin M.A., Bellefleur M.A., Provençal A., Cloutier P., Pucci B., Roy R., Gabius H.J.: Inhibitory potential of chemical substitutions at bioinspired sites of β-D-galactopyranose on neoglycoprotein/cell surface binding of two classes of medically relevant lectins. J. Bioorg. Med. Chem. Lett. 19, 3280–3287 (2011)
Cumpstey I., Sundin A., Leffler H., Nilsson U.J.: C2-symmetrical thiodigalactoside bis-benzamido derivatives as high-affinity inhibitors of galectin-3: efficient lectin inhibition through double arginine-arene interactions. J. Angew. Chem. Int. Ed. 44, 5110–5112 (2005)
Cumpstey I., Salomonsson E., Sundin A., Leffler H., Nilsson U.J.: Double affinity amplification of galectin-ligand interactions through arginine-arene interactions: synthetic, thermodynamic, and computational studies with aromatic diamido thiodigalactosides. Chem. 14, 4233–4245 (2008)
Aragão-Leoneti V., Campo V.L., Gomes A.S., Field R.A., Carvalho I.: Application of copper(I)-catalysed azide/alkyne cycloaddition (CuAAC) ‘click chemistry’ in carbohydrate drug and neoglycopolymer synthesis. Tetrahedron. 66, 9475–9492 (2010)
Tiwari V.K., Mishra B.B., Mishra K.B., Mishra N., Singh A.S., Chen X.: Cu-catalyzed click reaction in carbohydrate chemistry. Chem. Rev. 116, 3086–3240 (2016)
Blanchard H., Yu X., Collins P.M., Bum-Erdene K.: Galectin-3 inhibitors: a patent review (2008-present). Expert Opin. Ther. Pat. 24, 1053–1065 (2008)
Téllez-Sanz R., García-Fuentes L., Vargas-Berenguel A.: Human galectin-3 selective and high affinity inhibitors. Present state and future perspectives. Curr. Med. Chem. 20, 2979–2990 (2013)
Yang R.-Y., Rabinovich G.A., Liu F.-T.: Galectins: structure, function and therapeutic potential. Expert Rev. Mol. Med. 10, e17 (2008)
Nakahara S., Raz A.: Biological Modulation by lectins and their ligands in tumor progression and metastasis. Anti Cancer Agents Med. Chem. 8, 22–36 (2008)
Klyosov A.A., Traber P.G.: Galectins and disease implications for targeted therapeutics. ACS Symposium Series, American Chemical Society, Washington (2012)
Radosavljevic G.D., Pantic J., Jovanovic I., Lukic M.L., Arsenijevic N.: The two faces of galectin-3: roles in various pathological conditions. Ser. J. Exp. Clin. Res. 17, 1–1 (2016)
Giguére D., Patnam R., Bellefleur M.-A., St-Pierre C., Sato S., Roy R.: Carbohydrate triazoles and isoxazoles as inhibitors of galectins-1 and −3. Chem. Commun. 14, 2379–2381 (2006)
Tejler J., Skogman F., Lefflerc H., Nilsson U.J.: Synthesis of galactose-mimicking 1H-(1,2,3-triazol-1-yl)- mannosides as selective galectin-3 and 9 N inhibitors. Carbohydr. Res. 342, 1869–1875 (2007)
Compagno D., Jaworski F.M., Gentilini L., Contrufo G., González Pérez I., Elola M.T., Pregi N., Rabinovich G.A., Laderach D.J.: Galectins: major signaling modulators inside and outside the cell. Curr. Mol. Med. 14, 630–651 (2014)
Bum-Erdene K., Gagarinov I.A., Collins P.M., Winger M., Pearson A.G., Wilson J.C., Leffler H., Nilsson U.J., Grice I.D., Blanchard H.: Investigation into the feasibility of thioditaloside as a novel scaffold for galectin-3-specific inhibitors. ChemBiolChem. 14, 1331–1342 (2013)
Salameh B.A., Cumpstey I., Sundin A., Leffler H., Nilsson U.J.: 1H-1,2,3-triazol-1-yl thiodigalactoside derivatives as high affinity galectin-3 inhibitors. Bioorg. Med. Chem. 18, 5367–5378 (2010)
Fort S., Kim H.-S., Hindsgaul O.: Screening for galectin-3 inhibitors from synthetic lacto-N-biose libraries using microscale affinity chromatography coupled to mass spectrometry. J. Organomet. Chem. 71, 7146–7154 (2006)
Öberg C., Blanchard H., Leffler H., Nilsson U.J.: Protein subtype-targeting through ligand epimerization: talose-selectivity of galectin-4 and galectin-8. Bioorg. Med. Chem. Lett. 18, 3691–3694 (2008)
Öberg C., Noresson A.-L., Leffler H., Nilsson U.J.: Synthesis of 3-amido-3-deoxy-beta-D-talopyranosides: all-cis-substituted pyranosides as lectin inhibitors. Tetrahedron. 67, 9164–9172 (2011)
Collins P.M., Oberg C.T., Leffler H., Nilsson U.J., Blanchard H.: Taloside inhibitors of galectin-1 and galectin-3. Chem. Biol. Drug Des. 79, 339–346 (2012)
van Hattum H., Branderhorst H.M., Moret E.E., Nilsson U.J., Leffler H., Pieters R.J.: Tuning the preference of thiodigalactoside- and lactosamine-based ligands to galectin-3 over galectin-1. J. Med. Chem. 56, 1350–1354 (2013)
Cumpstey I., Solomonsson E., Sundin A., Leffler H., Nilsson U.J.: Double affinity amplification of galectin–ligand interactions through arginine–arene interactions: synthetic, thermodynamic, and computational studies with aromatic diamido thiodigalactosides. Chem. Eur. J. 14, 4233–4245 (2008)
Öberg C.T., Leffler H., Nilsson U.J.: Arginine binding motifs: design and synthesis of galactose-derived arginine tweezers as galectin-3 inhibitors. J. Med. Chem. 51, 2297–2301 (2008)
Öberg C.T., Leffler H., Nilsson U.J.: Inhibition of galectins with small molecules. Chimia. 65, 18–23 (2011)
Guigere D., Sato S., St-Pierre C., Sirois S., Roy R.: Aryl O- and S-galactosides and lactosides as specific inhibitors of human galectins-1 and −3: role of electrostatic potential at O-3. Bioorg. Med. Chem. Lett. 16, 1668–1672 (2006)
Rauthu S.R., Shiao T.C., André S., Miller M.C., Madej E., Mayo K.H., Gabius H.-J., Roy R.: Defining the potential of aglycone modifications for affinity/selectivity enhancement against medicallyrelevant lectins: synthesis, activity screening, and HSQCBased NMR analysis. Chembiochem. 16, 126–139 (2015)
Bianchet M.A., Ahmed H., Vasta G.R., Amzel L.M.: Soluble β-galactosyl-binding lectin (galectin) from toad ovary: crystallographic studies of two protein-sugar complexes. Proteins: Struct., Funct., Bioinf. 40, 378–388 (2002)
Pieters R.J.: Inhibition and Detection of Galectins. ChemBioChem. 7, 721–728 (2006)
van Scherpen zeel M., Moret E.E., Ballell L., Liskamp R.M.J., Nilsson U.J., Leffler H., Pieters R.J.: Synthesis and Evaluation of New Thiodigalactoside-Based Chemical Probes to Label Galectin-3. ChemBioChem. 10, 1724–1733 (2009)
André S., Kövér K.E., Gabius H.-J., Szilágyi L.: Thio- and selenoglycosides as ligands for biomedically relevant lectins: Valency–activity correlations for benzene-based dithiogalactoside clusters and first assessment for (di)selenodigalactosides. Bioorg. Med. Chem. Lett. 25, 931–935 (2015)
Wagner G., Nuhn P.: Synthese von Selenoglykosiden mit Acetyl-glykosyl-isoselenuronium-bromiden. Arch. Pharm. 297, 461–473 (1964)
Galante E., Geraci C., Sciuto S., Campo V.L., Carvalho I., Sesti-Costa R., Guedes P.M.M., Silva J.S., Hill L., Nepogodiev S.A., Field R.A.: Glycoclusters presenting lactose on calix[4]arene cores display trypanocidal activity. Tetrahedron. 67, 5902–5912 (2011)
Campo, V. L., Carvalho, I.: Click chemistry applied to carbohydrate-based drug discovery click chemistry in Glycoscience. New developments and strategies. In: Witczak, Z. J., Bielski, R., (eds); Wiley; Vol. 1, Chapter 13, pp 325–358 (2013)
Gouin S.G., Fernández J.M.G., Vanquelef E., Dupradeau F.-Y., Salomonsson E., Leffler H., Ortega-Muñoz M., Nilsson U.J., Kovensky J.: Multimeric lactoside “click clusters” as tools to investigate the effect of linker length in specific interactions with peanut lectin, galectin-1, and −3. Chembiochem. 11, 1430–1442 (2010)
Tejler J., Tullberg E., Frejd T., Leffler H., Nilsson U.J.: Synthesis of multivalent lactose derivatives by 1,3-dipolar cycloadditions: selective galectin-1 inhibition. Carbohydr. Res. 341, 1353–1362 (2006)
Salomonsson E., Larumbe A., Tejler J., Tullberg E., Rydberg H., Sundin A., Khabut A., Frejd T., Lobsanov Y.D., Rini J.M., Nilsson U.J., Leffler H.: Monovalent interactions of galectin-1. Biochemistry. 49, 9518–9532 (2010)
Mackeviča, J., Ostrovskis, P., Leffler, H., Nilsson, U. J., Rudovica, V., Viksna, A., Belyakov, S., Turks, M.: Synthesis of 1,2,3-triazole-linked galactohybrids and their inhibitory activities on galectins. ARKIVOC (iii) 90–112 (2014)
Glinsky G.V., Mossine V.V., Price J.E., Bielenberg D., Glinsky V.V., Ananthaswamy H.N., Feather M.S.: Inhibition of colony formation in agarose of metastatic human breast carcinoma and melanoma cells by synthetic glycoamine analogs. Clin. Exp. Metastasis. 14, 253–267 (1996)
Rabinovich G.A., Cumashi A., Bianco G.A., Ciavardelli D., Iurisci I., D’Egidio M., Piccolo E., Tinari N., Nifantiev N., Iacobelli S.: Synthetic lactulose amines: novel class of anticancer agents that induce tumor-cell apoptosis and inhibit galectin-mediated homotypic cell aggregation and endothelial cell morphogenesis. Glycobiology. 16, 210–220 (2006)
Dominique R., Liu B., Das S.K., Roy R.: Synthesis of `molecular Asterisks' via sequential cross-metathesis, Sonogashira and Cyclotrimerization Reactions. Synthesis. 6, 862–868 (2000)
Roy R., Trono M.C., Giguère D.: Effects of linker rigidity and orientation of mannoside cluster for multivalent interactions with proteins. ACS Symp. Ser. 896, 137–150 (2005)
Roy R., Trono M.C., Giguère D.: Synthesis of stable and selective inhibitors of human galectins-1 and −3. Bioorg. Med. Chem. 16, 7811–7823 (2008)
Vrasidas I., André S., Valentini P., Böck C., Lensch M., Kaltner H., Liskamp R.M.J., Gabius H.-J., Pieters R.J.: Rigidified multivalent lactose molecules and their interactions with mammalian galectins: a route to selective inhibitors. Org. Biomol. Chem. 1, 803–810 (2003)
Bian C.-F., Zhang Y., Sun H., Li D.-F., Wang D.-C.: Structural basis for distinct binding properties of the human galectins to Thomsen-Friedenreich antigen. PLoS One. 6, (2011). doi:10.1371/journal.pone.0025007
Wang H., Huang W., Orwenyo J., Banerjee A., Vasta G.R., Wang L.-X.: Design and synthesis of glycoprotein-based multivalent glyco-ligands for influenza hemagglutinin and human galectin-3. Bioorg. Med. Chem. 21, 2037–2044 (2013)
Campo V.L., Riul T.B., Carvalho I., Baruffi M.D.: Antibodies against mucin-based Glycopeptides affect Trypanosoma cruzi cell invasion and tumor cell viability. Chembiochem. 15, 1495–1507 (2014)
Zhao Q., Barclay M., Hilkens J., Guo X., Barrow H., Rhodes J.M., Yu L.-G.: Interaction between circulating galectin-3 and cancer-associated MUC1 enhances tumour cell homotypic aggregation and prevents anoikis. Mol. Cancer. 9, (2010). doi:10.1186/1476–4598–9-154
André S., Ortega P.J.C., Perez M.A., Roy R., Gabius H.-J.: Lactose-containing starburst dendrimers: influence of dendrimer generation and binding-site orientation of receptors (plant/animal lectins and immunoglobulins) on binding properties. Glycobiology. 9, 1253–1261 (1999)
Michel A.K., Nangia-Makker P., Raz A., Cloninger M.J.: Lactose-functionalized dendrimers arbitrate the interaction of galectin-3/muc1 mediated cancer cellular aggregation. Chembiochem. 15, 2106–2112 (2014)
Böcker S., Laaf D., Elling L.: Galectin binding to neo-glycoproteins: LacDiNAc conjugated BSA as ligand for human galectin-3. Biomolecules. 5, 1671–1696 (2015)
Li J., Li W., Jinga J., Yua W.W.: Influencing factors on the synthesis of magnetically responsive lipases. J. Mol. Catal. B Enzym. 101, 47–55 (2014)
Rech C., Rosencrantz R.R., Křenek K., Pelantová H., Bojarová P., Römer C.E., Hanisch F.-G., Křen V., Elling L.: Combinatorial one-pot synthesis of poly-n-acetyllactosamine oligosaccharides with leloir-glycosyltransferases. Adv. Synth. Catal. 353, 2492–2500 (2011)
Takenaka Y., Fukumori T., Raz A.: Galectin-3 and metastasis. Glycoconj. J. 19, 543–549 (2004)
Ballell L., van Scherpenzeel M., Buchalova K., Liskamp R.M.J., Pieters R.J.: A new chemical probe for the detection of the cancer-linked galectin-3. Org. Biomol. Chem. 4, 4387–4394 (2006)
Acknowledgments
We acknowledge the financial support and fellowships from FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo - Brazil) and CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Campo, V.L., Marchiori, M.F., Rodrigues, L.C. et al. Synthetic glycoconjugates inhibitors of tumor-related galectin-3: an update. Glycoconj J 33, 853–876 (2016). https://doi.org/10.1007/s10719-016-9721-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-016-9721-z