
Abstract
The majority of cell-surface and secreted proteins are glycosylated, which can directly affect their macromolecular interactions, stability, and localization. Investigating these effects is critical to developing a complete understanding of the role of glycoproteins in fundamental biology and human disease. The development of selective and unique chemical reactions have revolutionized the visualization, identification, and characterization of glycoproteins. Here, we review the chemical methods that have been created to enable the visualization of the major types of cell-surface glycoproteins in living systems, from mammalian cells to whole animals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
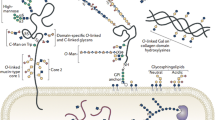
Many cell-surface proteins are modified by at least one type of glycosylation, which can have dramatic effects, including the alteration of the folding, stability, and subcellular localization of proteins [1]. Additionally, cell-surface glycans are well positioned to mediate interactions between the cell and its environment, including cell–cell interactions, pathogen infection, and immune system recognition. The most common types of cell-surface glycosylation in mammals (Fig. 1) are N-linked oligosaccharides [2, 3], which are attached to the amide side-chain of asparagine, and O-linked glycans, which occur on serine and threonine residues. The majority of these O-linked modifications involve the addition of GalNAc to the polypeptide chain, forming the core of mucin-type glycosylation [4]. Several other less-common forms of cell-surface O-linked glycosylation also exists [5], including O-GlcNAc, O-glucose and O-fucose. Previous research using traditional biological techniques (e.g., genetic knockouts, lectin staining, etc.) and analytical chemistry methods, such as mass spectrometry, have demonstrated that dynamic changes in glycosylation play critical roles during normal development and in human disease. However, the visualization of glycans in living systems remained challenging due to several methodological roadblocks. For example, because glycans are native members of the extracellular environment and heterogeneous in nature, the generation of anti-glycan antibodies for immunostaining has been very difficult. Additionally, lectins often display poor selectivity for individual types of glycans and in many cases have weak binding affinity.

Different types of cell-surface glycosylation in mammalian cells. Mammalian cells have several major (N-linked and mucin O-linked) and minor (O-GlcNAc, O-glucose, and O-fucose) types of cell-surface glycosylation
Fortunately, in the past 15 years, the creation of innovative chemical methods and imaging technologies have made significant progress towards addressing this fundamental technological gap. The vast majority of these techniques fall into three different categories: chemoselective chemical reactions that take advantage of the selective reactivity of certain carbohydrates, the installation of chemical probes (chemical reporters) that are absent from biological systems and can also undergo a range of bioorthogonal reactions for the installation of visualization tags, and imaging technologies that can directly detect chemical reporters without bioorthogonal tagging (Fig. 1). Here, we first provide a brief overview of these chemical methods, with a specific emphasis on the different strategies for incorporation of chemical reporters, and review the application of these technologies for the visualization of the major classes of glycoproteins in mammalian cells and common model organisms. For a more broad review of the applications of chemical reporters or the available toolkit of bioorthogonal reactions, we direct readers to other comprehensive reviews [6–8]. We end with a summary of the remaining challenges and opportunities for the glycoprotein imaging field.
Chemical methods for glycoprotein visualization
As stated above, there are three broad chemical categories for the installation of glycoprotein visualization probes. The first of these takes advantage of unique chemical reactivities imparted by the endogenous structure of particular monosaccharides (Fig. 2a). The most well-established example of this is the oxidation of sialic acid residues to aldehydes by periodate, which transforms cis-hydroxyl groups into aldehydes. These aldehydes can then undergo selective reactions with hydrazide- and aminooxy-functionalized visualization tags [7]. In these cases, the bioorthogonality of the method relies on the lack of endogenous aldehydes or ketones on the cell surface. The second method involves the installation of abiotic chemical groups that can be specifically reacted with a growing range of bioorthogonal reactions (Fig. 2b). These bioorthogonal groups are typically incorporated into glycans in two ways. The first, which we have termed metabolic chemical reporters (MCRs), involves the chemical synthesis of analogs of naturally-occurring monosaccharides that contain bioorthogonal reactivity (most commonly azide or alkyne groups). MCRs can then be added to living systems where they are metabolized into activated sugar-donors that are directly incorporated into glycans by endogenous glycosyltransferases. Alternatively, bioorthogonal-analogs of the sugar donors can be prepared chemically or chemoenzymatically and added to cell-surface glycans by exogenous addition of an appropriate glycosyltransferase. The final technique utilizes the chemical information inherent in bioorthogonal tags to enable their direct visualization without the need for the secondary installation of tags (Fig. 2c). Currently, the most powerful of these methods uses either stimulated Raman scattering or surface enhanced Raman scattering (SERS) to directly image the vibrational stretching of abiotic functionalities, such as alkynes, that reside in the “cell-silent region” of the Raman spectrum [9, 10]. Again, these types of probes could be installed either metabolically or enzymatically. To date, a range of MCRs have been developed for the visualization of cell-surface glycoproteins, including those outlined in Fig. 3. Each of these techniques has their own positives and negatives, but all have been successfully applied to the visualization of different classes of glycans as follows.

Chemical methods for the visualization of cell-surface glycans. a Some monosaccharides (e.g., sialic acid) can directly undergo chemoselective reactions for the installation of visualization tags. b Chemical reporters can be incorporated into glycans for subsequent installation of visualization tags using bioorthogonal chemistries. These chemical reporters can be incorporated using cellular metabolism or enzymatic modification of endogenous glycans. c Some chemical reporters can be directly detected using advanced spectroscopic techniques (e.g., stimulated Raman scattering)

Examples of metabolic chemical reporters. A range of chemical reporters have been synthesized for the visualization of cell-surface glycans
Sialic acid
Sialic acids are a class of nine-carbon backbone monosaccharides that are often found at the capping end of many oligosaccharide structures on the cell surface. As mentioned above, the inherent chemical properties of sialic acids allow them to undergo chemoselective reactions for the direct installation of visualization tags. For example, aldehydes can be introduced onto the cell surface using mild periodate oxidation chemistry which selectively oxidizes the polyhydroxy side-chain of already existing sialic acids. This reaction is fast, can occur at a neutral pH, and uses low concentrations of common, inexpensive reagents; however, the subsequent bioorthogonal reaction of aldehydes with hydrazide- and animooxy-visualization tags is typically slow and can be reversible. To overcome this limitation, Paulson and co-workers took advantage of analine-catalyzed oxime ligation (PAL) to increase the reaction efficiency with an aminooxy-biotin tag [11]. They utilized this technique to first specifically label cell-surface sialic acids at low temperature, then track the endocytosis of labeled glycoproteins in the endosomal/lysosomal pathway upon elevation to normal culture temperature, suggesting that PAL can be valuable for the visualization of glycan trafficking [12].
In addition to direct chemical methods, sialic acid residues were the first to be visualized by treating cells with an analog of the metabolic precursor, N-acetylmannosamine (ManNAc), which created the concept of MCRs. Bertozzi and co-workers first demonstrated that a chemically reactive ketone could be incorporated onto the N-acetyl side chain of ManNAc, termed ManLev. When living cells were treated with this compound it was metabolized to the corresponding CMP-sialic acid donor and then incorporated into cell-surface glycans as a sialic acid derivative. The ketone could be subsequently detected using biotin-hydrazide and flow cytometry [13]. While this method was the first to take advantage of the promiscuity of the sialic acid pathway to incorporate ManNAc analogs into cell-surface glycans, the ketone functionality suffers from the same reaction limitations as aldehydes and is not truly bioorthogonal, as there are endogenous keto-metabolites. To overcome this issue, the Bertozzi lab created the Staudinger ligation, which generates an amide bond upon reaction of two totally abiotic groups, an ester-functionalized triarylphosphine and an azide [14]. To take advantage of this reaction, living cells were treated with an azide containing peracetylated ManNAc analog, termed Ac4ManNAz. Following deacetylation of the acetate protecting-groups by endogenous lipases/hydrolases, ManNAz was metabolized and incorporated as SiaNAz onto the cell surface, where it could be readily detected using the Staudinger ligation. Notably, using the same concentrations of reagents, metabolism of Ac4ManNAz and subsequent reaction with a biotin-phosphine probe and incubation with fluorescein isothiocyanate (FITC)-avidin revealed two-fold higher signal compared to that of the ketosugar/hydrazide combination. Incredibly, Prescher et al. then demonstrated that injection of living mice with Ac4ManNAz resulted in incorporation of SiaNAz into a variety of tissues, which could be detected by both ex vivo and in vivo Staudinger reactions with a phosphine FLAG-tag followed by Western blotting, demonstrating cell surface visualization in a living organism for the first time [15]. This experiment set the stage for the application of Ac4ManNAz visualization in human disease, as increases in sialic acid on the cell-surface has been shown to correlate with the malignancy of many cancer types [16], raising the possibility that sialic acid visualization could be used as a non-invasive way to potentially monitor tumor progression and differentiate tumors from healthy tissue. Towards this goal, the Brindle lab injected Ac4ManNAz intraperitoneally into mice with lung carcinoma xenografts and reacted any labeled tissues in vivo with phosphine-biotin, followed by injection of either far-red fluorescent or radionucleotide-labeled avidin [17]. Healthy tissue had significantly lower Ac4ManNAz-dependent signal compared to the xenografts using both imaging technologies, indicated that sialic acid MCRs could be used for the detection of changes in glycosylation and determination of disease prognosis in vivo. Finally, bioorthogonal labeling typically requires the use of excess biotin or fluorescent tag to drive the kinetics of the reaction and ensure completion. This extra tag necessitates a wash-out step to remove unreacted material before addition of avidin reagents or fluorescence visualization. In an effort to sidestep this issue, Bertozzi and co-workers created a “fluorescently silent” probe comprised of a phosphine-tethered fluorophore attached to an intramolecular quencher [18]. Reaction with an azide during the Staudinger ligation liberates the quencher, resulting in fluorescence signal. This technique was applied to live HeLa cells for the visualization of cell surface SiaNAz residues, as well as those internalized by the Golgi.
Despite these key applications, the Staudinger ligation suffers from slow reaction rates and oxidation of the phosphine tags that renders them unreactive. The other available bioorthogonal reaction at the time, Copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) [19, 20] has a increased rate, but displays cytotoxicity. Therefore a new bioorthogonal reaction for dynamic, live-cell imaging was needed. The Bertozzi lab first reported the bioorthogonal reaction between an azide and a non-substituted, strain-activated cyclooctyne, termed strain-promoted azide-alkyne cycloaddition (SpACC) [21]. Unfortunately, this reaction proved to be no faster than the Staudinger ligation. In an attempt to increase the reaction rate of the reaction and therefore sensitivity of azide-detection, various cyclooctyne reagents were developed by both the Bertozzi and Boons lab, including a difluoronated cyclooctyne (DIFO) and a series of dibenzocyclooctynes [22]. Both of these compounds are highly reactive, for example, DIFO was reported to have a second-order rate constant 17–63 times greater in vitro than the rate constants previously reported for the Staudinger ligation and the previously reported, first generation cyclooctyne reagents [23]. Similarly, the use of DIBO, a dibenzyocyclooctyne, increases reactions rates through the enhancement of ring strain, which resulted in an approximately 1000-fold increase in the second order rate constant compared to unactivated cyclooctyne [24, 25]. After 1 h of bioorthogonal reaction, the rate enhancement provided by DIFO resulted in a 20-fold increase in the sensitivity of SiaNAz detection of cells treated with Ac4ManNAz compared to the Staudinger ligation. The dramatically improved reaction rate of DIFO was then used to achieve dual color time-lapse imaging of cell-surface glycosylation. Specifically, cells previously labeled with Ac4ManNAz were subjected to SpACC with DIFO-488 before being re-incubated with Ac4ManNAz to install additional SiaNAz residues. Subsequent reaction with a red-shifted DIFO-568 revealed the DIFO-488-conjugated population had migrated almost entirely to the lysosomes while the DIFO-568-population was present on the cell surface, indicating that metabolic labeling does not grossly alter glycan trafficking on the time scale of the experiment and demonstrating that improved bioorthogonal reactions in combination with MCRs provide a means to observe the kinetics of glycan internalization. Interestingly, Boons and co-workers took advantage of the improved reaction rate to monitor glycan localization and movement as it relates Niemann-Pick type C (NPC) disease. NPC is characterized by mutations in NPC-1 and NPC-2 proteins that normally mediate the passage of cholersterol from the lysosomes. Build up of cholesterol in this manner within neurons and other cell types, results in neurodegeneration and hepatosplenomegaly [26]. Remarkably, treatment of either NPC-1 null or NPC-2 deficient fibroblasts with Ac4ManNAz or Ac4GalNAz, followed by SpAAC with DIBO-biotin and subsequent visualization using streptavidin-AlexaFluor showed a disease-specific accumulation of a large set of glycans within endocytic compartments. Furthermore, using the endocytic uptake marker dextran, they were able to show that endosomal build up of glycoproteins in NPC cells is likely due to impaired recycling [27]. Increasing evidence has also linked glycosylation to a variety of pathological and physiological processes in the heart. Specifically, it has been shown that N-linked glycans, mucin-type O-linked glycans and sialic acids are required for regulating the electric cardiac signaling and heart rhythm [28]; however, visualization of such glycosylation events has been challenging. The Chen lab took advantage of SpAAC with another cyclooctyne-tag, azadibenzocyclooxtyen (DBCO), to visualize sialic acid modification in whole, intact rat hearts [29]. Specifically, rats were treated with Ac4ManNAz for 7 days at which time whole hearts were isolated, subjected to Langendorff-perfusion to permit the visualization and manipulation of intact hearts without the complications involved in whole-animal experiments. Upon close inspection, intense fluorescent signal was observed at the cell-cell junctions, in-line with previous reports of sialylated glycans, and also at the transverse tubule (T-tubule) networks that are responsible for synchronizing cell-wide excitation and excitation-contraction coupling. Furthermore, their studies showed an increase in the biosynthesis of sialic acid after rat hearts had been exposed to cardiac hypertrophy.
The MCRs used for visualization of sialic acids discussed thus far have been functionalized solely at the N-acetyl side chain of the metabolic precursor N-acetylmannosamine, resulting in the incorporation of an abiotic azide at the C5 position of the sialic acid product. Although the fate of these MCRs are now well characterized, less work has been invested in the metabolic fate of other substituted ManNAc analogs. Recently, for example, Hackenberger and co-workers investigated the C4-substituted ManNAc analog, termed Ac3-4-azido-ManNAc and its incorporation into glycan structures [30]. Interestingly, they found that Ac3-4-azido-ManNAc was being metabolically accepted by the biosynthetic machinery and was converted into the C7-azide of the resulting sialic acid analog. Remarkably, treatment with PNGase F, an enzyme that specifically cleaves N-glycans resulted in no loss of labeling, indicating that Ac3-4-azido-ManNAc was being specifically incorporated in to sialic acids of O-glycosylated proteins. To visualize the sialic residues of O-glycosylated cell surface proteins in live zebrafish, organisms were injected with Ac3-4-azido-ManNAc at 24 h post fertilization (hpf) and visualized following SpAAC with AlexaFluor 488-DIBO at 72 hpf. Interestingly, strong labeling was seen in regions of the central nervous system.
Unfortunately, first generation cyclooctyne SpAAC reagents suffer from poor water solubility, limiting their use in biological environments. To overcome this, the chemical structure of SpAAC reagents can be fine-tuned. For example, the Bertozzi lab synthesized 6,7-dimethoxyazacyclooct-4-yne (DIMAC), which contains a nitrogen atom within the ring structure as well as two methoxy-substituents, interrupting the hydrophobic surface resulting in improved polarity and water solubility [31]. In a similar fashion, refining of dibenzocyclooctyne structure can aide in improving the reactivity without compromising the stability of the reagent. To this end, Bertozzi and co-workers incorporated an amide within the ring structure, reasoning that an amide has double bond character due to its resonance structures [32]. The resulting bizarylazacyclooctyne termed BARAC, resulted in high signal-to-noise at nanomolar concentrations, therefore eliminating the need for extensive washing steps after reaction. When compared in cell-labeling experiments following treatment with Ac4ManNAz, BARAC-biotin showed 10-fold higher signal after 1 min compared to DIFO- and DIBO-biotin reagents, consistent with BARACs ~12-fold higher rate constant. Further improving on this chemistry, a click-activated fluorogenic coumarin-conjugated-BARAC (coumBARAC) was later developed [33]. Later, Boons and co-workers improved the solubility of DIBO by developing a polar, highly sulfated version, termed S-DIBO, that was subsequently found to be selective for only cell-surface modification as it cannot pass through the cell membrane [34].
As described, azide-containing MCRs have been extensively explored; however, Wong and co-workers were the first to develop an alkyne-modified ManNAc analog called N-4-pentynoylmannosamine (Ac4ManNAlk), for visualization of fixed cells using CuAAC [35]. As the cellular sialic acid biomachinery has been shown to tolerate variations at the N-acetate position of ManNAc up to five carbons in length [36, 37], Hsu et al. reasoned that Ac4ManNAlk could be metabolically incorporated and transformed in to SiaNAlk residues displayed on the cell surface. Indeed, treatment of Hep3B cells with Ac4ManNAlk followed by standard CuAAC with a fluorogenic azido-hydroxycoumarin probe resulted in a 15-fold increase in labeling over background and required less reagent for efficient labeling when compared to azido ManNAc. To expand the metabolic labeling strategy, Chen and co-workers synthesized sialic acid analogs with two bioorthogonal moieties, allowing for dual-color labeling or enrichment of sialic acid binding proteins through a cross-linker [38]. To this end, two bifunctional unnatural sialic acids were synthesized, 9AzSiaNAlk and 9AzSiaDAz which contain an azide at the C9 position and an alkyne or diazirine photo-cross-linker on the N-acyl side chain. Notably, a sialic acid bearing only the cross-linker had been previously used to analyze glycoprotein interactions [39]. In an elegant proof-of-principle experiment, the dimerization of CD22 (sialic acid-binding immunoglobulin-like lectin 2) was captured using photo-crosslinking. Subsequent reaction of cell lysates with an alkyne-biotin tag allowed for the enrichment of CD22. Analysis by Western blot using an anti-CD22 antibody revealed photo-crosslinked CD22 in 9AzSiaDAz treated cells while only monomeric CD22 was enriched in 9AzSia treated cells. Furthermore, 9AzSiaNAlk can be utilized for two-color and FRET-imaging.
Although improvements were achieved with the transition from the Staudinger ligation to SpACC, alternative chemistries have recently been explored to further enhance reaction rates. For example, tetrazines are known to react irreversibly with a variety of alkenes and alkynes through inverse electron-demand Diels-Alder (IED-DA) reactions. To increase the rate of IED-DA for its use in a bioorthogonal setting, examination of various reaction partners have been explored. For example, using the highly strained olefin trans-cyclooctene resulted in a second order rate constant in aqueous solution of 6000 M−1 s−1, orders of magnitude higher than SpACC and CuACC [40]. Additionally, tetrazines are inherently fluorogenic, capable of quenching several common fluorescent probes showing a significant “turn-on” signal after reaction through IED-DA, therefore lowering background caused by nonspecific binding or accumulation making them beneficial for intracellular imaging where removal of excess reagents can be problematic. Devaraj et al. first utilized tetrazines in combination with a trans-cyclooctene to visualize EGFR on the surface of live A549 lung carcinoma cells by employing an cyclooctene-conjugated EGFR-antibody followed by reaction with a tetrazine fluorophore [40]. Unfortunately, metabolic incorporation demands small functional groups, limiting the use of the large cyclooctene. To overcome this, methylcyclopropenes were explored as a smaller alternative for live cell imaging. For example, methylcyclopropene-tagged phospholipids were shown to label live human breast cancer cells, opening up the possibility for the incorporation of methylpropene moieties onto MCRs [41]. Due to their small and abiotic nature, it was reasoned that methylcyclopropenes would be useful in cellular labeling studies as modified sialic acids are known to be metabolized and incorporated on to the cell-surface. To this end, a methylcyclopropene-sialic acid conjugate, termed 9-Cp-NeuAc, was synthesized and labeling of cells was confirmed by flow cytometry. Interestingly, to investigate whether cyclopropene- and azido-sugars could be used together for live-cell labeling studies, 9-Cp-NeuAc and 9-Az-NeuAc were successfully used in tandem demonstrating IED-DA and SpACC chemistries could be used simultaneously [42]. Further building on this idea, the Devaraj lab incorporated methylcyclopropene into an N-acyl mannosamine analog termed Ac4ManNCyc [43]. Later, the Prescher lab introduced a N-cyclopropenyl carbamate linked version called Ac4ManCCp, which provided enhanced cellular fluorescence by flow cytometry [44]. Additionally, Wittmann and co-workers demonstrated that the alkene-modified derivative, Ac4ManPtl, could also be metabolized to sialic acid residues and visualized using fluorescent tetrazines [45]. More recently, Ac4ManCCp has been used simultaneously with Ac4GlcNAz (see Mucins section below) for the visualization of sialic acid and core mucin O-linked glycosylation [46].
Unfortunately, all cell-types with sialic acid residues are labeled following treatment with an MCR limiting their use for cell- or tissue-specific visualization. To overcome this issue, Bertozzi and co-workers developed a strategy in which ManNAz was delivered and enzymatically released in a cell-specific manner [47]. Specifically, they set out to selectively label cancer cells that overexpress a cancer specific prostate-specific antigen (PSA) protease compared to healthy tissue. To accomplish this, ManNAz was modified at its 6-hydroxyl with a hexapeptide-PSA substrate by way of a p-aminobenzyl alcohol linker. Following cleavage of the peptide substrate by PSA protease, ManNAz is liberated and metabolically incorporated into the immediately surrounding cells resulting in selective cell-surface labeling was as visualized using SpACC with DIFO reagents. Another method for the specific labeling of cell-types is to take advantage of specific ligand/receptor binding. For example, Chen and co-workers demonstrated that 9-azido sialic acid (9-AzSia) could be caged in ligand-displaying liposomes [48]. Termed ligand-targeted liposomal delivery, recognition of the liposomal folate ligands by cellular folate receptors promotes the endocytosis and intracellular delivery of 9-AzSia acid in a cell specific manner. This is particularly important because folate receptors have been shown to be overexpressed in a variety of cancers. Furthermore, liposomal delivery opened the door for the introduction of many different ligands that target numerous receptors for cell-specific delivery of MCRs for glycan labeling. In fact, recent work by the Chen lab has extended this technique for the visualization of sialylated glycans in living mice with melanoma xenografts, which are known to overexpress integrin αVβ3 [49]. Similar to above, 9-AzSia was encapsulated in a liposome bearing an integrin αVβ3 cyclic pentapeptide targeting ligand on its surface for its specific delivery, endocytosis and subsequent visualization. The dynamic changes in glycan synthesis within the tumors were observed over a 21-day period.
Even in these cell-specific labeling experiments, treatment of cells with MCRs results in the non-specific incorporation of analogs into the large variety of cell-surface glycans, offering no glycoprotein specific information. To gain another level of specificity and to probe at the specific glycoprotein level, intramolecular FRET probes have been developed that involve intra-protein FRET transfer from an MCR-installed donor molecule to the acceptor molecule on a protein of interest. Several methods to install the FRET acceptor onto the protein of interest have been explored, including genetic tagging with green fluorescent protein (GFP) [50]. Specifically, following protein GFP-tagging and expression, treatment of cells with Ac4ManNAz results in the display of SiaNAz residues. Following SpACC click chemistry with a fluorescent tag, FRET signal between the GFP-tagged protein and the newly installed fluorophore is observed, allowing for protein-specific visualization. As a proof-of-principle, this strategy was used to visualize sialylated GLUT4 over time, demonstrating that following insulin stimulation GLUT4 accumulates at the cell-surface and is subsequently internalized after insulin removal [50]. Unfortunately, this technique is hampered by low FRET efficiency due to the spacial separation of the FRET donor/acceptor through the membrane. Additionally, the large size of GFP limits its use to the tagging of cytosolic tails of proteins as fusion of GFP to the extracellular end can result in difficulty in protein expression and translocation [51]. An alternative FRET based method was therefore developed in which the FRET donor/acceptor pair are located on the same side of the membrane to enhance signal [52]. The FRET acceptor was installed using CuAAC and the MCR Ac4ManNAlk to incorporate SiaNAlk residues on the cell surface. The FRET donor is installed by first genetically fusing the protein of interest to a 13-amino acid peptide called LpIA Acceptor Peptide (LAP). A mutant engineered enzyme from E. Coli called lipoic acid ligase (LpIA) then recognizes the LAP and ligates an azide-containing lipoic acid derivative. Subsequent CuAAC with an alkyne-FRET acceptor allows for glycoprotein-specific visualization. This technique was used for the visualization of integrin αXβ2 glycoforms and revealed a ~40 % increase in FRET efficiency when compared to the transmembrane/GFP-FRET method.
Unfortunately, the scope of these FRET techniques is limited to proteins that can be genetically tagged and successfully expressed, thereby excluding endogenous glycoproteins. This can be overcome through the use of fragment-antigen binding (Fab)-delivered FRET probes. Again, the FRET acceptor is installed by metabolic labeling with Ac4ManNAz and subsequent click chemistry with a cyclooctyne-conjugated FRET acceptor. Next, the FRET donor is selectively delivered to the protein of interest using a protein-specific small fragment-antigen binding (Fab) domain [53]. Specifically, Bertozzi and co-workers demonstrated this technique in both cell culture and tissue samples from human adenocarcinomas to visualize integrin αVβ3, a cell-adhesion protein that is known to be upregulated in prostate cancer.
Although synthetic mannosamine analogs have shown a great deal of success in live-cell visualization, their incorporation relies on their ability to be converted into their sialic acid products. To avoid this potential bottleneck, the direct metabolic and chemoenzymatic installation of sialic acid analogs has been explored [37]. Notaby, sialic acid analogs with relatively bulky groups at the C-9 position, including aryl-azide for photo-crosslinking applications [54], can be metabolically incorporated into cells. Building upon these data, a fluorescein-isothiocyanate (FITC) substituted sialic acid (FITC-SA) was synthesized and intravenously injected into the tail vein of mice containing hepatocellular carcinoma xenografts. Interestingly, FITC-SA was shown to selectively illuminate tumorous tissue with high signal-to-noise and a resolution that is significantly below the minimal residual cancer limits pursued by surgeons [55]. Unfortunately, FITC-SA exhibits green fluorescence which has limited tissue penetration and suffers from a short in vivo lifetime making it difficult to visualize during a surgical procedure. To address these shortcomings, a pH-activatable, near-infrared profluoregenic sialic acid probe (SA-pNIR) was subsequently developed [56]. During cell-surface glycoprotein turnover, SA-pNIR containing glycans are relocalized to the acidic lysosome, where the dye becomes fluorescent. In addition to displaying long-term retention by tumors, compared to FITC-SA, SA-pNIR converts NIR irradiation into heat, causing cell death, which could be useful in tumor-specific visualization and ablation. Chemoenzymatic methods have also been developed that take advantage of the inherent specificity of a recombinant sialyltransferase to deliver sialic acid-chemical reporters to the cell surface of living cells for subsequent reaction with a visualization tag. For example, Boons and co-workers utilized a recombinant rat ST6Gal I sialyltransferase to transfer CMP-Neu6Ac9N3 to N-linked glycoconjugates on the cell surface of human skin fibroblasts. SpAAC with S-DIBO-biotin and incubation with an Alexafluor-streptavidin conjugate inabled the specific visualization of cell surface N-linked glycans over time [57].
Traditional labeling using chemical reporters involves a two-step procedure in which a bioorthogonal moiety is first installed by metabolic incorporation through the endogenous cellular machinery. In a second step, visualization is then achieved by reaction with a bioorthogonal fluorescent probe. While this method has been fruitful in numerous aspects, its success is dependent upon not only the incorporation of abiotic chemical groups small enough to be tolerated by the cell but also the rate and efficiency of the secondary bioorthogonal reaction. Therefore methods that allow for the direct visualization of unnatural glycans within cells without further derivatization are an attractive alternative. Raman spectroscopy uses Raman scattering to detect specific vibrational signals of molecules. Common chemical reporters contain abiotic functional groups such as the azide and the alkyne that possess unique Raman scattering compared to the multitude of small molecules found within the cell making them inertly bioorthogonal with Raman scattering at around 2100 cm−1 [58]. Passive Raman spectroscopy requires high concentrations of functional groups in cells because of the intrinsic weakness of the signal. Chen and co-workers overcame this obstacle by using surface-enhanced Raman spectroscopy (SERS). Specifically, gold plasmonic nanoparticles (AuNPs) were used as the Raman signal enhancer and were decorated with 4-mercaptophenylboronic acids (MPBA) that show high affinity for sialic acids. Following treatment with Ac4ManNAlk, and incubation with MPBA-AuNPs, incorporated SiaNAlk residues can be easily distinguished in signal from endogenous sialic acids and visualized using dark-field microscopy. This method is not limited by the constraints of bioorthogonal chemistry and thus enabled its expansion to include other unnatural mannosamine derivatives including peracetylated N-(3-cyano-propanoyl)mannosamine (Ac4ManNCy) and n-trideutero-acetylmannosamine ([D3]Ac4ManNAc) [58]. Unfortunately, although SERS simplifies traditional labeling methods by eliminating the second visualization tag, the use of metallic nanoparticle substrates to enhance the Raman signal limits the use of the technique for intracellular glycan tracking and complicates the procedure. Later work by the Chen lab demonstrated that this could be overcome by using stimulated Raman scattering (SRS) microscopy that utilizes two beams to detect the vibrational frequency of the alkyne, eliminating the use for additional materials [59]. This improved Raman technique was used with Ac4ManNAlk to visualize cell-surface modification of K20 cells.
Mucins
Mucin type O-linked glycosylation is the most abundant type of cell-surface O-linked glycosylation and is characterized by the linkage of α-N-acetyl-galactosamine (GalNAc) to serine and threonine side chains. The core GalNAc residue of mucins is installed by enzymes called polypeptide N-acetyl-α-galactosaminyltransferases (ppGalNAcTs) by way of a high-energy UDP-donor sugar, UDP-GalNAc, in the Golgi. After this initial modification event, the core GalNAc residue is typically elaborated by other monosaccharides to generate an oligosaccharide chain. Like sialic acids, the labeling of core GalNAc residues can be achieved through metabolic incorporation of unnatural GalNAc residues using MCRs. Bertozzi and co-workers were the first to demonstrate that an azide-containing GalNAc, termed Ac4GalNAz, could be incorporated into the core position of mucins in living cells [60]. Its presence was successfully detected by flow cytometry following the Staudinger ligation with phosphine-FLAG and subsequent incubation with a FITC-conjugated antibody. While monitoring glycosylation in cell culture is useful, many physiologically relevant changes in glycosylation can be attributed to small changes in a tissue microenvironment. Thus, labeling of mucin modification of cells in culture with GalNAz was soon expanded to the labeling of live organisms. To this end, Ac4GalNAz was injected into live mice for 7 days at which time spleens and other organs were harvested and were probed for mucin modification ex vivo using the Staudinger reaction with phosphine-FLAG [61]. Interestingly, harvested organs exhibited differential labeling, and specifically, splenocytes from Ac4GalNAz treated mice displayed two distinct populations consisting of high- or low-labeling efficiency and it was later elucidated that the glycoproteins of B-cells in mice are preferentially labeled over those of T-cells, allowing the use of GalNAz for the targeting of specific splenocyte populations.
Labeling cells using GalNAz selects for mucins that are newly synthesized, which allows for a unique snapshot of not only glycan structures but also the activity of the biosynthetic machinery. Therefore, the changes in incorporation of Ac4GalNAz into glycans on the cell surface can serve as a readout of different cellular states. Taking advantage of these properties, Laughlin et al. sought to monitor such changes in mucin glycosylation during the developmental stages of zebrafish [62]. First, zebrafish were treated with Ac4GalNAz to label their cell-surface glycans starting at 3 hpf. Subsequent reaction with a DIFO-fluorescent tag yielded robust signal, even after one minute of reaction. Longer time course experiments lasting up to 72 hpf resulted in a strong fluorescence intensity around the jaw region, pectoral fins and olfactory organs in as little as 24 hpf. To further resolve distinct populations of glycans during the stages of development, two- and three-color detection experiments in which Ac4GalNAz treatments were administered at different times using different DIFO dyes were conducted. Remarkably, visualization of distinct zones of de novo glycan synthesis was achieved that would have otherwise gone unobserved with conventional visualization methods. Soon after, Ac4GalNAz labeling was applied to Caenorhabditis elegans, another common model organism [63]. The worms were treated with the MCR to visualize glycans during three stages of development. Importantly, while single color labeling in adult hermaphrodite worms revealed concentrated areas of glycans during development, it did not give insight into newly synthesized glycans that are needed to study their dynamics. Dual color labeling with Ac4GalNAz 12 h apart followed by SpAAC with DIFO-dyes resulted two populations of glycans representing the two treatment periods. Worms in the second stage of development contained about equal amounts of the new and old glycoprotein populations, while worms that were in the adult stages showed pronounced labeling pharynx region, which is known to have motive capabilities, indicating that O-linked mucins could play a role in the lubrication required for these processes.
Fucose
Fucose is a six-carbon deoxyhexose, a monosaccharide that is commonly present at the terminal end of N- and O-linked glycans and glycolipids synthesized by mammalian cells. Like sialic acids, fucosylation has also been implicated in cell-cell and cell-pathogen interactions and fucose levels are usually elevated during development [64]. Fucose is added onto glycans by enzymes called fucosyltransferases through an activated GDP-fucose. The labeling of fucosylated proteins takes advantage of endogenous biosynthetic enzymes in the fucose salvage pathway to incorporate fucose MCRs. For example, similar to analogs of mannosamine and galactosamine previously described, a modified azido-fucose analog was synthesized by Wong and co-workers. They confirmed that the subsequently formed GDP-activated azido-fucose donor sugar (FucAz) was accepted by human-1,3-fucosyltransferases (II-VII) in vitro and was subsequently demonstrated to be metabolically accepted by live cells and incorporated onto cell surface fucosylated glycans. Fucose modified cell-surface oligosaccharides of fixed cells were visualized following CuAAC with an alkyne-containing fluorescent tag [65]. Simultaneously, the Bertozzi lab published the development of additional 2-, 4- and 6-substituted fucose analogs and determined that 2- and 4-azido fucose were not metabolically incorporated, while 6-azido fucose showed labeling over background but was also unfortunately cytotoxic at high concentrations in cell culture, limiting its use [66]. Interestingly, an C-5 alkyne containing fucose analog (FucAlk) introduced by the Wong lab [35] was later shown to exhibit far less cytotoxicity compared to FucAz, and visualization studies of fixed cells using alkynyl fucose resulted in less background signal compared to FucAz under CuAAC conditions. Wu and co-workers then extended this technique to visualize the developmental cycles of zebrafish whose growth is known to be accompanied by distinct changes in the glycan structure profile [62]. Using chemoenzymatically synthesized GDP-FucAlk, zebrafish embryos were microinjected at the one-cell stage before being subjected to CuAAC with a fluorescent probe [67]. Remarkably, the development of a biocompatible Cu(I)-complex, bis(tert-butyltriazoly) ligand (BTTES), which prevents cell permeability of the coordinated copper, allowed for the use of copper catalyzed click chemistry in the presence of living cells and organisms. In as little as 2.5 hpf, the visualization of fucosylated glycans within the enveloping layer was observed. Following this study, Bertozzi and co-workers utilized GDP-FucAz and SpAAC for the visualization of fucosylated cell-surface glycans in zebrafish during development. In addition, this study also tested the labeling efficiency of synthetic FucAz-1-phosphate, the metabolic precursor to GDP-FucAz and revealed that although zebrafish fucosyltransferases accept GDP-FucAz, FucAz-1-phosphate did not result in labeling, indicating that the salvage-pathway biosynthetic enzymes are not tolerant the C-6 azido modification [68]. Improving upon their initial chemistry, the Wu lab recently published the use of GDP-FucAlk with a rate accelerating, chelating azide-assisted CuAAC reaction that allowed for the visualization of fucosylated proteins in zebrafish development at the two-cell stage for the first time [69]. It is worth noting that similar reagents containing an internal Cu(I)-chelating site within the azide partner have been utilized to effectively increase the copper concentration at the site of reaction allowing the use of less copper catalyst. Developed by the Ting lab, these picolyl azide reagents have recently been used to label transmembrane LAP-neurexin-1B proteins on live HEK cells as well as LAP-neuroligin-1 on the surface of living rat hippocampal neurons following installment of the alkyne by an E. coli derived lipoic acid lipase [70].
In addition to global metabolic labeling experiments, fucose analogs have been used to probe the presence of particular glycans on the cell surface by taking advantage of chemoenzymatic modification. These experiments used a H. pylori-1,3-fucosyltransferase that catalyzes the transfer of fucose from GDP-fucose to the 3-hydroxy of GlcNAc in LacNAc, a ubiquitous cell-surface glycan present in vertebrates, enveloped viruses, and certain pathogenic bacteria. The resulting FucAz on the surface of the living cells was then visualized using SpAAC with an fluorescent cyclooctyne (DIFO) [71]. This method allowed for the discrimination of LacNAc levels on the surface of mouse splenocytes, indicating that the degree of unmodified LacNAcylation correlates with a cell’s activation status. The use of the chemoenzymatic modification also allows for the observation of specific glycan structures, such as those upregulated in cancer, presenting a new strategy for biomarker detection. Specifically, the Hsieh-Wilson lab utilized a bacterial homologue of the human blood group A antigen glycosyltransferase (BgtA), to catalyze the transfer of an azide- or ketone-bearing GalNAc residue onto the C3 position of the glycan fucose-(α1-2)-galactose, a glycan known to be upregulated in cancer [72]. This technique was used to successfully label and visualize live MCF-7 cells after SpAAC. Furthermore, this technique was utilized to assay the levels of Fuc-(α1-2)-Gal in a panel of cancer and non-cancer cell lines by flow cytometry. Notably LnCAP cells (prostate cancer) showed a 53 % increase in display of Fuc-(α1-2)-Gal compared to healthy tissue.
Conclusions and future outlook
Chemical methods have been uniquely enabling for the visualization of glycans in living systems. In particular, the development of metabolic chemical reporters has fundamentally changed the types of imaging and characterization experiments that can be performed. Through the creation of ever improved bioorthogonal reactions, this method has moved from the analysis of glycans on cells in culture to ex vivo and in vivo applications in a variety of organisms. The more recent development of stimulated Raman scattering, lays the groundwork for the development of imaging systems that no longer require the bioorthogonal attachment of visualization tags. Despite the great progress that has been made in these areas, several challenges remain. None of the direct visualization techniques are completely specific for one type of glycoprotein, nor do they necessarily exclude the labeling of other glycoconjugates (i.e., glycolipids). For example, oxidation of sialic acid residues generates aldehyde reactivity on both N-linked and mucin O-linked glycoproteins, as well as sialic acid containing glycolipids. Any subsequent visualization will necessarily be a readout on this heterogeneous mixture of glycans. MCRs are likely to suffer from this same issue, and we and others have also demonstrated that the majority of MCRs for glycosylation are also not selective for one type of carbohydrate [73–76]. For example, treatment with Ac4GalNAz can result in the labeling of certain glycophosphatidylinositol (GPI) anchor-modified proteins [77]. Not necessarily surprising, it now also appears that the majority of N-acetyl-modified MCRs (e.g., GalNAz) can be metabolically interconverted to other monosaccharides, resulting in at least some “off-target” labeling. We recently demonstrated that a different MCR, 6-azido-6-deoxy-GlcNAc, is highly selective for intracellular O-GlcNAc modifications [76], which should encourage the continued development and characterization of new MCRs that may be selective for another type of glycosylation. Simultaneous to continued work on chemical reporters, future effort should also continue to focus on the development of improved bioorthogonal reactions and visualization tags that can improve in vivo labeling and imaging in mammals (e.g., positron electron tomography). Finally, it is also possible that direct imaging techniques, such as stimulated Raman scattering, could be improved to the point where the requirement for the bioorthogonal attachment of imaging tags would be obviated for in vivo experiments.
References
Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E. (eds.): Essentials of Glycobioloy. Cold Spring Harbor Laboratory Press, Cold Spring Harbor (2009)
Helenius, A., Aebi, M.: Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 (2004)
Schwarz, F., Aebi, M.: Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 21, 576–582 (2011)
Hang, H.C., Bertozzi, C.R.: The chemistry and biology of mucin-type O-linked glycosylation. Bioorg. Med. Chem. 13, 5021–5034 (2005)
Rana, N.A., Haltiwanger, R.S.: Fringe benefits: functional and structural impacts of O-glycosylation on the extracellular domain of Notch receptors. Curr. Opin. Struct. Biol. 21, 583–589 (2011)
Grammel, M., Hang, H.C.: Chemical reporters for biological discovery. Nat. Chem. Biol. 9, 475–484 (2013)
Patterson, D.M., Nazarova, L.A., Prescher, J.A.: Finding the right (bioorthogonal) chemistry. ACS Chem. Biol. 9, 592–605 (2014)
Chuh, K.N., Pratt, M.R.: Chemical methods for the proteome-wide identification of posttranslationally modified proteins. Curr. Opin. Chem. Biol. 24, 27–37 (2015)
Wei, L., Hu, F., Shen, Y., Chen, Z., Yu, Y., Lin, C.-C., Wang, M.C., Min, W.: Live-cell imaging of alkyne-tagged small biomolecules by stimulated Raman scattering. Nat. Methods 11, 410–412 (2014)
Hong, S., Lin, L., Xiao, M., Chen, X.: Live-cell bioorthogonal Raman imaging. Curr. Opin. Chem. Biol. 24C, 91–96 (2015)
Dirksen, A., Hackeng, T.M., Dawson, P.E.: Nucleophilic catalysis of oxime ligation. Angew. Chem. Int. Ed. Engl. 45, 7581–7584 (2006)
Zeng, Y., Ramya, T.N.C., Dirksen, A., Dawson, P.E., Paulson, J.C.: High-efficiency labeling of sialylated glycoproteins on living cells. Nat. Methods 6, 207–209 (2009)
Mahal, L.K., Yarema, K.J., Bertozzi, C.R.: Engineering chemical reactivity on cell surfaces through oligosaccharide biosynthesis. Science 276, 1125–1128 (1997)
Saxon, E., Bertozzi, C.: Cell surface engineering by a modified Staudinger reaction. Science 287, 2007–2010 (2000)
Prescher, J., Dube, D., Bertozzi, C.: Chemical remodelling of cell surfaces in living animals. Nature 430, 873–877 (2004)
Hedlund, M., Ng, E., Varki, A., Varki, N.M.: 2-6 linked sialic acids on N-glycans modulate carcinoma differentiation in vivo. Cancer Res. 68, 388–394 (2008)
Neves, A.A., Stockmann, H., Harmston, R.R., Pryor, H.J., Alam, I.S., Ireland-Zecchini, H., Lewis, D.Y., Lyons, S.K., Leeper, F.J., Brindle, K.M.: Imaging sialylated tumor cell glycans in vivo. FASEB J. 25, 2528–2537 (2011)
Hangauer, M., Bertozzi, C.: A FRET-based fluorogenic phosphine for live-cell imaging with the Staudinger ligation. Angew. Chem. Int. Ed. 47, 2394–2397 (2008)
Tornøe, C.W., Christensen, C., Meldal, M.: Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 67, 3057–3064 (2002)
Rostovtsev, V.V., Green, L.G., Fokin, V.V., Sharpless, K.B.: A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 41, 2596–2599 (2002)
Agard, N., Prescher, J., Bertozzi, C.: A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 126, 15046–15047 (2004)
Jewett, J.C., Bertozzi, C.R.: Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 39, 1272–1279 (2010)
Baskin, J., Prescher, J., Laughlin, S., Agard, N., Chang, P., Miller, I., Lo, A., Codelli, J., Bertozzi, C.: Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. U. S. A. 104, 16793–16797 (2007)
Ning, X., Guo, J., Wolfert, M., Boons, G.: Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angew. Chem. Int. Ed. 47, 2253–2255 (2008)
Mbua, N.E., Guo, J., Wolfert, M.A., Steet, R., Boons, G.-J.: Strain-promoted alkyne-azide cycloadditions (SPAAC) reveal new features of glycoconjugate biosynthesis. ChemBioChem 12, 1912–1921 (2011)
Rosenbaum, A.I., Maxfield, F.R.: Niemann-Pick type C disease: molecular mechanisms and potential therapeutic approaches. J. Neurochem. 116, 789–795 (2011)
Mbua, N.E., Flanagan-Steet, H., Johnson, S., Wolfert, M.A., Boons, G.-J., Steet, R.: Abnormal accumulation and recycling of glycoproteins visualized in Niemann-Pick type C cells using the chemical reporter strategy. Proc. Natl. Acad. Sci. U. S. A. 110, 10207–10212 (2013)
Montpetit, M.L., Stocker, P.J., Schwetz, T.A., Harper, J.M., Norring, S.A., Schaffer, L., North, S.J., Jang-Lee, J., Gilmartin, T., Head, S.R., Haslam, S.M., Dell, A., Marth, J.D., Bennett, E.S.: Regulated and aberrant glycosylation modulate cardiac electrical signaling. Proc. Natl. Acad. Sci. U. S. A. 106, 16517–16522 (2009)
Rong, J., Han, J., Dong, L., Tan, Y., Yang, H., Feng, L., Wang, Q.-W., Meng, R., Zhao, J., Wang, S.-Q., Chen, X.: Glycan imaging in intact rat hearts and glycoproteomic analysis reveal the upregulation of sialylation during cardiac hypertrophy. J. Am. Chem. Soc. 136, 17468–17476 (2014)
Möller, H., Böhrsch, V., Bentrop, J., Bender, J., Hinderlich, S., Hackenberger, C.P.R.: Glycan-specific metabolic oligosaccharide engineering of C7-substituted sialic acids. Angew. Chem. Int. Ed. 51, 5986–5990 (2012)
Sletten, E.M., Bertozzi, C.R.: A hydrophilic azacyclooctyne for Cu-free click chemistry. Org. Lett. 10, 3097–3099 (2008)
Jewett, J.C., Sletten, E.M., Bertozzi, C.R.: Rapid Cu-free click chemistry with readily synthesized biarylazacyclooctynones. J. Am. Chem. Soc. 132, 3688–3690 (2010)
Jewett, J.C., Bertozzi, C.R.: Synthesis of a fluorogenic cyclooctyne activated by Cu-Free click chemistry. Org. Lett. 13, 5937–5939 (2011)
Friscourt, F., Ledin, P.A., Mbua, N.E., Flanagan-Steet, H.R., Wolfert, M.A., Steet, R., Boons, G.-J.: Polar dibenzocyclooctynes for selective labeling of extracellular glycoconjugates of living cells. J. Am. Chem. Soc. 134, 5381–5389 (2012)
Hsu, T.-L., Hanson, S.R., Kishikawa, K., Wang, S.-K., Sawa, M., Wong, C.-H.: Alkynyl sugar analogs for the labeling and visualization of glycoconjugates in cells. Proc. Natl. Acad. Sci. U. S. A. 104, 2614–2619 (2007)
Keppler, O.T., Horstkorte, R., Pawlita, M., Schmidt, C., Reutter, W.: Biochemical engineering of the N-acyl side chain of sialic acid: biological implications. Glycobiology 11, 11R–18R (2001)
Luchansky, S.J., Goon, S., Bertozzi, C.R.: Expanding the diversity of unnatural cell-surface sialic acids. ChemBioChem 5, 371–374 (2004)
Feng, L., Hong, S., Rong, J., You, Q., Dai, P., Huang, R., Tan, Y., Hong, W., Xie, C., Zhao, J., Chen, X.: Bifunctional unnatural sialic acids for dual metabolic labeling of cell-surface sialylated glycans. J. Am. Chem. Soc. 135, 9244–9247 (2013)
Tanaka, Y., Kohler, J.: Photoactivatable crosslinking sugars for capturing glycoprotein interactions. J. Am. Chem. Soc. 130, 3278–3279 (2008)
Devaraj, N., Upadhyay, R., Haun, J., Hilderbrand, S., Weissleder, R.: Fast and sensitive pretargeted labeling of cancer cells through a tetrazine/trans-cyclooctene cycloaddition. Angew. Chem. Int. Ed. Engl. 48, 7013–7016 (2009)
Yang, J., Šečkutė, J., Cole, C.M., Devaraj, N.K.: Live-cell imaging of cyclopropene tags with fluorogenic tetrazine cycloadditions. Angew. Chem. Int. Ed. Engl. 51, 7476–7479 (2012)
Patterson, D.M., Nazarova, L.A., Xie, B., Kamber, D.N., Prescher, J.A.: Functionalized cyclopropenes as bioorthogonal chemical reporters. J. Am. Chem. Soc. 134, 18638–18643 (2012)
Cole, C.M., Yang, J., Šečkutė, J., Devaraj, N.K.: Fluorescent live-cell imaging of metabolically incorporated unnatural cyclopropene-mannosamine derivatives. ChemBioChem 14, 205–208 (2013)
Patterson, D.M., Jones, K.A., Prescher, J.A.: Improved cyclopropene reporters for probing protein glycosylation. Mol. BioSyst. 10, 1693 (2014)
Niederwieser, A., Späte, A.-K., Nguyen, L.D., Jüngst, C., Reutter, W., Wittmann, V.: Two-color glycan labeling of live cells by a combination of Diels-Alder and click chemistry. Angew. Chem. Int. Ed. 52, 4265–4268 (2013)
Späte, A.-K., Bußkamp, H., Niederwieser, A., Schart, V.F., Marx, A., Wittmann, V.: Rapid labeling of metabolically engineered cell-surface glycoconjugates with a carbamate-linked cyclopropene reporter. Bioconjug. Chem. 25, 147–154 (2014)
Chang, P.V., Dube, D.H., Sletten, E.M., Bertozzi, C.R.: A strategy for the selective imaging of glycans using caged metabolic precursors. J. Am. Chem. Soc. 132, 9516–9518 (2010)
Xie, R., Hong, S., Feng, L., Rong, J., Chen, X.: Cell-selective metabolic glycan labeling based on ligand-targeted liposomes. J. Am. Chem. Soc. 134, 9914–9917 (2012)
Xie, R., Dong, L., Huang, R., Hong, S., Lei, R., Chen, X.: Targeted imaging and proteomic analysis of tumor-associated glycans in living animals. Angew. Chem. Int. Ed. 53, 14082–14086 (2014)
Haga, Y., Ishii, K., Hibino, K., Sako, Y., Ito, Y., Taniguchi, N., Suzuki, T.: Visualizing specific protein glycoforms by transmembrane fluorescence resonance energy transfer. Nat. Commun. 3, 907 (2012)
Brock, R., Hamelers, I.H., Jovin, T.M.: Comparison of fixation protocols for adherent cultured cells applied to a GFP fusion protein of the epidermal growth factor receptor. Cytometry 35, 353–362 (1999)
Lin, W., Du, Y., Zhu, Y., Chen, X.: A cis-membrane FRET-based method for protein-specific imaging of cell-surface glycans. J. Am. Chem. Soc. 136, 679–687 (2014)
Belardi, B., de la Zerda, A., Spiciarich, D.R., Maund, S.L., Peehl, D.M., Bertozzi, C.R.: Imaging the glycosylation state of cell surface glycoproteins by two-photon fluorescence lifetime imaging microscopy. Angew. Chem. Int. Ed. Engl. 52, 14045–14049 (2013)
Han, S., Collins, B.E., Bengtson, P., Paulson, J.C.: Homomultimeric complexes of CD22 in B cells revealed by protein-glycan cross-linking. Nat. Chem. Biol. 1, 93–97 (2005)
Wu, X., Tian, Y., Yu, M., Lin, B., Han, J., Han, S.: A fluorescently labelled sialic acid for high performance intraoperative tumor detection. Biomater. Sci. 2, 1120 (2014)
Wu, X., Yu, M., Lin, B., Xing, H., Han, J., Han, S.: A sialic acid-targeted near-infrared theranostic for signal activation based intraoperative tumor ablation. Chem. Sci. 6, 798–803 (2014)
Mbua, N.E., Li, X., Flanagan-Steet, H.R., Meng, L., Aoki, K., Moremen, K.W., Wolfert, M.A., Steet, R., Boons, G.-J.: Selective exo-enzymatic labeling of N-glycans on the surface of living cells by recombinant ST6Gal I. Angew. Chem. Int. Ed. Engl. 52, 13012–13015 (2013)
Lin, L., Tian, X., Hong, S., Dai, P., You, Q., Wang, R., Feng, L., Xie, C., Tian, Z.-Q., Chen, X.: A bioorthogonal Raman reporter strategy for SERS detection of glycans on live cells. Angew. Chem. Int. Ed. Engl. 52, 7266–7271 (2013)
Hong, S., Chen, T., Zhu, Y., Li, A., Huang, Y., Chen, X.: Live-cell stimulated raman scattering imaging of alkyne-tagged biomolecules. Angew. Chem. Int. Ed. 53, 5827–5831 (2014)
Hang, H.C., Yu, C., Kato, D.L., Bertozzi, C.R.: A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc. Natl. Acad. Sci. U. S. A. 100, 14846–14851 (2003)
Dube, D., Prescher, J., Quang, C., Bertozzi, C.: Probing mucin-type O-linked glycosylation in living animals. Proc. Natl. Acad. Sci. U. S. A. 103, 4819–4824 (2006)
Laughlin, S., Baskin, J., Amacher, S., Bertozzi, C.: In vivo imaging of membrane-associated glycans in developing zebrafish. Science 320, 664–667 (2008)
Laughlin, S.T., Bertozzi, C.R.: In vivo imaging of caenorhabditis elegans glycans. ACS Chem. Biol. 4, 1068–1072 (2009)
Becker, D., Lowe, J.: Fucose: biosynthesis and biological function in mammals. Glycobiology 13, 41R–53R (2003)
Sawa, M., Hsu, T.-L., Itoh, T., Sugiyama, M., Hanson, S.R., Vogt, P.K., Wong, C.-H.: Glycoproteomic probes for fluorescent imaging of fucosylated glycans in vivo. Proc. Natl. Acad. Sci. U. S. A. 103, 12371–12376 (2006)
Rabuka, D., Hubbard, S., Laughlin, S., Argade, S., Bertozzi, C.: A chemical reporter strategy to probe glycoprotein fucosylation. J. Am. Chem. Soc. 128, 12078–12079 (2006)
Soriano Del Amo, D., Wang, W., Jiang, H., Besanceney, C., Yan, A.C., Levy, M., Liu, Y., Marlow, F.L., Wu, P.: Biocompatible copper(I) catalysts for in vivo imaging of glycans. J. Am. Chem. Soc. 132, 16893–16899 (2010)
Dehnert, K.W., Baskin, J.M., Laughlin, S.T., Beahm, B.J., Naidu, N.N., Amacher, S.L., Bertozzi, C.R.: Imaging the sialome during zebrafish development with copper-free click chemistry. ChemBioChem 13, 353–357 (2012)
Jiang, H., Zheng, T., Lopez-Aguilar, A., Feng, L., Kopp, F., Marlow, F.L., Wu, P.: Monitoring dynamic glycosylation in vivo using supersensitive click chemistry. Bioconjug. Chem. 25, 698–706 (2014)
Uttamapinant, C., Tangpeerachaikul, A., Grecian, S., Clarke, S., Singh, U., Slade, P., Gee, K.R., Ting, A.Y.: Fast, cell-compatible click chemistry with copper-chelating azides for biomolecular labeling. Angew. Chem. Int. Ed. 51, 5852–5856 (2012)
Zheng, T., Jiang, H., Gros, M., Soriano Del Amo, D., Sundaram, S., Lauvau, G., Marlow, F., Liu, Y., Stanley, P., Wu, P.: Tracking N-acetyllactosamine on cell-surface glycans in vivo. Angew. Chem. Int. Ed. Engl. 50, 4113–4118 (2011)
Chaubard, J.-L., Krishnamurthy, C., Yi, W., Smith, D.F., Hsieh-Wilson, L.C.: Chemoenzymatic probes for detecting and imaging fucose-α(1-2)-galactose glycan biomarkers. J. Am. Chem. Soc. 134, 4489–4492 (2012)
Boyce, M., Carrico, I.S., Ganguli, A.S., Yu, S.-H., Hangauer, M.J., Hubbard, S.C., Kohler, J.J., Bertozzi, C.R.: Metabolic cross-talk allows labeling of O-linked {beta}-N-acetylglucosamine-modified proteins via the N-acetylgalactosamine salvage pathway. Proc. Natl. Acad. Sci. U. S. A. 108, 3141–3146 (2011)
Zaro, B.W., Yang, Y.-Y., Hang, H.C., Pratt, M.R.: Chemical reporters for fluorescent detection and identification of O-GlcNAc-modified proteins reveal glycosylation of the ubiquitin ligase NEDD4-1. Proc. Natl. Acad. Sci. U. S. A. 108, 8146–8151 (2011)
Bateman, L.A., Zaro, B.W., Chuh, K.N., Pratt, M.R.: N-propargyloxycarbamate monosaccharides as metabolic chemical reporters of carbohydrate salvage pathways and protein glycosylation. Chem. Commun. 49, 4328–4330 (2013)
Chuh, K.N., Zaro, B.W., Piller, F., Piller, V., Pratt, M.R.: Changes in metabolic chemical reporter structure yield a selective probe of O-GlcNAc modification. J. Am. Chem. Soc. 136, 12283–12295 (2014)
Vainauskas, S., Cortes, L.K., Taron, C.H.: In vivo incorporation of an azide-labeled sugar analog to detect mammalian glycosylphosphatidylinositol molecules isolated from the cell surface. Carbohydr. Res. 362, 62–69 (2012)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chuh, K.N., Pratt, M.R. Chemistry-enabled methods for the visualization of cell-surface glycoproteins in Metazoans. Glycoconj J 32, 443–454 (2015). https://doi.org/10.1007/s10719-015-9589-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-015-9589-3