
Abstract
Miamiensis avidus is a parasitic pathogen that causes the disease scuticociliatosis in teleost fish species. It is a ciliate and a free-living marine protozoan belonging to the order Philasterida, subclass Scuticociliatida, class Oligohymenophorea, and phylum Ciliophora. The complete mt-genome of M. avidus was linear and 38,695 bp in length with 47 genes, including 40 protein-coding genes, two ribosomal RNA (rRNA) genes, and five transfer RNA (tRNA) genes. Of these, 20 genes typically belong to the clusters of orthologous groups, playing roles in energy production and conversion, translation, ribosomal structure and biogenesis, and defense mechanisms. This is the first report of sequencing and characterization of the mt-genome of M. avidus, which was observed to be linear and possessing the typical ciliate mitochondrial genome organization and phylogenetic relationships. Remarkable differences were observed between M. avidus and other ciliates in the mitochondrially encoded rRNAs, extensive gene loss in ribosomal genes and tRNAs, terminal repeat sequences, and stop codon usage. A comparative and phylogenetic analysis of M. avidus and Uronema marinum of the order Hymenostomatida, which is most closely related to the order Philasterida, signified the promise of the mitogenome data of M. avidus as a valuable genetic marker in species detection and taxonomic research. The present study has potential applications in epidemiological studies and host-parasite interaction investigations facilitating disease control.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Scuticociliatosis, a parasitic disease of fish and crustaceans is caused by invasive scuticociliates, which are free-living marine protozoa belonging to the subclass Scuticociliatida of the phylum Ciliophora (Gao et al. 2016). In the last decade, scuticociliate infection became a prominent parasitological problem in mariculture worldwide. There have been many reports of severe disease outbreaks caused by several scuticociliates species including Uronema marinum, Philasterides dicentrarchi, Pseudocohnilembus persalinus, Anophryoides haemophila, and Miamiensis avidus, which have led to serious economic loss (Ragan et al. 1996; Kim et al. 2004a, b; Harikrishnan et al. 2012; Stidworthy et al. 2014; Iglesias et al. 2018; Li et al. 2018). Scuticociliates are characterized by their high potential for systemic invasion, infecting not only the surface of the body but also internal organs, including the brain, kidney, spleen, and the spinal cord, leading to high host mortality rates (Puig et al. 2007; Harikrishnan et al. 2010). The major clinicopathological manifestations of scuticociliatosis in infected fish are dark coloration, loss of scales, excessive body mucus, hemorrhagic and bleached spots on the skin, and severe dermal necrotic lesions that ultimately destroy tissues, leading to mortality (Jung et al. 2007; Jin et al. 2009; Moustafa et al. 2010).
Previous reports of scuticociliate infection, including pathological and chemotherapeutic studies, have focused on the prevention and control of scuticociliatosis, species identification for taxonomy, investigation on diversity, and on the diagnosis of scuticociliatosis. Little attention has been paid to the molecular mechanisms of pathogenicity, mainly due to a lack of basic research on areas such as the life cycle, genetics, and the genome of scuticociliates. Although morphological analysis of scuticociliates performed by a series of methods such as scanning electron microscopy, protein silver staining, and silver impregnation is an useful approach for identification, it is time-consuming and laborious, and thus, is not suitable for the identification of closely related species and for scuticociliatosis treatment (Jung et al. 2011; Huang et al. 2021).
Although molecular identification of scuticociliate species, such as Pseudocohnilembus persalinus (Jones et al. 2010), Phiasterides dicentrachi (DE Felipe et al. 2017), Uronema marinum (Smith et al. 2009) and Miamiensis avidus (Jung et al. 2005), has been performed through the DNA identification of mitochondrial cytochrome oxidase c subunit I (COI) sequences, mitochondrial small subunit ribosomal DNA (mtSSU-rDNA) sequences, and nuclear small subunit ribosomal DNA (nSSU-rDNA) sequences (Whang et al. 2013; Zhang et al. 2019), and through phylogenetic analyses based on SSU rDNA or the ITS1-5.8 S-ITS2 region sequences of related parasite species, these methods are limited in their identification of closely related species (Jung et al. 2011). In addition, due to the presence of different serotypes of scuticociliate species (Piazzón et al. 2008; Song et al. 2009), molecular detection in hosts with low parasite numbers and vaccine-mediated controls is difficult (Jung et al. 2011). Mitochondrial (mt) genomes are increasingly used to study the evolution and molecular epidemiology of scuticociliates such as Pseudocohnilembus persalinus isolated from Turbot (Scophthalmus maximus L.) (Gao et al. 2018) and Uronema marinum isolated from Takifugu rubripes (Li et al. 2018). The mt-genome of Miamiensis avidus causing flatfish scuticociliatosis has been largely uninvestigated.
Here, we sequenced and annotated the first complete mt-genome of Miamiensis avidus, in order to determine the nucleotide and amino acid similarities between the mt-genomes of Miamiensis avidus and other related ciliates.
Materials and methods
Ciliates isolation and cultivation
M. avidus strain was obtained from the Pathology Research Division of the National Institute of Fisheries Science, which were identified as M. avidus using species-specific oligonucleotide primers (Seo et al. 2013) were isolated from the ascitic fluids of olive flounders in a local farm (Pohang-si, Gyeongsangbuk-do, 2008). In a recent publication we isolated 32 clones harboring peptidase gene sequences from 1,265 EST clones of the M. avidus cDNA library that related to infection of M. avidus by comparison of expression level between the cell-fed and the starved ciliates. Chinook salmon epithelia-214 cells were incubated at 20 °C in Eagle’s minimum essential medium (Sigma) supplemented with 10% heat-inactivated fetal bovine serum and were used to grow the ciliates. The ciliates were then harvested by centrifugation at 200 × g for 5 min and washed three times by centrifugation at 150 × g for 5 min in 10 ml of phosphate-buffered saline (Sigma). The washed ciliates were counted using a hemocytometer.
DNA isolation, library preparation, and sequencing
The M. avidus ciliates were harvested by centrifugation at 200 × g for 5 min and washed more than three times by centrifugation at 150 × g for 5 min in Hanks’ balanced salt solution (Sigma) or filtered seawater. The genomic DNA of M. avidus was extracted using a HiGene™ Genomic DNA Prep Kit (Biofact, Daejeon, Korea). Using a Covaris G-tube, fragments of 20 kb were generated by shearing the genomic DNA according to the manufacturer’s protocol. The AMpureXP bead purification system was used to remove small fragments. A total of 5 µg of each sample was used as the input for the preparation of the library to be used in PacBio sequencing (Pacific Biosciences). The SMRTbell library was constructed using a SMRTbell™ Template Prep Kit 1.0 (PN 100-259-100). Using the BluePippin Size selection system, small fragments were removed from the large-insert library. After a sequencing primer was annealed to the SMRTbell template, DNA polymerase was bound to the complex (Sequel Binding Kit 2.0). Purification was performed using SMRTbell clean up columns (SMRTbell® Clean Up Columns v2 Kit-Mag: PN 01-303-600). The purification step was performed after polymerase binding to remove the unbound polymerases and the polymerase molecules that are bound to small DNA inserts. A MagBead Kit was used for the binding of the library complex with MagBeads before sequencing, since MagBead-bound complexes provide more reads per single-molecule real-time (SMRT) cell. This polymerase-SMRTbell-adaptor complex was then loaded into zero-mode waveguides. The SMRTbell library was sequenced using three SMRT cells (Pacific Biosciences, Sequel™ SMRT® Cell 1 M v2) using a sequencing kit (Sequel Sequencing Kit 2.1) and 1 × 600 min movies were captured for each SMRT cell using the PacBio sequencing platform (Pacific Biosciences).
Genomic DNA (gDNA) (200 ng) for Miseq was sheared using an S220 Ultra sonicator (Covaris) (peak incident power 175 W, duty factor 5%, 200 cycles per burst, treatment time 35 s). Library preparation was performed using an Illumina TruSeq Nano DNA sample prep kit (Illumina) according to the manufacturer’s instructions. Briefly after clean-up of fragmented gDNA using sample purification beads, the fragmented gDNA was end-repaired at 30°C for 30 min, followed by size selection for 500 bp insert size using sample purification beads. A single ‘A’ nucleotide was added to the 3’ ends of the blunt fragments using the a-tailing mix reagent by incubation at 37 °C for 30 min followed by incubation at 70 °C for 5 min. Indexing adapters were ligated to the ends of the DNA fragments using ligation mix 2 reagents at 30 °C for 10 min. After washing twice with sample purification beads, a PCR was performed to enrich the DNA fragments with adapter molecules on both ends. The thermocycler conditions were as follows: 95 °C for 3 min, 8 cycles of 98 °C for 20 s, 60 °C for 15 s, and 72 °C for 30 min, with a final extension at 72 °C for 5 min. Eventually, the quality and the band size of the libraries were assessed using an Agilent 2100 Bioanalyzer (Agilent). The libraries were quantified by qPCR using a CFX96 Real-Time System (Bio-Rad). After normalization, sequencing of the prepared library was performed using the MiSeq system (Illumina) with 300 bp paired-end reads.
Illumina Miseq sequencing, PacBio SMRT sequencing, and hybrid assembly
A total of 16.1 Gb of paired-end fastq files were produced using Illumina MiSeq paired-end sequencing, and Raw sequence data were deposited into Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/sra/) with the accession no. PRJNA763762. Using these data, the genome size of coconut was estimated by k-mer distribution using the KmerGenie program (Chikhi and Medvedev 2014). Pre-assembled short-read next generation sequencing contigs were used to correct and derive a compact representation of the 7.8 Gb SMRT (Pacific Biosciences) long reads using a DBG2OLC hybrid assembler (Ye et al. 2016). The DBG2OLC assembler was used to assemble the raw PacBio SMRT sequence data, with the Illumina Miseq contig sequence assembly utilized as an anchor for error correction. The overlap and the consensus steps were executed with the following parameters: k-mer value: 19, adaptive k-mer matching threshold: 0.0001, fixed k-mer matching threshold: 2, minimum overlap score between a pair of long reads: 8, removal of chimeric reads: allow. The quality of the resulting assembly was assessed using a local Perl script (https://github.com/aubombarely/GenoToolBox/blob/master/SeqTools/FastaSeqStats). The assembled contigs were scanned for M. avidus mitochondrial sequence using local Basic Local Alignment Search Tool (BLAST) (Altschul et al. 1997) with the Tetrahymena thermophila mtDNA sequence (NC_003029) as the search query. The complete M. avidus mitochondrial sequence with terminal repeat sequences at both ends was selected.
Annotation of the M. avidus mtDNA genome and phylogenetic analysis
Open reading frame (ORF) finding and gene prediction were performed using BLAST (BLASTn and BLASTx), GeneMarkS (Besemer et al. 2001), and Geneious (ver. R11). The large and small subunits of ribosomal RNA (rRNA) were identified using BLASTn with published ciliate rRNAs as queries and the transfer RNAs (tRNAs) were identified using the tRNAscan-SE search server (Lowe and Eddy 1997) (http://lowelab.ucsc.edu/tRNAscan-SE/). All protein-coding, rRNA, and tRNA genes, and genes in the novel mt-genomes were confirmed by multiple sequence alignment with published ciliate data using the T-Coffee package (Di Tommaso et al. 2011) (http://www.tcoffee.org). A graphical linear map of the complete mt-genome was produced using Circos v0.67 (Krzywinski et al. 2009). The whole mt-genome alignment of M. avidus was compared with that of other related ciliates (Table 1) using the MAUVE program (Mauve version 20,150,226 build 10)(Darling et al. 2010). To compare the M. avidus mt-genome with other ciliate mt-genomes, the respective amino acid sequences of the protein-encoding gene rpl14 were aligned using ClustalX and neighbor-joining (NJ) analysis was conducted using MEGA 7.0 with 1000 bootstrap replicates (Kumar et al. 2016). In addition, the distance tree of the mitochondrial cytochrome c oxidase subunit 1 (cox1) genes was constructed using MEGA 7.0 with 1000 bootstrap replicates. The list of the taxa used in the cox1 alignments is listed in Table 10. The additional sequences used for comparison and their GenBank accession numbers were as follows: Philasterides dicentrarchi cox1_a (accession number: MN531306.1), P. dicentrarchi cox1_b (GQ342957.1), Pseudocohnilembus persalinus cox1 from freshwater-reared rainbow trout, Oncorhynchus mykiss (GU584095), P. persalinus cox1 from Turbot (Scophthalmus maximus) (MH608212.2), P. persalinus cox1 (GQ500579), Uronema marinum isolate ZZF20170302 cox1 from Turbot (MG001901.1), and Uronema heteromarinum isolate FXP08082901 cox1 (MG001901.1).
Results
Complete mt-genome organization and composition
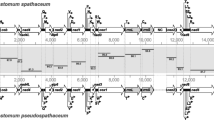
The mt-genome of M. avidus was observed to be linear with two terminal repeat sequences (TRS; 30 bp) located at both ends and 38,695 bp in length with 47 genes, including 40 protein-coding genes (29 coding sequences, four large subunit ribosomal proteins [rpl], seven small subunit ribosomal proteins [rps]), two ribosomal RNA (rRNA) genes, and five transfer RNA (tRNA) genes (Fig. 1). The mt-genome organization of M. avidus, including a list of gene order, gene length, alternative start codons, and intergenic spacer regions, is given in Table 2. The nucleotide composition of the entire M. avidus mtDNA sequence was found to be 40.8% thymine, 39.1% adenine, 10.4% guanine, and 9.7% cytosine, while the complete mtDNA sequence had a high AT content of 79.87% (Table 3). Clusters of orthologous protein-coding genes (COGs) located in the M. avidus mt-genome included genes encoding 13 energy pathway proteins, including ATP synthase subunit 9 (atp9), two subunits of cytochrome c oxidase (cox1 and cox2), apocytochrome b (cob), and nine NADH dehydrogenase subunits (nad1a, nad1b, nad2, nad3, nad4, nad5, nad7, nad9, and nad10), genes for translation, ribosomal structure, and biogenesis, including six ribosomal proteins (rpl2, rps12, rps13, rpl14, rpl16, and rps19), and genes encoding proteins involved in defense mechanisms, such as putative mitochondrial protein orf386 (orf386) (Table 4).

Visual representation of the linear mitochondrial genome of Miamiensis avidus. Protein-coding genes (40) are purple, tRNAs (5) are dark blue, rRNAs (2) are red, and Terminal Repeat Sequences (TRS; 30 bp) at both ends are not shown. Gene composition in the whole mitochondrial genome of the M. avidus is indicated by a table below the mtDNA map
Codon usage
Seven alternative start codons were found to be used in the M. avidus mt-genome, in which 37 of the 40 protein-coding genes have been shown to use TAA as a termination codon, and three genes (yejR, rps13, and rps3_a) were shown to use TAG as a termination codon. On the whole, 27.5% of the genes were found to have an ATG start codon; 27.5%, ATT (rpl2, rps12, rpl6/orf176, orf73, rpl16, nad4L/orf119, nad9, cob, nad5, nad1_a, and rps13); 20%, TTA (rps3_b/orf149, rps19, atp9, nad2_a/orf371, orf159, rpl14, orf143, and rps3_a/orf320); 12.5%, ATA (orf437, rps14/orf107, rps7/orf229, cox2, and nad6/orf246); 5%, ATC (nad4 and yejR/orf571); 5%, TTG (orf195 and orf492); and only 2.5%, a GTG start codon (nad2_b) in the M. avidus mt-genome (Table 2).
Comparative analysis of the complete mitogenomes of ciliates
The genome structure and organization of the mt-genomes (mitogenomes), the mitochondrially encoded gene content, and the TRS in the mitochondrial genome of M. avidus determined in the present study were compared to that of the mt-genomes of other ciliates reported previously (Table 1). As shown in Table 5, with the exception of a few gene losses, ciliate mitochondrial genomes share largely the same complement of known protein-coding genes. The content of the identified protein-coding genes was almost identical in M. avidus and U. marinum mtDNAs, both organisms belonging to the order Philasterida. Genes for the small and large subunit ribosomal RNAs (rnl_b_1 and rns b, respectively) were found in M. avidus mtDNA in contrast to the split large and small subunit ribosomal RNA genes and the duplicated large subunit rRNA genes in the Tetrahymena genus (Table 6). The rnl and rns genes in M. avidus were found to be not duplicated, similar to those in T. pyriformis and I. multifiliis.
The M. avidus mitogenome was also found to contain 12 M. avidus and ciliate-specific ORFs, called Ymf genes, for which function a cannot be assigned, due to lack of sufficient sequence similarity to strongly indicate homology (Table 8). Three of these ORFs (orf386, orf437, and orf492) were also observed in Tetrahymena, Ichthyophthirius, and Paramecium mtDNAs (Ymf67, Ymf66, and Ymf68 in I. multifiliis). The linear mitogenomes of ciliates have between 18 and 35 bp repeat sequences and have symmetrical ends forming TRS (Table 9). The terminal repeat regions in M. avidus and U. marinum were found to be 30 and 32 nt long, respectively. In six ciliate species, the terminal repeats at both ends of the mitochondrial DNA were observed to be completely different.
In general, Miamiensis, Uronema, Tetrahymena, and Ichthyophthirius mtDNAs had almost the same preference for relative synonymous codons, with only a slight difference (Fig. 2). The most frequent amino acids in the M. avidus mitogenome were leucine (Leu), phenylalanine (Phe), and isoleucine (Ile), with percentages of occurrence of 13.4%, 11.37%, and 10.51%, respectively. The least frequent amino acids were cysteine (Cys), histidine (His), and proline (Pro), with percentages of occurrence of 0.95%, 1.47%, and 1.72%, respectively.

Relative synonymous codon usage (RSCU) of 8 ciliate representatives. Codon families are labelled on the x-axis. Values on the top of the bars denote amino acid usage
Mitochondrial genome organization and phylogenetic relationships
To assess the extent and comparison of ciliate mitochondrial genome arrangements more closely, mt-genomes were aligned using the MAUVE program. The mt-genomes of Miamiensis and Uronema were found to be largely collinear. Those of Tetrahymena and Ichthyophthirius were largely collinear as well. In the large subunit ribosomal protein (rpl14)-based phylogeny, class Oligohymenophorea was monophyletic with the orders Philasterida, Hymenostomatida, and Peniculida clustered together, supported by significant statistical values (Fig. 3). Considering the order Philasterida to which M. avidus and U. marinum belong as a benchmark, the order Hymenostomatida, which is most closely related to Philasterida, revealed only slight differences in the order of common genes (Table 5).

Whole mitochondrial genome alignment of other ciliate genomes by the MAUVE program (Mauve version 20,150,226 build 10). Color blocks of the same color represent homologous regions between different mitogenomes. GenBank accession numbers are provided (in parentheses) for all reference sequences
Two major clusters were observed in the phylogenetic tree of the cox1 gene in the M. avidus isolates (Fig. 4; Table 10). Cluster I included 11 strains (GJ01, Mie0301, WDB-0708, SJF-06 A, YS2, WD4, JJ4, JJ3, SJF-03 A, WS1, and Nakajima) and cluster II contained 8 strains (Iyo1, xiapu1, SJF-03B, YK1, YK2, JF05To, RF05To, and SK05Kyo). While the highest bootstrap values were observed in two major clusters of cox1 gene (100%) including M. avidus SCUTICA2 strain, the low bootstrap values were observed in Philasterides dicentrarchi strains (33–52%).

Phylogenetic tree of Miamiensis avidus strains based on the Cox 1 gene. Abbreviation: Cox 1, Cytochrome c oxidase I
Discussion
The complete mt-genome of M. avidus was observed to be remarkably compact when compared to other ciliate mitogenomes in similar taxonomic positions (Burger et al. 2000; Brunk et al. 2003; de Graaf et al. 2009; Harikrishnan et al. 2010; Gao et al. 2018; Power et al. 2019; Retallack et al. 2019) and had the shortest genome length and the least coding sequence (CDS) of genes. The most remarkable observation in the order Philasterida and Hymenostomatida mt-genomes is the very low GC content observed in certain species, for instance, in the five Tetrahymena species (18.5 ~ 21.3%), U. marinum (19.0%), and I. multifiliis (16.7%), while species such as N. ovalis and P. Aurelia had relatively high GC contents of 41.5% and 41.2%, respectively (Johri et al. 2019). The low GC content of Paramecium mitogenomes is marked by a highly biased codon usage, with most synonymous positions exhibiting a strong bias for A or T nucleotides (Barth and Berendonk 2011). Only TAA was found to terminate protein-coding genes, whereas TAG never occurred in P. caudatum and Tetrahymena. In the M. avidus mitogenome, 37 of the 40 protein-coding genes were shown to use TAA as the termination codon, and three genes (yejR, rps13, and rps3_a) were shown to use TAG as the termination codon.
COGs located in the M. avidus mitogenome showed that most of the functional genes were related to secondary metabolite biosynthesis, transport, and catabolism, as has been observed in the mitochondrial genomes of 14 related ciliates (Huang et al. 2021). The M. avidus mitogenome shares a number of structural features with the existing ciliate mt-genome showing molecular affinity and similarities in encoded gene content with other members of the order, such as Philasterida, Hymenostomatida, and Peniculida. Indeed, M. avidus had extensive gene loss, especially for ribosomal proteins, compared to species within Hymenostomatida, with only two ribosomal genes in its mitochondrial genome and only five tRNAs.
In all ciliate mitogenomes, including that of M. avidus, there is either a central in T. pyriformis (Burger et al. 2000)d minuta (de Graaf et al. 2009), and the hydrogenosome of Nyctotherus ovalis (de Graaf et al. 2011) or terminal in P. aurelia (Pritchard et al. 1990)d avidus, which bear low sequence complexity repeats. In the P. aurelia mitogenome, pure AT repeats were observed in the terminal region (AAATATTAATATATTTATTTTTTTATTTTAAT), and. P. aurelia terminal repeat sequences are considerably more GC-rich (51.4% AT) than all other ciliate mitochondrial genome repeats.
A previous report on the analysis of cox1 genes from the serotypes of 21 strains of M. avidus isolated from diseased olive flounder (Paralichthys olivaceus), ridged-eye flounder (Pleuronichthys cornutus), and spotted knifejaw (Oplegnathus fasciatus) indicated that the 21 strains can be classified to have five cox1 types (two heterogeneous clusters and three individual branches). Among the species-specific regions flanked by conserved sequences, such as ribosomal RNA (SSU and LSU) and cox1 genes, cox1 was similar to SSU and better than LSU in discriminating between M. avidus and related scuticociliates, which has been used previously in investigations of a wide range of human and nonhuman infectious diseases. These cox1 types have not been found to reflect geographical origins and host species (Jung et al. 2011). In the present study, we performed a comparative and phylogenetic analysis of cox1 genes from the mitochondrial genomes of M. avidus and the serotypes of M. avidus strains. The mitochondrial cox1 gene of M. avidus which belongs to the order Philasterida of the subclass Scuticociliatida showed promise as a valuable genetic marker for species detection. Based on previous studies, intraspecific variations in cox1 are not attributed to the infectivity and virulence demonstrated in some strains (Nakajima, WS1, YK1, Iyo1, and Mie 0301) of M. avidus that are highly pathogenic to olive flounder (Song et al. 2009). However, the cox1 cluster I and II types in the serotypes A and B of the M. avidus strains showed cross-immobilization/agglutination activities with the anti-sera against serotypes A and B, respectively. The present study can be utilized in epidemiological studies by informing detection, taxonomic research, strain identification, geographical spread, and disease control.
Miamiensis avidus is a dangerous parasitic pathogen that causes scuticociliatosis in fish and high mortality rates in mariculture worldwide. Methods to identify this species from among closely related species were limited. Mitochondrial DNA sequences can be valuable genetic markers for species detection and diagnostics, which are increasingly used in molecular epidemiology and surveillance tool. This is the first report in which the mitogenome of M. avidus was thoroughly compared to seven related ciliate mitogenomes by analyzing the nucleotide composition, codon usage, genome organization, protein-coding genes, and the terminal repeat sequences. The results of this study could facilitate better understanding of scuticociliate infection, aid in the development of control measures against scuticociliatosis, and provide insights into the molecular epidemiology of scuticociliates.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402. https://doi.org/10.1093/nar/25.17.3389
Barth D, Berendonk TU (2011) The mitochondrial genome sequence of the ciliate Paramecium caudatum reveals a shift in nucleotide composition and codon usage within the genus Paramecium. BMC Genomics 12:272. https://doi.org/10.1186/1471-2164-12-272
Besemer J, Lomsadze A, Borodovsky M (2001) GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29(12):2607–2618. https://doi.org/10.1093/nar/29.12.2607
Brunk CF, Lee LC, Tran AB, Li J (2003) Complete sequence of the mitochondrial genome of Tetrahymena thermophila and comparative methods for identifying highly divergent genes. Nucleic Acids Res 31(6):1673–1682. https://doi.org/10.1093/nar/gkg270
Burger G, Zhu Y, Littlejohn TG, Greenwood SJ, Schnare MN, Lang BF, Gray MW (2000) Complete sequence of the mitochondrial genome of Tetrahymena pyriformis and comparison with Paramecium aurelia mitochondrial DNA. J Mol Biol 297(2):365–380. https://doi.org/10.1006/jmbi.2000.3529
Chikhi R, Medvedev P (2014) Informed and automated k-mer size selection for genome assembly. Bioinformatics 30(1):31–37. https://doi.org/10.1093/bioinformatics/btt310
Darling AE, Mau B, Perna NT (2010) progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5(6):e11147. https://doi.org/10.1371/journal.pone.0011147
DE Felipe AP, Lamas J, Sueiro RA, Folgueira I, Leiro JM (2017) New data on flatfish scuticociliatosis reveal that Miamiensis avidus and Philasterides dicentrarchi are different species. Parasitology 144(10):1394–1411. https://doi.org/10.1017/S0031182017000749
de Graaf RM, Ricard G, van Alen TA, Duarte I, Dutilh BE, Burgtorf C, Kuiper JW, van der Staay GW, Tielens AG, Huynen MA, Hackstein JH (2011) The organellar genome and metabolic potential of the hydrogen-producing mitochondrion of Nyctotherus ovalis. Mol Biol Evol 28(8):2379–2391. https://doi.org/10.1093/molbev/msr059
de Graaf RM, van Alen TA, Dutilh BE, Kuiper JW, van Zoggel HJ, Huynh MB, Görtz HD, Huynen MA, Hackstein JH (2009) The mitochondrial genomes of the ciliates Euplotes minuta and Euplotes crassus. BMC Genomics 10:514. https://doi.org/10.1186/1471-2164-10-514
Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang JM, Taly JF, Notredame C (2011) T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. https://doi.org/10.1093/nar/gkr245. 39(Web Server issue):W13-7
Gao F, Warren A, Zhang Q, Gong J, Miao M, Sun P, Xu D, Huang J, Yi Z, Song W (2016) The All-Data-Based Evolutionary Hypothesis of Ciliated Protists with a Revised Classification of the Phylum Ciliophora (Eukaryota, Alveolata). Sci Rep 29(6):24874. https://doi.org/10.1038/srep24874
Gao YQ, Jin SB, Dang HF, Ye SG, Li RJ (2018) Mitochondrial genome sequencing of notorious scuticociliates (Pseudocohnilembus persalinus) isolated from Turbot (Scophthalmus maximus L.). Mitochondrial DNA Part B 3:1077–1078. https://doi.org/10.1080/23802359.2018.1508388
Harikrishnan R, Balasundaram C, Heo MS (2010) Scuticociliatosis and its recent prophylactic measures in aquaculture with special reference to South Korea Taxonomy, diversity and diagnosis of scuticociliatosis: Part I Control strategies of scuticociliatosis: Part II. Fish Shellfish Immunol 29(1):15–31. https://doi.org/10.1016/j.fsi.2010.02.026
Harikrishnan R, Jin CN, Kim JS, Balasundaram C, Heo MS (2012) Philasterides dicentrarchi, a histophagous ciliate causing scuticociliatosis in olive flounder, Philasterides dicentrarchi-histopathology investigations. Exp Parasitol 130(3):239–245. https://doi.org/10.1016/j.exppara.2011.12.007
Huang YX, Wang S, Gao YQ, Chen JH, Wang XL, Li RJ (2021) Comparison of mitochondrial genome and development of specific PCR primers for identifying two scuticociliates, Pseudocohnilembus persalinus and Uronema marinum. Parasit Vectors 14(1):318. https://doi.org/10.1186/s13071-021-04821-3
Iglesias R, Paramá A, Alvarez MF, Leiro J, Fernández J, Sanmartín ML (2018) Philasterides dicentrarchi (Ciliophora, Scuticociliatida) as the causative agent of scuticociliatosis in farmed turbot Scophthalmus maximus in Galicia (NW Spain). Dis Aquat Organ 46(1):47–55. https://doi.org/10.3354/dao046047
Jin CN, Harikrishnan R, Moon YG, Kim MC, Kim JS, Balasundaram C, Azad IS, Heo MS (2009) Histopathological changes of Korea cultured olive flounder, Paralichthys olivaceus due to scuticociliatosis caused by histophagous scuticociliate, Philasterides dicentrarachi. Vet Parasitol 161(3–4):292–301. https://doi.org/10.1016/j.vetpar.2009.01.033
Johri P, Marinov GK, Doak TG, Lynch M (2019) Population Genetics of Paramecium Mitochondrial Genomes: Recombination, Mutation Spectrum, and Efficacy of Selection. Genome Biol Evol 11(5):1398–1416. https://doi.org/10.1093/gbe/evz081
Jones SR, Prosperi-Porta G, LaPatra SE (2010) First isolation of Pseudocohnilembus persalinus (Ciliophora: Scuticociliatida) from freshwater-reared rainbow trout, Oncorhynchus mykiss. J Parasitol 96(5):1014–1016. https://doi.org/10.1645/GE-2500.1
Jung SJ, Im EY, Strüder-Kypke MC, Kitamura S, Woo PT (2011) Small subunit ribosomal RNA and mitochondrial cytochrome c oxidase subunit 1 gene sequences of 21 strains of the parasitic scuticociliate Miamiensis avidus (Ciliophora, Scuticociliatia). Parasitol Res 108(5):1153–1161. https://doi.org/10.1007/s00436-010-2157-7
Jung SJ, Kitamura S, Song JY, Joung IY, Oh MJ (2005) Complete small subunit rRNA gene sequence of the scuticociliate Miamiensis avidus pathogenic to olive flounder Paralichthys olivaceus. Dis Aquat Organ 64(2):159–162. https://doi.org/10.3354/dao064159
Jung SJ, Kitamura S, Song JY, Oh MJ (2007) Miamiensis avidus (Ciliophora: Scuticociliatida) causes systemic infection of olive flounder Paralichthys olivaceus and is a senior synonym of Philasterides dicentrarchi. Dis Aquat Organ 73(3):227–234. https://doi.org/10.3354/dao073227
Kim SM, Cho JB, Kim SK, Nam YK, Kim KH (2004a) Occurrence of scuticociliatosis in olive flounder Paralichthys olivaceus by Phiasterides dicentrarchi (Ciliophora: scuticociliatida). Dis Aquat Organ 62(3):233–238. https://doi.org/10.3354/dao062239
Kim SM, Cho JB, Lee EH, Kwon SR, Kim SK, Nam YK, Kim KH (2004b) Pseudocohnilembus persalinus (Ciliophora: Scuticociitida) is an additional species causing scuticociliatosis in olive flounder Paralichthys olivaceus. Dis Aquat Organ 62(3):239–244. https://doi.org/10.1101/gr.092759.109
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19(9):1639–1645. https://doi.org/10.1093/molbev/msw054
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 33(7):1870–1874. https://doi.org/10.1080/23802359.2018.1483757
Li R, Gao Y, Hou Y, Ye S, Wang L, Sun J, Li Q (2018) Mitochondrial genome sequencing and analysis of scuticociliates (Uronema marinum) isolated from Takifugu rubripes. Mitochondrial DNA B Resour 3(2):736–737. https://doi.org/10.1093/nar/25.5.955
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25(5):955–964. https://doi.org/10.1371/journal.pone.0000650
Moradian MM, Beglaryan D, Skozylas JM, Kerikorian V (2007) Complete mitochondrial genome sequence of three Tetrahymena species reveals mutation hot spots and accelerated nonsynonymous substitutions in Ymf genes. PLoS ONE 2(7):e650. https://doi.org/10.1292/jvms.10-0214
Moustafa EM, Tange N, Shimada A, Morita T (2010) Experimental Scuticociliatosis in Japanese flounder (Paralichthys olivaceus) infected with Miamiensis avidus: pathological study on the possible neural routes of invasion and dissemination of the scuticociliate inside the fish body. J Vet Med Sci 72(12):1557–1563. https://doi.org/10.1016/j.fsi.2008.05.012
Piazzón C, Lamas J, Castro R, Budiño B, Cabaleiro S, Sanmartín M, Leiro J (2008) Antigenic and cross-protection studies on two turbot scuticociliate isolates. Fish Shellfish Immunol 25(4):417–424. https://doi.org/10.1093/nar/18.1.173
Power C, Balli-Garza J, Evans D, Nowak FB, Bridle RA, Nathan J (2019) Detection of Miamiensis avidus (Ciliophora: Scuticociliatia) and Cardicola spp. (Trematoda: Aporocotylidae) DNA in biofouling from Southern Bluefin Tuna, Thunnus maccoyii pontoons off Port Lincoln, South Australia. Aquaculture 502:128–133. https://doi.org/10.1016/j.aquaculture.2018.12.027
Pritchard AE, Seilhamer JJ, Mahalingam R, Sable CL, Venuti SE, Cummings DJ (1990) Nucleotide sequence of the mitochondrial genome of Paramecium. Nucleic Acids Res 18(1):173–180. https://doi.org/10.3354/dao076131
Puig L, Traveset R, Palenzuela O, Padrós F (2007) Histopathology of experimental scuticociliatosis in turbot Scophthalmus maximus. Dis Aquat Organ 76(2):131–140. https://doi.org/10.1111/j.1550-7408.1996.tb03998.x
Ragan MA, Cawthorn RJ, Despres B, Murphy CA, Singh RK, Loughlin MB, Bayer RC (1996) The lobster parasite Anophryoides haemophila (Scuticociliatida: Orchitophryidae): nuclear 18S rDNA sequence, phylogeny and detection using oligonucleotide primers. J Eukaryot Microbiol 43(4):341–346. https://doi.org/10.1186/1746-6148-9-10
Retallack H, Okihiro MS, Britton E, Sommeran SV, DeRisi JL (2019) Metagenomic next-generation sequencing reveals Miamiensis avidus (Ciliophora: Scuticociliatida) in the 2017 epizootic of leopard sharks (Triakis semifasciata) in San Francisco Bay, California. USA. J Wildl Dis 55(2):375–386. https://doi.org/10.7589/2018-04-097
Seo JS, Jeon EJ, Jung SH, Park MA, Kim JW, Kim KH, Woo SH, Lee EH (2013) Molecular cloning and expression analysis of peptidase genes in the fish-pathogenic scuticociliate Miamiensis avidus. BMC Vet Res 9:10. https://doi.org/10.1186/1746-6148-9-10
Smith DR, Keeling PJ (2013) Gene conversion shapes linear mitochondrial genome architecture. Genome Biol Evol 5(5):905–912. https://doi.org/10.3354/dao02113
Smith PJ, McVeagh SM, Hulston D, Anderson SA, Gublin Y (2009) DNA identification of ciliates associated with disease outbreaks in a New Zealand marine fish hatchery. Dis Aquat Organ 86(2):163–167. https://doi.org/10.1111/j.1365-2761.2009.01089.x
Song JY, Sasaki K, Okada T, Sakashita M, Kawakami H, Matsuoka S, Kang HS, Nakayama K, Jung SJ, Oh MJ, Kitamura SI (2009) Antigenic differences of the scuticociliate Miamiensis avidus from Japan. J Fish Dis 32(12):1027–1034. https://doi.org/10.1177/0300985813492800
Stidworthy MF, Garner MM, Bradway DS, Westfall BD, Joseph B, Repetto S, Guglielmi E, Schmidt-Posthaus H, Thornton SM (2014) Systemic Scuticociliatosis (Philasterides dicentrarchi) in sharks. Vet Pathol 51(3):628–663. https://doi.org/10.1093/gbe/evr136
Swart EC, Nowacki M, Shum J, Stiles H, Higgins BP, Doak TG, Schotanus K, Magrini VJ, Minx P, Mardis ER, Landweber LF (2012) The Oxytricha trifallax mitochondrial genome. Genome Biol Evol 4(2):136–154. https://doi.org/10.1093/oxfordjournals.molbev.a026234
van Hoek AH, Akhmanova AS, Huynen MA, Hackstein JH (2000) A mitochondrial ancestry of the hydrogenosomes of Nyctotherus ovalis. Mol Biol Evol 17(1):202–206
Whang I, Kang HS, Lee J (2013) Identification of scuticociliates (Pseudocohnilembus persalinus, P. longisetus, Uronema marinum and Miamiensis avidus) based on the cox1 sequence. Parasitol Int 62(1):7–13. https://doi.org/10.1016/j.parint.2012.08.002
Ye C, Hill CM, Wu S, Ruan J, Ma ZS (2016) DBG2OLC: Efficient Assembly of Large Genomes Using Long Erroneous Reads of the Third Generation Sequencing Technologies. Sci Rep 6:31900. https://doi.org/10.1038/srep31900
Zhang T, Fan X, Gao F, Al-Farraj SA, El-Serehy HA, Song W (2019) Further analyses on the phylogeny of the subclass Scuticociliatia (Protozoa, Ciliophora) based on both nuclear and mitochondrial data. Mol Phylogenet Evol 139:106565. https://doi.org/10.1016/j.ympev.2019.106565
Acknowledgements
We thanks to Dr. Moo Sang Kim for his valuable and constructive suggestions during the planning and development of this research work.
Funding
This research was supported by a grant from the National Institute of Fisheries Science, Republic of Korea (R2022071).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: Na Young Kim and Sang Jung Ahn wrote the main manuscript. Jung Soo Seo and Eun Ji Jeon were conducted the resources and data curation. Mi Young Cho and Hye Sung Choi were reviewed and edited the manuscript. All authors reviewed the manuscript. Especially, Na Young Kim and Sang Jung Ahn have contributed equally to this work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval:
Not applicable.
Consent for publication
Not applicable.
Consent to participate:
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kim, N.Y., Ahn, S.J., Seo, J.S. et al. Characterization of the complete mitochondrial genome of Miamiensis avidus causing flatfish scuticociliatosis. Genetica 150, 407–420 (2022). https://doi.org/10.1007/s10709-022-00167-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-022-00167-5