
Abstract
Barley shrunken endosperm mutants have been extensively reported. However, knowledge of the underlying molecular mechanisms of these mutants remains limited. Here, a pair of near isogenic lines (normal endosperm: Bowman and shrunken endosperm: sex1) was subjected to transcriptome analysis to identify mRNAs and lncRNAs related to endosperm development to further dissect its mechanism of molecular regulation. A total of 2123 (1140 up- and 983 down-regulated) unique differentially expressed genes (DEGs) were detected. Functional analyses showed that these DEGs were mainly involved in starch and sucrose metabolism, biosynthesis of secondary metabolites, and plant hormone signal transduction. A total of 343 unique target genes were identified for 57 differentially expressed lncRNAs (DE lncRNAs). These DE lncRNAs were mainly involved in glycerophospholipid metabolism, starch and sucrose metabolism, hormone signal transduction, and stress response. In addition, key lncRNAs were identified by constructing a co-expression network of the target genes of DE lncRNAs. Transcriptome results suggested that mRNA and lncRNA played a critical role in endosperm development. The shrunken endosperm in barley seems to be closely related to plant hormone signal transduction, starch and sucrose metabolism, and cell apoptosis. This study provides a foundation for fine mapping, elucidates the molecular mechanism of shrunken endosperm mutants, and also provides a reference for further studies of lncRNAs during the grain development of plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As one of the first food crops and feed crops to be domesticated and utilized in human history, barley (Hordeum vulgare L.) ranks fourth in global cereal crop (http://faostat.fao.org). Compared with most other cereal crops, barley is more tolerant to drought, salinity, aluminum, and other abiotic stresses (Dai et al. 2013). However, the increasing human population is expected to reach 8.5 billion by 2030 (Desa 2015; Qi et al. 2019), which highlights the need for a significant increase of crop productivity. The thousand-grain weight is one of the three factors that are used to determine crop yield (the others being number of ears per m2 and number of grains per spike) (Khahani et al. 2019; Schmalenbach et al. 2008). Both grain size and plumpness exert significant effects on the thousand-grain weight. The dissection of barley shrunken endosperm traits plays a role toward increasing grain weight and grain plumpness.
The barley shrunken endosperm trait was first reported in 1975, and it is typically caused by two reasons. It can either be caused by a mutation in a gene that is expressed in maternal tissue (i.e., shrunken endosperm genetic, seg) (Jarvi 1975; Ramage and Scheuring 1976), which is independent of pollen sources, or it is controlled by the kernel itself (i.e., shrunken endosperm xenia, sex) (Cook et al. 2018; Ma et al. 2014a). Eight mutants have been reported to date (officially named seg (seg1–seg8) and sex (sex1–sex8)), and each of these types is controlled by a different single gene (Franckowiak et al. 2010). The shrunken endosperm mutant sex1 (Eslick and Ries 1976) (formerly known as se6 (Jarvi 1975)) usually exhibits xenia effects (Röder et al. 2006). Studies on shrunken endosperm in barley focused on grain growth and grain filling (Djarot and Peterson 1991; Felker et al. 1983), changes of grain composition (Felker et al. 1984a, b; Nese et al. 2010), starch structure (De Arcangelis et al. 2019; Ma et al. 2014a; Schulman et al. 1995), cell morphology (Bosnes et al. 1987; Cook et al. 2018; Felker et al. 1985; Solomon and Drea 2019), and gene expression (Clarke et al. 2008; Ma et al. 2014a; Patron et al. 2004). However, little is known about the molecular mechanisms underlying shrunken endosperm. Morell et al. (2010) analyzed sex6 and identified a nucleotide mutation in the coding region of the starch synthase gene SSIIa. This mutation resulted in the appearance of a stop codon, thus inactivating SSIIa.
RNA sequencing (RNA-seq) offers high accuracy and sensitivity and is one of the most suitable techniques to study the whole transcriptome (Jain 2012; Wang et al. 2009). It offers advantages such as the identification of novel genes or transcripts, the detection of transcripts with low abundance, the detection of genetic variants, and the detection of more differentially expressed genes (DEGs) with higher fold-change (FC) compared with microarrays (Shanrong et al. 2014; Wang et al. 2009).
In addition to messenger RNA (mRNA) in the transcriptome, there are also several types of non-coding RNA (ncRNA), including miRNA, siRNA, tRNA, and long non-coding RNA (lncRNA) (Archak and Nagaraju 2007). LncRNA represents a class of RNA transcripts that are more than 200 nt in length, lack a complete open reading frame, and do not encode proteins (Guttman et al. 2009; Rinn and Chang 2012; Zhu 2012). Numerous lncRNAs can regulate gene expression at epigenetic, transcriptional, and post-transcriptional levels. Studies on plant lncRNA have been conducted in Arabidopsis (Wang et al. 2014), rice (Yuan et al. 2017; Zhang et al. 2014), wheat (Díaz et al. 2019; Xin et al. 2011), maize (Li et al. 2014), and Chlamydomonas reinhardtii (Ou et al. 2017). LncRNA is mainly involved in flowering, pollen development, and a number of other processes related to reproductive development in plants (Cho and Hsieh 2015; Karlik et al. 2019; Kim and Sung 2012; Zhang and Chen 2013). Recently, lncRNA involvement in response to biotic and abiotic stresses has also been reported (Csorba et al. 2014; Datta and Paul 2019; Tian et al. 2016; Xin et al. 2011). To date, little focus has been directed on the lncRNAs of gramineous plants and information on barley is particularly limited. This study used RNA-seq to identify mRNAs and lncRNAs related to shrunken endosperm to understand the mechanism of its molecular regulation.
Materials and methods
Plant materials
This study selected the previously identified barley seg4 (Bowman *5/BGS380 seg4) shrunken mutant (Ma et al. 2014a). The following mapping analysis showed that the gene underlying this mutant was mapped on chromosome 6H, which is inconsistent with a previous study where seg4 was located on 7H (Jarvi 1975). The seg4 mutant reported by sex1 was apparently due to mislabeling (Röder et al. 2006).
For mRNA and lncRNA analyses, a pair of near isogenic lines (NILs) was used: Bowman (wild type) and the backcrossed NIL Bowman*5/sex1 (shrunken endosperm, which was named sex1 for convenience, Fig. 1a–e). The NILs were planted in Wenjiang (103° 51′ E, 30° 43′ N), Sichuan, China. Each line was in a single 2 m row with 0.3 m between rows, and the sowing density was 15 seeds per row with 0.1 m between plants within a row. Nitrogen and superphosphate fertilizers were applied at rates of 80 and 100 kg/ha, respectively, at sowing (Yu et al. 2018). Grains on the spikelet were extracted 10, 15, and 20 days after flowering (DAF) using four biological replicates. Immediately after sampling, samples were frozen in liquid nitrogen and stored at − 80 °C for RNA extraction. Two of the four biological replicates for each time-point were used for mRNA analyses only (dataset A), while the other two replicates were used for both mRNA and lncRNA analyses (dataset B). Datasets A and B were obtained in different runs.

Phenotype comparison between shrunken endosperm mutants sex1 and Bowman. a Plants, b young spike, c mature spike, d thousand-grain weight, *** significance level at P < 0.001. e Grains at 5, 10, 15, 20, 25, 30, 35 DAF and mature stages
Chromosomal mapping of single-nucleotide polymorphisms
The clean reads of each line-time point sample of both datasets A and B were pooled. According to Ma et al. (2014b), single-nucleotide polymorphisms (SNPs) between two samples were identified: Bowman_vs_sex1. Alignment of reads to high-confidence and low-confidence barley genes from Mascher et al. (2017) (http://webblast.ipk-gatersleben.de/barley_ibsc/downloads/) was conducted with a maximum of two mismatches per read. Minimum coverage for the declaration of an SNP was four reads. SNPs were identified using the Biokanga snpmarkers sub-process with a minimum score of 80%, i.e. the percentage of a given nucleotide at the SNP position in each sample was at least 80%.
Expressed genes that contain SNP (SNP-EGs) and SNPs were mapped on chromosomes by BLAST against the 6H pseudomolecule sequence (Mascher et al. 2017). All sequence alignments were conducted using the BLASTN 2.2.26 + algorithm with an e-value < 1e− 5 and a length > 100 bp.
RNA extraction, cDNA library construction, and Illumina sequencing
Total RNA was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. For dataset B with lncRNA sequencing, the Ribo-Zero rRNA Removal kit (Epicentre, Madison, WI, USA) was used to remove sample rRNA. The purity, concentration, integrity, and contamination with genomic DNA of the RNA samples were detected prior to sequencing. Libraries were constructed and sequenced using an Illumina HiSeqTM 2500 sequencing platform (Illumina Inc., San Diego, CA, USA). Clean reads (clean data, 150 pair ends) were obtained by removing adaptor contaminants, empty reads, and low-quality reads. All of these processes were conducted by the BioMarker Technologies Corporation in Beijing. The obtained RNA-seq reads were deposited to the NCBI Sequence Read Archive (SRA) database under the accession number SRP217977.
Identification and analysis of differentially expressed genes
Clean reads were aligned to the reference genome PGSB_r1 (https://webblast.ipk-gatersleben.de/barley_ibsc/downloads/) (Mascher et al. 2017) using HISAT2 (http://ccb.jhu.edu/software/hisat2/index.shtml) (Kim et al. 2015) and Cufflinks (Trapnell et al. 2010). Mapped reads were spliced using String Tie (1.3.1) (https://ccb.jhu.edu/software/stringtie/index.shtml) (Pertea et al. 2015) and were aligned with the annotated information (both high- and low-confidence barley genes, https://webblast.ipk-gatersleben.de/barley_ibsc/downloads) (Mascher et al. 2017) to detect new transcripts. These new transcripts were further obtained by filtering out sequences that encode short peptide chains (of less than 50 amino acid residues) or that contain only a single exon.
Clean reads were aligned against high- and low-confidence barley genes (https://webblast.ipk-gatersleben.de/barley_ibsc/downloads) (Mascher et al. 2017) and newly identified transcripts to estimate the gene expression level using the Fragments per Kilobase of transcript per Million fragments mapped (FPKM) method. Differential expression analysis of both samples was performed using the DESeq fit (http://www.bioconductor.org/packages/release/bioc/html/DESeq.html) to duplicate biological samples (Wang et al. 2010). Differentially expressed genes (DEGs) were determined with a false discovery rate (FDR) threshold < 0.05 and an absolute value of log2FoldChange > 1 or ‘inf’ (the value of one comparative object is zero and the other is not). DEGs were identified accordingly for datasets A and B. Unique DEGs were further obtained using reciprocal BLASTn of DEGs from datasets A and B.
Functional analysis of DEGs
Following the procedure of Zeng et al. (2014), to confirm putative biological functions and biochemical pathways for DEGs, Blast2GO (Ana et al. 2005) was used for Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. GO enrichment analysis of DEGs was conducted using agriGov2.0 (Tian et al. 2017) (P ≤ 0.05) with single enrichment analysis (SEA) by comparing the list of DEGs with the customized annotation reference from the IPK Barley Blast server (Mascher et al. 2017). KOBAS 3.0 was used to define significant enrichment of DEGs in KEGG pathways (P ≤ 0.05).
Identification of lncRNAs
Clean reads of each library were aligned with the barley reference genome PGSB_r1 (https://webblast.ipk-gatersleben.de/barley_ibsc/downloads/) using HISAT2. Mapped reads were assembled into transcripts using String Tie (1.3.1) (Pertea et al. 2015) and the expression levels were calculated as FPKM, also using String Tie (1.3.1). To identify lncRNAs, a three-step pipeline was adopted as described by Qiu et al. (2019) and Ding et al. (2019): (1) transcripts with class codes “i”, “x”, “u”, “o”, and “e” were selected in the NONCODE database (Bu et al. 2012; Jie et al. 2013); (2) transcripts that belonged to known protein-coding genes on the same strand were removed if they met the following criteria: length < 200 bp, exon number < 2, and FPKM < 0.1 (Kelley and Rinn 2012); (3) transcripts with coding potential were excluded according to the evaluation of the Coding Potential Calculator (CPC) (Kong et al. 2007), the Coding-Non-Coding Index (CNCI) (Sun et al. 2013), and the Pfam (Finn et al. 2014) and Coding-Potential Assessment Tool (CPAT) (Wang et al. 2013). Differential expression analysis of both groups was performed using the DESeq R package. Differentially expressed lncRNAs (DE lncRNAs) were identified via FDR < 0.05 and |log2FC| > 1.
Prediction of target genes and construction of lncRNA and mRNA co-expression networks
Based on the action mode of lncRNAs and their target genes, two prediction methods were used to identify target genes: (1) Since lncRNAs typically act on their target genes through cis or trans acting (Kornienko et al. 2013), the genes spaced 100 kb up- and down-stream of lncRNAs were considered as target genes (Wang et al. 2018); (2) lncRNAs interact with mRNA through formation of two-paired RNAs. LncTar was used to calculate the free energy and the normalized free energy of paired RNAs. The cutoff of calculated normalized free energy was set as to − 0.1 to determine target genes (Li et al. 2015).
As Wang et al. (2018) described, based on the FPKM values of both target genes and lncRNAs, a lncRNA-target gene co-expression network was constructed using Cytoscape_v3.6.1 software (Saito et al. 2012). According to the results, for each lncRNA-target gene pair, the lncRNA with a degree > 10 and P < 0.05 was defined as hub lncRNA in the regulatory network. Functional analysis of the target genes associated with hub lncRNAs was then performed to identify candidate lncRNAs.
Functional analysis of differentially expressed lncRNAs
To better understand the function of DE lncRNAs, clusters in orthologous groups of protein (COG), KEGG, and GO annotation were investigated on target genes for DE lncRNAs using BLAST. The COG database was constructed based on the phylogenetic relationships among bacteria, algae, and eukaryotes. Genes in an orthologous relationship can be classified using the resulting COG database (Tatusov 2000). GO enrichment analysis of target genes was performed with the topGO R packages. KOBAS software was used to test the statistical enrichment of target genes in KEGG pathways (Kanehisa et al. 2004).
Quantitative real-time PCR verification
Seven DEGs and five DE lncRNAs were randomly selected and verified by quantitative real-time PCR (qRT-PCR). The plant RNA Kit R6827 (Omega Bio-Tek, Norcross, GA, USA) was used to extract RNAs and the TaKaRa Prime Script TMRT reagent Kit with gDNA Eraser (Perfect Real Time; TaKaRa, Shiga, Japan) was used to synthesize cDNA according to the manufacturer’s instructions. QRT-PCR was performed using SYBR Premix Ex Taq™ II (TaKaRa, Shiga, Japan). The 10 µl mixtures for PCR reactions consisted of 5 µl of 2× SYBR Green II Mix, 0.5 µl of each forward and reverse primers, 2 µl of cDNA, and 2 µl of ddH2O. The PCR program applied 94 °C for 5 min, followed by 35 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s. Three biological replicates were conducted for each sample. The relative expression levels were calculated using the 2−ΔΔCt method (Ma et al. 2014b). Specific primers for qRT-PCR were designed using Primer 5.0 software (Table S1). GAPDH was used as reference gene (Rapacz et al. 2012).
Results
Phenotype of sex1
Plant height, spike length, and spikelet number per spike were not significantly different between NILs of sex1 and Bowman (Fig. 1a, b). Grain shape and weight showed significant morphological differences between sex1 and Bowman after 15 DAF (Fig. 1e). The grains of sex1 were shrunken, while those of Bowman were plump (Fig. 1c, e). Sex1 had significantly lower thousand-grain weight than Bowman (P < 0.001) (Fig. 1d).
Sequence assembly of transcriptome and lncRNA sequencing data
Totals of 73.67 Gb (≥ 6.01 Gb/sample) and 145.66 Gb (≥ 12.03 Gb/sample) of clean data were generated for datasets A and B, respectively. The GC content of each sample ranged from 52.97 to 56.34% for dataset A, from 50.31 to 55.83% for dataset B, and the Q30 (a 0.1% chance of error and 99.9% confidence) values were > 89.67% and 88.77% for datasets A and B, respectively. Approximately 82.58–89.20% (dataset A) and 77.02–89.20% (dataset B) of the clean reads were successfully mapped on the barley genome reference sequence (Table S2). These results indicated that the sequence quality was sufficiently high to enable further analysis.
Distribution of SNPs and SNP-EGs on the barley genome
A total of 1949 SNPs and 585 SNP-EGs were detected, and chromosome 6H had significantly more SNPs and SNP-EGs than other chromosomes (Fig. 2a). SNPs were mainly distributed in three regions of the 6H chromosome: 25–165 Mb, 345–465 Mb, and 550–565 Mb (Fig. 2b).

Distribution of SNPs and SNP-EGs across the seven chromosomes (a) and along the 6H pseudomolecule in barley (b)
Identification of DEGs
A total of 1292 (308 newly assembled and 984 known) and 1290 (320 newly assembled and 970 known) DEGs were identified from dataset A and dataset B, respectively (Table S3); furthermore, both datasets had 105 new and 354 known genes. Finally, 2123 unique DEGs were found between Bowman and sex1 during different flowering periods. Of these, 1140 were up-regulated and 983 were down-regulated. Among the 2123 DEGs, seven were expressed across three periods (Fig. 3). In addition, there were 188, 207, and 1517 unique DEGs at 10 DAF, 15 DAF, and 20 DAF, respectively (Fig. 3).

Venn diagram of DEGs in sex1 compared with Bowman at 10 DAF, 15 DAF, and 20 DAF
GO and KEGG pathway functional enrichment analysis of DEGs
The 2123 DEGs were classified into three main categories (Cellular Component, Molecular Function and Biological Process) by GO annotation. At 10 DAF, the terms of organelle part, nucleosome, chromatin, and chromosome were dominant in Cellular Component, binding and catalytic activity were dominant in Molecular Function, and cellular process, nucleosome organization, and chromatin assembly were dominant in Biological Process (Fig. 4a, Table S4). At 15 and 20 DAF, the terms of cell part, cell, and organelle were dominant in cellular component, binding and catalytic activity were dominant in molecular function, and metabolic process, cellular process and single-organism process were dominant in biological process (Figs. S1a, S2a, Table S4). GO terms demonstrated that genes related to barley endosperm development encoded diverse regulators and proteins.

Functional categories of DEGs. The a GO and b KEGG enrichment analysis of DEGs between Bowman and sex1 at 10 DAF. For a, the Y and X axes correspond to GO terms and the number of DEGs, respectively. For b, the Y axis corresponds to KEGG pathway, and the X axis shows the enrichment ratio between the numbers of DEGs and all unigenes enriched in a particular pathway. The color of the dot represents q-value, and the size of the dot represents the number of DEGs in a given pathway
To further elucidate the biological functions and interactions of genes related to shrunken endosperm, pathway analysis was conducted using the KEGG pathway database. This showed that 51, 104, and 567 DEGs were mapped to 31, 39, and 98 KEGG pathways at 10 DAF, 15 DAF, and 20 DAF, respectively (Table S5). Most of these DEGs were expressed differently in secondary metabolic synthesis and protein synthesis in the endoplasmic reticulum, photosynthesis-antenna proteins, carbon metabolism, and metabolic pathways. At 10 DAF, 51 DEGs were mainly enriched in ribosomes, RNA transcription, RNA degradation, oxidative phosphorylation, brassinosteroid (BR) biosynthesis, as well as starch and sucrose metabolism (Fig. 4b). At 15 DAF, 104 DEGs were mainly concentrated in the biosynthesis of secondary metabolites, carbon metabolism, and protein procession in the endoplasmic reticulum, as well as metabolic pathways (Fig. S1b). At 20 DAF, 567 DEGs were mainly enriched in starch and sucrose metabolism, and protein processing in endoplasmic reticulum, metabolic pathways, biosynthesis of secondary metabolites, and galactose metabolism (Fig. S2b).
Identification and characterization of lncRNAs
According to the three-step screening method, 3109 lncRNAs (10 known, Table S7) were predicted, including 2361 (75.9%) long intervening noncoding RNAs (lincRNAs), 318 (10.2%) antisense lncRNAs, 197(6.3%) intron lncRNAs, and 233 (7.5%) sense lncRNAs (Fig. S3a, b, Table S6). To understand the differences in structure and sequence between mRNA and lncRNA, their length, exon number, and expression level were compared. The results showed that 36.57% of lncRNAs had > 400 nt, while 18.57% of mRNAs had > 3000 nt (Fig. S3c, d). Furthermore, 74.17% of lncRNAs had two exons and 77.6% of mRNAs had more than two exons (Fig. S3e, f).
Differential expression analysis of lncRNA
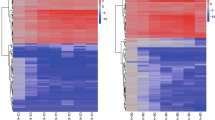
Fifty-seven unique DE lncRNAs were detected at three time-points (Table S8). Thirty-three lncRNAs were DE at 10 DAF, with 17 up-regulated and 16 down-regulated; seven lncRNAs were DE at 15 DAF, with 3 up-regulated and 4 down-regulated; 20 lncRNAs were DE at 20 DAF, with 14 up-regulated and 6 down-regulated; and there were 30, 7, and 17 specifically expressed lncRNAs during each period, respectively (Fig. 5a). Hierarchical clustering analyses of the gene expression patterns of the DE lncRNAs were conducted. The results showed that compared with Bowman, most DE lncRNAs were up-regulated and few DE lncRNAs were down-regulated in sex1 (Fig. 5b).

Venn diagram (a) and cluster map (b) of DE lncRNAs at 10 DAF, 15 DAF and 20 DAF. For b, the X axis represents the sample name and the clustering result of the samples, and the Y axis represents the clustering result of the differential lncRNAs and the lncRNA. The color represents the level of expression of the lncRNA. Red represents highly expressed lncRNAs, green represents lower expressed lncRNAs, and each row represents an individual lncRNA
Functional analysis of lncRNA target genes
For the 57 DE lncRNAs, 343 unique target genes were predicted (Table S8). Among these target genes, 225 were subjected to lncRNA trans-acting regulation, and 118 were subjected to lncRNA cis-acting regulation. LncRNAs have recently been identified as regulatory factors; however, no direct annotation exists for them in the database (Qiu et al. 2019). Thus, only the function of their target genes was analyzed using COG, KEGG, and GO. The target genes (mRNA) were clustered into different categories by COG analysis. Most were associated with categories of ‘general function prediction only’ and ‘amino acid transport and metabolism’ (Figs. 6a, S4a, S5a, Table S9).

Functional categories of the target genes of DE lncRNAs between Bowman and sex1 at 10 DAF. The a COG classify, b GO and c KEGG enrichment analysis. For a, the X and Y axes correspond to the classification content of COG and the number of target genes of lncRNA, respectively. For b, the Y and X axes correspond to GO terms and the number of target genes of lncRNA, respectively. For c, the Y axis corresponds to KEGG pathway, the X axis shows the enrichment ratio between the numbers of target genes of lncRNA and all unigenes enriched in a particular pathway. The color of the dot represents q-value, and the size of the dot represents the number of target genes of lncRNA mapped to the referent pathway
GO analysis indicated that the functions of the target genes from each time-point were similar: the cellular parts, cells, and organelles were dominant in Cellular Composition; binding and catalytic activity were dominant in Molecular Function; metabolic processes, cellular processes, and single biological processes were dominant in Biological Processes (Figs. 6b, S4b, S5b, Table S10).
KEGG analysis was conducted to investigate the involvement of target genes in metabolic processes. The results showed that these target genes participated in different metabolic pathways at various time-points (Fig. 6c, Table S11). At 10 DAF, 182 target genes (33 lncRNAs) were mainly involved in RNA degradation, amino acid biosynthesis, and the 2-oxocarboxylic acid metabolism pathway. At 15 DAF, 48 target genes (7 lncRNAs) were mainly involved in C5-branched diced acid metabolism, biosynthesis of valine, leucine and isoleucine, pantothenic acid, and CoA biosynthetic pathway (Fig. S4c, Table S11). At 20 DAF, a total of 138 target genes (20 lncRNAs) were involved in the pyruvate metabolic pathway (Fig. S5c, Table S11). This indicated that endosperm formation was associated with both the carbohydrate metabolism and the amino acid metabolism.
Co-expression network analysis of DE lncRNA and target genes
A co-expression network of DE lncRNA-target genes was constructed to identify potential interactions between DE lncRNAs and these target genes (Fig. S6, Table S12). This network identified that MSTRG.73.1, MSTRG.79665.1, MSTRG.30463.1, MSTRG.79382.1, and MSTRG.79382.1 (Fig. 7) as hub lncRNAs. These were associated with many target genes. This suggested that mRNA expression may be regulated by lncRNA, and that lncRNA was involved in grain development.

The network map of hub DE lncRNAs and their target genes. DE lncRNAs were represented by green cycles, inverted triangle represents down-regulated genes, and positive triangle represents up-regulated genes, and the color depth and size of green cycles were determined according to their degrees/connections to others
Analysis of candidate lncRNAs involved in endosperm development
The candidate lncRNA was determined based on the lncRNA-target gene co-expression network. Among the five hub lncRNAs, MSTRG.79382.1 (located on chromosome 6H) was specifically expressed in Bowman but not in sex1. To further investigate MSTRG.79382.1, functional analysis of its co-expressed target genes was conducted (Fig. S7, Table S13). COG analysis showed that its co-expressed target genes were associated with translation, ribosome structure, biological processes, inorganic ion transport, and metabolism (Fig. S7a). GO analysis showed that the functions of these genes were mainly involved in cellular parts, cells, and organelles were dominant in Cellular Components; binding and catalytic activity in Molecular Function; and metabolic processes, cellular processes, and single biological processes in Biological Processes (Fig. S7b). KEGG enrichment analysis indicated that the co-expressed target genes of MSTRG.79382.1 were mainly involved in nitrogen (N) metabolism, galactose metabolism, ribosome, as well as starch and sucrose metabolism (Fig. S7c).
Confirmation of RNA sequencing results by qRT-PCR
To confirm the expression results obtained by RNA-seq data, seven DEGs and five DE lncRNAs were selected for qRT-PCR. The qRT-PCR results were shown in Fig. 8. Although the qRT-PCR expressions of several lncRNAs and genes were not completely consistent with the sequencing data, the trends of their total expressions were similar between qRT-PCR and sequencing data (Fig. 8). High correlation coefficients (r = 0.604–0.921) were revealed between these two independent measurements. These results indicated that the RNA-seq results were reliable.

Expression validation of selected genes using qRT-PCR. The expression levels of seven DEGs and five DE lncRNAs are shown (S: sex1, B: Bowman). Error bar shows standard deviation, r value shows the goodness of fit in linear regression
Discussion
DEGs related to shrunken endosperm
Plant hormones including BR, auxin (IAA), abscisic acid (ABA), and cytokinin (CTK) play an important role during grain development (Li and Li 2015). For example, Tanabe et al. (2005) reported that the D11/CYP724B1 gene played a role in BR synthesis and regulated rice grain length. In this study, HORVU2Hr1G002230 was involved in BR biosynthesis and was down-regulated in sex1 compared with Bowman. Riefler et al. (2006) reported that CTK played an important role on embryo enlargement in Arabidopsis. Here, HORVU1Hr1G042360 and HORVU5Hr1G060870 participated in the biosynthesis of zeatin (a CTK, derived from adenine). Furthermore, their expression levels in Bowman were significantly higher than in sex1. These results suggested that the formation of shrunken endosperm in sex1 was likely closely related to the synthesis and expression of plant hormones, such as BR, CTK, and ABA. This is consistent with the results of previous studies (Basunia and Nonhebel 2019; Sreenivasulu et al. 2010).
Starch is the main component of grains, and starch biosynthesis involves both sucrose transport and its conversion to starch. In this study, DEGs involved in the starch and sucrose metabolism were enriched. For example, both up-regulated genes (HORVU1Hr1G091600, HORVU2Hr1G019180, and HORVU5Hr1G096390) and down-regulated genes (HORVU4Hr1G084390, HORVU2Hr1G109120, and HORVU2Hr1G116320) were involved in starch and sucrose metabolisms. In addition to the known genes, several newly assembled genes (i.e., 2_Hordeum_vulgare_newGene_22899, 1_Hordeum_vulgare_newGene_20615, and 2_Hordeum_vulgare_newGene_22901) were also enriched. Suppression of starch biosynthesis genes has been reported to decrease grain starch levels, thus leading to a wrinkled phenotype (Scofield et al. 2002). Consequently, it is likely that sucrose and starch metabolic pathways are also related to the formation of shrunken endosperm, thus providing a clue for the further identification of candidate genes underlying sex1.
DEGs were also enriched in fatty acid metabolism, tricarboxylic acid cycle, biosynthesis of various secondary metabolites, phenylalanine acid metabolism, and lysine degradation and autophagy regulation. Apoptosis is part of the normal development of all multicellular organisms (Bowen 1992), and autophagy is the most common form of apoptosis in both animal and plant cells. In this study, HORVU1Hr1G022190 was involved in the regulation of autophagy and was down-regulated in sex1 compared with Bowman. Starch biosynthesis is accompanied by the progression of endosperm apoptosis (Chun-Yan et al. 2010). Thus, it is likely that shrunken endosperm mutation typically produces severe apoptosis compared with plump endosperm. Differences in carbon metabolism are likely between shrunken and plump endosperm during grain development. Given the relationship between carbon metabolism and apoptosis (Azanza et al. 1996; Boyer and Shannon 1983), the abnormal carbon metabolism of sex1 likely accelerates the onset of apoptosis.
In summary, these results suggest that shrunken endosperm in barley is closely related to plant hormone signal transduction, starch and sucrose metabolism, and cell apoptosis. Plant hormones and apoptosis affected starch and sucrose metabolism, which resulted in shrunken endosperm.
Chromosomal interval of the sex1 candidate gene
This study selected the seg4 (Bowman *5/BGS380 seg4) shrunken mutant (Ma et al. 2014a) for further analysis, the candidate gene of which was previously mapped on 7H (Jarvi 1975). According to the SNP distribution, the SNP-EGs were mainly located on chromosome 6H of barley, suggesting that the candidate gene for the shrunken endosperm mutant is most likely on 6H. Further demonstration would indicate this means that this mutant might in fact be sex1.
The gene sex1 was previously genetically located in the centromeric region of chromosome 6H (Jarvi 1975). Due to the inhibition of recombination and the low SNP density in the centromeric region (Chen 2002; Fan et al. 2017; Li et al. 2019; Liu et al. 2018; Saintenac et al. 2009), further fine mapping of sex1 is required using genetic segregation populations.
To further identify sex1-related genes, RNA-seq data was combined with gene mapping to mine candidate genes. This identified 210 DEGs on 6H. According to gene expression and annotation information, 13 DEGs were related to genes expressed in the shrunken endosperm mutant sex1. Among them, HORVU6Hr1G022420, HORVU6Hr1G023770, and HORVU6Hr1G038700 were shown to be involved in leucine metabolism, and were up-regulated in sex1 compared with Bowman. This indicates that sex1 was related to high leucine content, which is consistent with previous studies (Ullrich and Eslick 1978). HORVU6Hr1G066330, HORVU6Hr1G071920, HORVU6Hr1G011110, HORVU6Hr1G078420, HORVU6Hr1G075240, HORVU6Hr1G062070, and HORVU6Hr1G037700 encoded enzymes related to the starch and sucrose metabolism. HORVU6Hr1G038760 and HORVU6Hr1G038750 encoded CYP450 proteins in the CYP450 family, and their expression levels in Bowman were significantly higher than in sex1, further indicating that the CYP450 protein played an important role in endosperm development. In addition, HORVU6Hr1G037950 encodes a mitochondrial substrate carrier family protein (MCF), and the expression was significantly higher in Bowman than in sex1. MCF is a protein with a solute carrier in the inner mitochondrial membrane. Brittle 1 (BT1) proteins are members of the MCF that only occur in plants. Zea mays Brittle 1–1 (ZmBT1-1) is an essential component of the starch biosynthetic mechanism of the maize endosperm, where it enables ADPglucose transport from the cytosol to the amyloplast, and MCF is closely related to the starch and sucrose metabolism (Pozueta-Romero et al. 2019). As described by Lee et al. (2007) specific AGPase genes could lead to the decrease of starch synthesis, thus resulting in a shrunken endosperm phenotype. Therefore, HORVU6Hr1G037950 likely participates in the regulation of the expression of the sex1 shrunken endosperm.
LncRNAs related to the shrunken endosperm
This study identified 3109 lncRNAs and suggested their possible roles in the regulation of grain development. Furthermore, 57 DE lncRNAs were identified in sex1 compared with its isogenic line Bowman. LncRNAs typically function by regulating target mRNAs. Thus, 343 target genes were predicted for these DE lncRNAs. Most lncRNAs were found to be highly conserved over three periods. Functional analysis of these target genes showed that these lncRNAs were mainly involved in the biosynthesis of leucine and isoleucine as well as pyruvate metabolism pathways. These DE lncRNA might play important roles in endosperm development of barley grains, and participated in various regulatory networks with target genes.
Co-expression network analysis identified a hub lncRNA (MSTRG.79382.1) located on chromosome 6H. It was specifically expressed in Bowman but not in sex1. Functional analysis indicated that its target genes were mainly involved in N metabolism, galactose metabolism, ribosome, as well as starch and sucrose metabolism. N metabolism provides energy for carbohydrate reserves, thus affecting the starch and sucrose metabolism (Numoto et al. 2019). Combined with functional analysis of DEGs, MSTRG.79382.1 was predicted to regulate shrunken endosperm through both N metabolism, as well as starch and sugar metabolism. This study demonstrated that in addition to mRNAs, lncRNAs play a vital role during endosperm development. The results presented here lay a foundation for further studies on the molecular mechanism of barley shrunken endosperm.
References
Ana C, Stefan GT, Juan Miguel GG, Javier T, Manuel T, Montserrat R (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Archak S, Nagaraju J (2007) Computational prediction of rice (Oryza sativa) miRNA targets. Genomics Proteomics Bioinform 5:196–206
Azanza F, Bar-Zur A, Juvik JA (1996) Variation in sweet corn kernel characteristics associated with stand establishment and eating quality. Euphytica 87:7–18
Basunia MA, Nonhebel HM (2019) Hormonal regulation of cereal endosperm development with a focus on rice (Oryza sativa). Funct Plant Biol 46:493–506
Bosnes M, Harris E, Aigeltinger L, Olsen O (1987) Morphology and ultrastructure of 11 barley shrunken endosperm mutants. Theor Appl Genet 74:177–187
Bowen I (1992) Apoptosis: the molecular basis of cell death (current-cominunications in cell & molecular biology). FEBS Lett 3:91–93. https://doi.org/10.1016/0014-5793(92)81160-n
Boyer CD, Shannon JC (1983) The use of endosperm genes for sweet corn improvement. Plant Breed Rev 1:139–161
Bu D et al (2012) NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic Acids Res 40:D210–D215
Chen M (2002) An integrated physical and genetic map of the rice genome. Plant Cell Online 3:537–545
Cho K-O, Hsieh J (2015) The lncRNA Pnky in the brain. Cell Stem Cell 16:344–345
Chun-Yan et al (2010) Starch synthesis and programmed cell death during endosperm development in triticale (x Triticosecale Wittmack). J Integr Plant Biol 52:602–615
Clarke B, Liang R, Morell MK, Bird AR, Jenkins CLD, Li Z (2008) Gene expression in a starch synthase IIa mutant of barley: changes in the level of gene transcription and grain composition. Funct Integr Genomics 8:211–221
Cook F, Hughes N, Nibau C, Orman-Ligeza B, Schatlowski N, Uauy C, Trafford K (2018) Barley lys3 mutants are unique amongst shrunken-endosperm mutants in having abnormally large embryos. J Cereal Sci 82:S073352101830167X
Csorba T, Questa JI, Sun Q (2014) Antisense COOLAIR mediates the coordinated switching of chromatin states at FLC during vernalization. Proc Natl Acad Sci 111:16160–16165
Dai H et al (2013) Comparative proteomic analysis of aluminum tolerance in tibetan wild and cultivated barleys. PLoS ONE 8:e63428
Datta R, Paul S (2019) Long non-coding RNAs: fine-tuning the developmental responses in plants. J Biosci 44:77
De Arcangelis E, Djurle S, Andersson AA, Marconi E, Messia MC, Andersson R (2019) Structure analysis of β-glucan in barley and effects of wheat β-glucanase. J Cereal Sci 85:175–181
Desa U (2015) World population prospects: the 2015 revision, key findings and advance tables. Working PaperNo
Díaz ML, Soresi DS, Basualdo J, Cuppari SJ, Carrera A (2019) Transcriptomic response of durum wheat to cold stress at reproductive stage. Mol Biol Rep 46:2427–2445
Ding Z et al (2019) Strand-specific RNA-seq based identification and functional prediction of drought-responsive lncRNAs in cassava. BMC Genom 20:214
Djarot IN, Peterson DM (1991) Seed development in a shrunken endosperm barley mutant. Ann Bot 68:495–499
Eslick R, Ries M (1976) Positioning sex1 on chromosome 6. Barley Genet Newsl 6:21–22
Fan C et al (2017) Identification of QTLs controlling grain protein concentration using a high-density SNP and SSR linkage map in barley (Hordeum vulgare L.). BMC Plant Biol 17:122
Felker F, Peterson D, Nelson O (1983) Growth characteristics, grain filling, and assimilate transport in a shrunken endosperm mutant of barley. Plant Physiol 72:679–684
Felker FC, Peterson DM, Nelson OE (1984a) Development of tannin vacuoles in chalaza and seed coat of barley in relation to early chalazal necrosis in the seg1 mutant. Planta 161:540–549
Felker FC, Peterson DM, Nelson OE (1984b) Sucrose uptake and labeling of starch in developing grains of normal and segl barley. Plant Physiol 74:43–46
Felker FC, Peterson DM, Nelson OE (1985) Anatomy of immature grains of eight maternal effect shrunken endosperm barley mutants. Am J Bot 72:248–256
Finn RD et al (2014) Pfam: the protein families database. Nucleic Acids Res 42:222–230
Franckowiak JD, Lundqvist U, NG B (2010) Descriptions of barley genetic stocks for 2010. Barley Genet Newsl 40:45–177
Guttman M et al (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAS in mammals. Nature 458(7235):223–227
Jain M (2012) Next-generation sequencing technologies for gene expression profiling in plants. Brief Funct Genom 11:63–70
Jarvi AJ (1975) Shrunken endosperm mutants in barley, Hordeum vulgare L. Cropence 15:363–366
Jie L et al (2013) Identification and characterization of long non-coding RNAs related to mouse embryonic brain development from available transcriptomic data. PLoS ONE 8:e71152
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res 32:D277–D280
Karlik E, Ari S, Gozukirmizi N (2019) LncRNAs: genetic and epigenetic effects in plants. Biotechnol Biotechnol Equip.https://doi.org/10.1080/13102818.2019.1581085
Kelley D, Rinn J (2012) Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol 13(11):R107
Khahani B, Tavakol E, Vahid SJ (2019) Genome-wide meta-analysis on yield and yield-related QTLs in barley (Hordeum vulgare L.). Mol Breed.https://doi.org/10.1007/s11032-019-0962-y
Kim ED, Sung S (2012) Long noncoding RNA: unveiling hidden layer of gene regulatory networks. Trends Plant Sci 17:16–21
Kim D, Langmead B, Steven LS (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357–360
Kong L, Zhang Y, Ye Z-Q, Liu X-Q, Zhao S-Q, Wei L, Gao G (2007) CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35:W345
Kornienko AE, Guenzl PM, Barlow DP, Pauler FM (2013) Gene regulation by the act of long non-coding RNA transcription. BMC Biol 11:59
Lee SK et al (2007) Identification of the ADP-glucose pyrophosphorylase isoforms essential for starch synthesis in the leaf and seed endosperm of rice (Oryza sativa L.). Plant Mol Biol 65:531–546
Li N, Li Y (2015) Maternal control of seed size in plants. J Exp Bot 66:1087–1097
Li L et al (2014) Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol 15:R40
Li J, Ma W, Zeng P, Wang J, Geng B, Yang J, Cui Q (2015) LncTar: a tool for predicting the RNA targets of long noncoding RNAs. Brief Bioinform 16:806
Li X et al (2019) Development of an integrated 200K SNP genotyping array and application for genetic mapping, genome assembly improvement and genome wide association studies in pear (Pyrus). Plant Biotechnol J 17:1582–1594
Liu J et al (2018) A 55K SNP array-based genetic map and its utilization in QTL mapping for productive tiller number in common wheat. Theor Appl Genet 131:2439–2450
Ma J et al (2014a) Characterization of shrunken endosperm mutants in barley. Gene 539:15–20
Ma J et al (2014b) Transcriptome and allele specificity associated with a 3BL locus for Fusarium crown rot resistance in bread wheat. PLoS ONE 9:e113309
Mascher M et al (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature 544:427–433
Morell MK et al (2010) Barley sex6 mutants lack starch synthase lla activity and contain a starch with novel properties. Plant J 34:173–185
Nese S et al (2010) De-regulation of abscisic acid contents causes abnormal endosperm development in the barley mutant seg. Plant J Cell Mol Biol 64:589–603
Numoto AY, Vidigal Filho PS, Scapim CA, Franco AAN, Ortiz AHT, Marques OJ, Pelloso MF (2019) Agronomic performance and sweet corn quality as a function of inoculant doses (Azospirillum brasilense) and nitrogen fertilization management in summer harvest. Bragantia 78:26–37
Ou L et al (2017) Noncoding and coding transcriptome analysis reveals the regulation roles of long noncoding RNAs in fruit development of hot pepper (Capsicum annuum L.). Plant Growth Regul 83:1–16
Patron N, Greber B, Fahy B, Laurie D, Parker M, Denyer K (2004) The lys5 mutations of barley reveal the nature and importance of plastidial ADP-Glc transporters for starch synthesis in cereal endosperm. Plant Physiol 135:2088–2097
Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL (2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33:290–295
Pozueta-Romero J et al (2019) Mitochondrial Zea mays Brittle1-1 Is a major determinant of the metabolic fate of incoming sucrose and mitochondrial function in developing maize endosperms. Front Plant Sci 10:242
Qi J et al (2019) The anther-specific CYP704B is potentially responsible for MSG26 male sterility in barley. Theor Appl Genet 132:2413–2423
Qiu C-W, Zhao J, Chen Q, Wu F (2019) Genome-wide characterization of drought stress responsive long non-coding RNAs in Tibetan wild barley. Environ Exp Bot 164:124–134
Ramage RT, Scheuring JF (1976) Shrunken endosperm mutants Seg6 and Seg7 [Barley]. Barley Genet Newsl 6:59–60
Rapacz M, Stępień A, Skorupa K (2012) Internal standards for quantitative RT-PCR studies of gene expression under drought treatment in barley (Hordeum vulgare L.): the effects of developmental stage and leaf age. Acta Physiol Plant 34:1723–1733
Riefler M, Novak O, Strnad M, Schmulling T (2006) Arabidopsis cytokinin receptor mutants reveal functions in shoot growth, leaf senescence, seed size, germination, root development, and cytokinin metabolism. Plant Cell 18:40–54
Rinn JL, Chang HY (2012) Genome regulation by long noncoding RNAs. Annu Rev Biochem 81:145–166
Röder MS, Kaiser C, Weschke W (2006) Molecular mapping of the shrunken endosperm genes seg8 and sex1 in barley (Hordeum vulgare L.). Genome 49:1209–1214
Saintenac C, Falque M, Martin OC, Paux E, Feuillet C, Sourdille P (2009) Detailed recombination studies along chromosome 3B provide new insights on crossover distribution in wheat (Triticum aestivum L.). Genetics 181:393–403
Saito R et al (2012) A travel guide to Cytoscape plugins. Nat Methods 9:1069–1076
Schmalenbach I, Jens L, Klaus P (2008) Identification and verification of QTLs for agronomic traits using wild barley introgression lines. Theor Appl Genet 118:483–497
Schulman AH, Tomooka S, Suzuki A, Myllärinen P, Hizukuri S (1995) Structural analysis of starch from normal and shx (shrunken endosperm) barley (Hordeum vulgare L.). Carbohydr Res 275:361–369
Scofield GN, Hirose T, Gaudron JA, Furbank RT, Ohsugi R (2002) Antisense suppression of the rice sucrose transporter gene, OsSUT1, leads to impaired grain filling and germination but does not affect photosynthesis. Funct Plant Biol 29:815–826
Shanrong Z, Wai-Ping FL, Anton B, Karen N, Xuejun L (2014) Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 9:e78644
Solomon CU, Drea S (2019) Delineation of post-phloem assimilate transport pathway into developing caryopsis of Brachypodium distachyon. BioRxiv:718569
Sreenivasulu N, Borisjuk L, Junker BH, Mock HP, Wobus U (2010) Barley grain development: toward an integrative view. Int Rev Cell Mol Biol 281:49–89
Sun L et al (2013) Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res 41:e166–e166
Tanabe S et al (2005) A novel cytochrome P450 is implicated in brassinosteroid biosynthesis via the characterization of a rice dwarf mutant, dwarf11, with reduced seed length. Plant Cell 17:776–790
Tatusov LR (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36
Tian J et al (2016) Population genomic analysis of gibberellin-responsive long non-coding RNAs in Populus. J Exp Bot 67:2467–2482
Tian T et al (2017) agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res 45:W122–W129
Trapnell C et al (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515
Ullrich SE, Eslick RF (1978) Lysine and protein characterization of spontaneous shrunken endosperm mutants of barley. Crop Sci 18:97–99
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Wang L, Feng Z, Wang X, Wang X, Zhang X (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26:136–138
Wang L, Park HJ, Dasari S, Wang S, Kocher J-P, Li W (2013) CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res 41:e74–e74. https://doi.org/10.1093/nar/gkt006
Wang H, Chung PJ, Liu J, Jang IC, Kean MJ, Xu J, Chua NH (2014) Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res 24:444–453
Wang Y et al (2018) Analysis of long-non-coding RNAs associated with ethylene in tomato. Gene 674:151–160
Xin M et al (2011) Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol 11:61–61
Yu M et al (2018) Analysis of contributors to grain yield in wheat at the individual quantitative trait locus level. Plant Breed.https://doi.org/10.1111/pbr.12555
Yuan J et al (2017) Stress-responsive regulation of long noncoding RNAs’ polyadenylation in Oryza sativa. Plant J 93:814–827
Zeng J et al (2014) Comparative transcriptome profiling of two tibetan wild barley genotypes in responses to low potassium. PLoS ONE 9:e100567
Zhang YC, Chen YQ (2013) Long noncoding RNAs: new regulators in plant development. Biochem Biophys Res Commun 436:111–114
Zhang YC et al (2014) Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol 15:512
Zhu W (2012) Molecular functions of long non-coding RNAs in plants. Genes 3:176–190
Acknowledgements
This work is supported by the National Natural Science Foundation of China (31970243 and 31971937), the International Science and Technology Cooperation and Exchanges Program of Science and Technology Department of Sichuan Province (2017HH0076), and the Key Projects of Scientific and Technological Activities for Overseas Students of Sichuan Province. We thank MogoEdit Bianji Company (http://www.mogoedit.com/) for editing the English text of this manuscript. We thank the anonymous referees for critical reading and revising of this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Functional categories of DEGs. The (a) GO and (b) KEGG enrichment analysis of DEGs between Bowman and sex1 at 15 DAF. Electronic supplementary material 1 (TIF 8026 kb)
Fig. S2
Functional categories of DEGs. The (a) GO and (b) KEGG enrichment analysis of DEGs between Bowman and sex1 at 20 DAF. Electronic supplementary material 2 (TIF 8238 kb)
Fig. S3
Characterization of all lncRNAs and mRNAs. (a) Venn diagrams of coding potential prediction of lncRNAs by CPC, CPAT, CNCI and pfam. (b) Classification of lncRNAs. X axis indicates different types of lncRNA, Y axis indicates the corresponding number of lncRNA. Distributions of lncRNAs and mRNAs in transcript length (c, d), and exon number (e, f), respectively. Electronic supplementary material 3 (TIF 12861 kb)
Fig. S4
Functional categories of the target genes of DE lncRNAs between Bowman and sex1 at 15 DAF. The (a) COG classify, (b) GO and (c) KEGG enrichment analysis. Electronic supplementary material 4 (TIF 11789 kb)
Fig. S5
Functional categories of the target genes of DE lncRNAs between Bowman and sex1 at 20 DAF. The (a) COG classify, (b) GO and (c) KEGG enrichment analysis. Electronic supplementary material 5 (TIF 12021 kb)
Fig. S6
The network map of DE lncRNAs and their target genes. Electronic supplementary material 6 (TIF 12304 kb)
Fig. S7
Functional analysis of MSTRG.79382.1. The (a) COG analysis, (b) GO annotation and (c) KEGG enrichment analysis of MSTRG.79382.1. Electronic supplementary material 7 (TIF 11757 kb)
Table S1
Primers used for qRT-PCR for selected DEGs and DE lncRNAs. Electronic supplementary material 8 (XLSX 9 kb)
Table S2
Summary of Illumina RNA-seq reads for 24 samples (dataset A and dataset B). Electronic supplementary material 9 (XLSX 12 kb)
Table S3
DEGs in dataset A and dataset B. Electronic supplementary material 10 (XLSX 65 kb)
Table S4
GO analysis of DEGs at 10, 15 and 20 DAF. Electronic supplementary material 11 (XLSX 22 kb)
Table S5
KEGG enrichment annotation of DEGs at 10, 15 and 20 DAF. Electronic supplementary material 12 (XLSX 22 kb)
Table S6
Details of classification of lncRNA. Electronic supplementary material 13 (XLSX 58 kb)
Table S7
Summary of 10 known lncRNAs. Electronic supplementary material 14 (XLSX 9 kb)
Table S8
Summary of DE lncRNAs and their target genes. Electronic supplementary material 15 (XLSX 17 kb)
Table S9
COG classification of target genes DE lncRNAs at 10, 15 and 20 DAF. Electronic supplementary material 16 (XLSX 14 kb)
Table S10
GO analysis of target genes of DE lncRNAs at 10, 15 and 20 DAF. Electronic supplementary material 17 (XLSX 15 kb)
Table S11
KEGG enrichment annotation of target genes of DE lncRNAs at 10, 15 and 20 DAF. Electronic supplementary material 18 (XLSX 14 kb)
Table S12
DE lncRNA and target gene co-expression network analysis. Electronic supplementary material 19 (XLSX 21 kb)
Table S13
Functional analysis of MSTRG.79382.1. Electronic supplementary material 20 (XLSX 15 kb)
Rights and permissions
About this article

Cite this article
Zou, Y., Tang, H., Li, T. et al. Identification and characterization of mRNAs and lncRNAs of a barley shrunken endosperm mutant using RNA-seq. Genetica 148, 55–68 (2020). https://doi.org/10.1007/s10709-020-00087-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-020-00087-2