
Abstract
To explore the spatio-temporal dynamics of endangered fin whales (Balaenoptera physalus) within the baleen whale (Mysticeti) lineages, we analyzed 148 published mitochondrial genome sequences of baleen whales. We used a Bayesian coalescent approach as well as Bayesian inferences and maximum likelihood methods. The results showed that the fin whales had a single maternal origin, and that there is a significant correlation between geographic location and evolution of global fin whales. The most recent common female ancestor of this species lived approximately 9.88 million years ago (Mya). Here, North Pacific fin whales first appeared about 7.48 Mya, followed by a subsequent divergence in Southern Hemisphere approximately 6.63 Mya and North Atlantic about 4.42 Mya. Relatively recently, approximately 1.76 and 1.42 Mya, there were two additional occurrences of North Pacific populations; one originated from the Southern Hemisphere and the other from an uncertain location. The evolutionary rate of this species was 1.002 × 10−3 substitutions/site/My. Our Bayesian skyline plot illustrates that the fin whale population has the rapid expansion event since ~ 2.5 Mya, during the Quaternary glaciation stage. Additionally, this study indicates that the fin whale has a sister group relationship with humpback whale (Meganoptera novaeangliae) within the baleen whale lineages. Of the 16 genomic regions, NADH5 showed the most powerful signal for baleen whale phylogenetics. Interestingly, fin whales have 16 species-specific amino acid residues in eight mitochondrial genes: NADH2, COX2, COX3, ATPase6, ATPase8, NADH4, NADH5, and Cytb.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The fin whale, Balaenoptera physalus (Linnaeus, 1758), is the second largest animal on our planet—about 27 m in body length and 75 tonnes in weight—after the blue whale. Its body shape is long and slender, and the body color is gray and white respectively for the dorsal and ventral side. To date, the fin whale has provided important benefits such as meat, blubber, and baleen to humans. However, this animal is unfortunately close to extinction due to commercial whaling; their global population estimates range from 100,000 to 119,000 (Reilly et al. 2008). In order to prevent extinction, the International Whaling Commission (IWC) recently started to control the commercial hunting of fin whales.
Conservation and management of the endangered species require an understanding of its demography, biogeography, ecology, behavior, genetics, and their interactions. When exploring the evolutionary phylogenetics of animals, the mitochondrial DNA is a very useful genetic marker largely due to its maternal inheritance, relatively low rate of genetic recombination, and the presence of strictly orthologous genes evolving at different rates (Saccone et al. 1999). To this end, many works have been published regarding cetacean phylogeny on the basis of mitochondrial genome (Archer et al. 2013; Morin et al. 2010) and particular gene regions, such as control region (D-loop) (Hatch et al. 2006; Jackson et al. 2014; Pomilla et al. 2014) and cytochrome b (Amaral et al. 2012; May-Collado and Agnarsson 2006; McGowen et al. 2009) sequences. Nevertheless, spatiotemporal dynamics of the fin whales remains inconclusive. There has been only one phylogenetic study focused on fin whale mitogenome sequences (Archer et al. 2013). In that report, authors suggested that the North Atlantic fin whales are more closely related to Southern Hemisphere than to the North Pacific regions. However, they did not address the population size changes and phylogenetic position of the fin whale within the baleen whale lineages; the authors dealt with only fin whale species of the 15 modern Mysticeti species and used only one phylogenetic analysis method—the concatenation in BEAST program.
Accordingly, to further explore the phylogeny and evolutionary dynamics of the fin whale, it is still needed to analyze more extensive sequence information from additional representative species, using more exhaustive analytical methods. We analyzed 148 published mitochondrial genome sequences of 14 baleen whale species using the Bayesian coalescent approach as well as Bayesian inferences (BI) and maximum likelihood (ML) methods. The present study has five specific objectives: (1) testing the monophyletic origin of the fin whale; (2) addressing the geographic influences on the heterogeneity of fin whales; (3) examining the phylogenetic relationships between fin whales and other baleen whale species; (4) estimating the time of the most recent common female ancestor of fin whales; (5) finally, assessing the changes in the population size of modern fin whales.
Materials and methods
Mitochondrial genome characterization of the fin whales
We analyzed the published mitochondrial genome sequences of 148 baleen whales (Mysticeti) from NCBI. In detail, 125 are fin whales (B. physalus) from the North Pacific (n = 72), North Atlantic (n = 12), and Southern Hemisphere (n = 41), while the remaining 23 whales are members of 13 other baleen whale species. The taxa utilized in the present study are listed in Table S1 with their GenBank accession numbers. The sequences were aligned using MAFFT v 6 (Katoh et al. 2002), and then visually edited. The final alignment for phylogenomic analyses comprised 16,454 nucleotide positions. Furthermore, we characterized 125 fin whale mitochondrial genome sequences. Here, we estimated the following values for both nucleotide and amino acid sequences of the 38 mitochondrial genome regions using BIOEDIT 7.053 (Hall 1999), Modeltest 3.7 (Posada and Crandall 1998), and PAUP 4.0b10 (Swofford 2003)—total sites (including gaps), variable sites, sequence identities, Ts/Tv ratios, base frequencies, and evolutionary models. The 38 partitions used in the analysis were divided according to sequence positions of a published mitochondrial genome (GenBank Accession No. AP006472). In addition, we plotted the number of nucleotide and amino acid variations at each position throughout the mitochondrial genomes. We first estimated the nucleotide differences by counting the number of minor nucleotides at each position across the sequence alignment. Next, we defined the most frequent nucleotide at each site as the major nucleotide (A, T, C, or G), and the rest of the nucleotides were considered minor nucleotides. As an example, at a specific position of the alignment, if the nucleotide C was major in 117 samples, while A, T, and G were observed as minor nucleotides in the remaining 31 samples, the nucleotide difference was 31. We applied the same principle for amino acid differences.
Phylogenomic analyses
Phylogenomic analyses were carried out using BI and ML. Kogia breviceps (NC005272) (Odontoceti; Kogiidae) was used as an outgroup as it is the closest related species to the Mysticeti. The best-fit model for the baleen whale mitochondrial genomes was determined using the standard Modeltest PAUP block in PAUP 4.0b10 (Swofford 2003) and Akaike’s information criterion (AIC) in Modeltest 3.7 (Posada and Crandall 1998); GTR+I+G was selected as the best evolutionary model.
BI analysis was conducted using MrBayes 3.2.1 (Ronquist and Huelsenbeck 2003) with the following parameters: Nst, 6; rates, gamma; number of generation, 30,000,000; sample frequency, 500; number of chains, 4; burn-in generation, 25%. Here, the Bayesian posterior probabilities (BPP) were calculated as a measure of node support.
ML phylogenetic analysis was performed in PHYML 3.0 (Guindon and Gascuel 2003) and was run under the following options: model of nucleotide substitution, GTR; initial tree, BIONJ; nonparametric bootstrap analysis, yes, 500 pseudoreplicates; proportion of invariable sites, estimated; number of substitution rate categories, 6; gamma shape parameter, estimated by program; optimize tree topology, yes. Bootstrap values shown on respective internal nodes indicated confidence of the phylogenomic analysis.
In addition to the phylogenomic analyses, we employed BI analyses at the mitochondrial gene level in order to screen the most significant phylogenetic marker among the 16 mitochondrial genes. The best-fit models for each region used in analyses are summarized in Table S2.
Estimation of time of the most recent common ancestor (tMRCA), substitution rate, and population size changes
BEAST 2.4.4 (Drummond and Rambaut 2007) was used to estimate the times of the most recent common ancestor (tMRCA), rates of nucleotide substitutions, and changes in population size under Bayesian coalescent approach. Crown Cetacea was calibrated based on the oldest mysticete fossil Llanocetus (Mitchell 1989; Steeman 2007) (34 Mya, 35 mean, 1.0 SD). The age of the basal of the crown Mysticeti was estimated on the basis of an unnamed balaenid from New Zealand (Hubbard et al. 2002) (28 Mya, 29.0 mean, 1.0 SD). Kogia breviceps (Odontoceti; Kogiidae) was used as an outgroup. The analysis was conducted under the GTR+I+G model, nst = 6, and rates = gamma derived from AIC in Modeltest 3.7 (Posada and Crandall 1998). We employed the relaxed uncorrelated lognormal for clock model and Yule process for tree topology prior. The data sets were each run for 500,000,000 generations to ensure convergence of all parameters (ESSs > 100) with discarded burn-in of 10%. Subsequently, we examined the effective population size changes of fin whales over the time and analyzed only 125 fin whale mitochondrial genome sequences using the Coalescent bayesian skyline for tree topology prior. For the calibration point of fin whale, we assumed an tMRCA of 9.88 My, which was estimated from entire 148 baleen whale mitochondrial genome sequences. The output files were analyzed together in Tracer 1.5 (http://beast.bio.ed.ac.uk/Tracer), and the statistical uncertainties were summarized in the 95% highest probability density (HPD) intervals. Trees were summarized as maximum clade credibility trees using the TreeAnnotator program in the BEAST package and were displayed using FigTree 1.4.2 (Rambaut 2012).
Results
Findings from the mitochondrial genome sequences of fin whales
Characterization of the entire mitochondrial genome and individual gene sequences of the 125 fin whales are reported in Table 1. The mitochondrial genome alignment (including insertions) is 16,402 bp in length, and 951 (5.8%) of the nucleotide sites are polymorphic. Our plot, which visualizes the number of both nucleotide and amino acid variations at each site throughout the alignment, illustrates that both nucleotide and amino acid alterations were evenly distributed throughout the mitochondrial genomes (Fig. 1). As a note, higher amino acid similarities were observed in three genes (COX1, COX2, and COX3). The nucleotide sequence identities among the fin whale samples ranged from 98.7 to 99.9% (average, 99.3%), corresponding to 97.9–100% (average, 99.1%) identity at the amino acid level. Of the 16 regions analyzed, NADH2 was the most variable (average sequence identities of 98.8), while both 16S rRNA and NADH1 were the most conserved (average sequence identities of 99.7% for both nucleotides). Interestingly, all fin whales had their specific amino acid sequences throughout these eight genes: NADH2, COX2, COX3, ATPase6, ATPase8, NADH4, NADH5, and Cytb (Table 2).

Plotting the nucleotide (a) and amino acid (b) differences throughout the mitochondrial genomes of 125 fin whales. The number of differences was estimated as the total number of altered nucleotides at each site compared with the multiple sequence alignment method. Both nucleotide and amino acid alterations were evenly distributed throughout the mitochondrial genomes. Especially, higher amino acid similarities were observed in three genes (COX1, COX2, and COX3)
Phylogenomic analyses
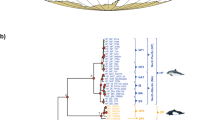
For phylogenomic inferences, we analyzed 148 mitochondrial genome sequences of baleen whales (125 fin whales and 23 other baleen whales). Our study focused on reliably aligned regions comprising a total of 16,454 nucleotide positions. BI and ML methods produced identical tree topologies and supported the configurations of the maximum clade credibility tree (Figs. 2, 3). Especially, the fin whale clade within the baleen whale lineages was enlarged as shown in Fig. 3.

Bayesian maximum clade credibility phylogenomic tree on the ground of mitochondrial genome sequences of 148 baleen whales. Kogia breviceps was used as an outgroup. The data set (16,454 base pairs) was also analyzed phylogenetically with BI and ML methods which showed identical topologies. The scale bar represents time in million years before present. The robustness of the phylogenetic analysis is presented above the nodes: left numbers are Bayesian posterior probabilities (≥ 0.80) and right ones are ML bootstrap values (≥ 60%)

Distribution of the fin whale (B. physalus Linnaeus, 1758) within the Bayesian maximum clade credibility phylogenetic tree derived from complete mitochondrial genome sequences of 148 baleen whales. The robustness of the phylogenetic analysis is showed above the nodes: left numbers are Bayesian posterior probabilities (≥ 0.80) and right ones are ML bootstrap values (≥ 60%). Divergence times (in My) are positioned below the nodes; the 95% HPD intervals are indicated in brackets. Groups are marked by a “G”
The topologies of the phylogenomic trees indicated that fin whale had a monophyletic origin and its members were classified into one of four major clades with high confidence for each node. There were geographic groupings within the global fin whale trees. Group 1 consisted of 50 fin whales, Southern Hemisphere (n = 41) and North Pacific (n = 9); here, North Pacific whales had a single origin. Group 2 (n = 12) were from the North Atlantic, whereas both Group 3 (n = 2) and Group 4 (n = 61) were individuals from the North Pacific regions. The phylogenetic trees also strongly supported the fin whale clade as the sister group to humpback whale (Megaptera novaengliae) within the baleen whale lineages.
In addition to phylogenomic analyses we carried out BI analyses at gene level under the best-fit evolutionary model to identify strongest phylogenetic marker among the 16 regions (Table S2). Here, all individual trees were compared to the complete mitochondrial genome trees. From the comparison, NADH5 gene tree (Fig. 4) is the most similar to the genomic tree; thus, the NADH5 gene was selected as the most significant phylogenetic marker for baleen whales. The overall tree topologies of both control region (Fig. S1) and Cytochrome b gene (Fig. S2), which have been used as the markers of Mysticeti phylogeny, were very different from those of mitochondrial genome tree (Fig. 2); most groupings were collapsed in both control region and cytochrome b trees.

The BI tree based on NADH5 sequences of 148 fin whales. Posterior probabilities (≥ 0.80) are indicated above the branches. K. breviceps was specified as the outgroup
Divergence times, substitution rates, and population size changes
The most recent common female ancestor of the global fin whales existed approximately 9.88 Mya, which is much younger than 29.86 Mya of modern baleen whales (Fig. 2). North Pacific fin whales emerged first (7.48 Mya), followed by a subsequent segregation of Southern Hemisphere (6.63 Mya) and North Atlantic (4.42 Mya). Notably, 1.76 Mya, some Southern Hemisphere females migrated to the North Pacific (Fig. 3), and then North Pacific fin whales appeared 1.42 Mya, independently. The mitochondrial evolutionary rate of this whale is 1.002 × 10−3 substitutions/site/My, which is much lower than 2.252 × 10−3 substitutions/site/My of modern baleen whales. According to the Bayesian skyline plot (BSP) (Fig. 5), fin whale population size has increased from about 2.5 Mya to the present day, which coincides with the Quaternary glaciation stage.

BSP on the basis of mitochondrial genome sequences from 125 fin whales. The dark line in the BSP shows the estimated effective population size through time. The blue area represents the 95% highest posterior density confidence intervals for this estimate. The BSP analysis indicated that the fin whale population experienced two rapid expansion events about 2 and 0.25 Mya during the Pleistocene
Discussion
Our results indicate a monophyletic origin of fin whale, which supported its synapomorphic characters such as tall spout, long back, prominent dorsal fin, and asymmetrical color pattern. These characters enable distinguishment of fin whale from three balaenopterid relatives (blue, sei, and Bryde’s whale). The spout of fin whales is much more pointed than that of blue whales, and the prominent dorsal fin is positioned more posterior than those of sei or Bryde’s whales. Most of all, the most distinctive character of the fin whale is the asymmetrical coloration of their body. The monophyly of this species was in line with the presence of its species-specific amino acid sequences in eight genes: NADH2, COX2, COX3, ATPase6, ATPase8, NADH4, NADH5, and Cytb.
Next, our phylogenomic study focused on the geographic influences on the evolution of fin whales. To date, on the basis of the geographic distribution, many experts have sub-divided this species into two subspecies, Baleana physalus physalus Linnaeus, 1758 for Northern Hemisphere and Baleana physalus quoyi Fischer, 1829 for Southern Hemisphere fin whales (Lönnberg 1931; Lockyer and Waters 1986; Tomilin 1946). Lönnberg (1931) mentioned that there were significant differences between the vertebral features of the two subspecies. A few years later, Tomilin (1946) claimed that the body sizes of Southern Hemisphere whales are much larger than those from Northern Hemisphere. This viewpoint was confirmed by Lockyer and Waters (1986), who conducted morphological examinations using a larger series of specimens from the North Atlantic and Antarctica. In their report, the maximum body length of Antarctic fin whales was about 3–4 m greater than those in the Northern Hemisphere. Recently, there was an additional suggestion relating the subspecies classification of fin whale; Clarke (2004) added a “pygmy” subspecies (B. p. patachonica, Burmeister, 1865) living in the Southern Hemisphere to the previous two subspecies. The form is small (approximately 18–24 m) and dark with black baleen. Today, the Society of Marine Mammalogy accepts these three subspecies: B. p. physalus Linnaeus, 1758 in the Northern Hemisphere, B. p. quoyi Fischer, 1829 in the Southern Hemisphere, and the pygmy fin whale, B. p. patachonica Burmeister, 1865 (2012). However, there was a contradicting article on this topic, on the basis of mitochondrial genome sequences; Archer et al. (2013) noted that North Pacific and North Atlantic fin whales were not the same subspecies. Our mitochondrial genome sequences supported that there is a significant correlation between geographic location and evolution of global fin whales, and indicated that North Pacific fin whales might be classified into three geographic stocks: Group 3, Group 4, and a clade within Group 1. Accordingly, our findings propose six subspecies system of this species: three from North Pacific and one each from North Atlantic, Southern Hemisphere, and “pygmy” form. As a note, the presence of North Pacific clade within the Southern Hemisphere Group 1 revealed that some Southern Hemisphere females returned to the North Pacific.
Our trees also clearly depict that the fin whale and humpback whale (M. novaengliae) are sister group taxa within the baleen whale lineages. This observation is in line with the results from cytochrome b (LeDuc and Dizon 2002), control region (Hatch et al. 2006), and mitochondrial genome (Dornburg et al. 2011) sequences. Several workers, however, have disagreed on this matter. Molecular investigators, based on the control region sequences, reported that fin whale has stronger affinities to blue whale (Balaenoptera musculus) than humpback whale (Arnason et al. 1993). Bouetel and de Muizon (2006), on the grounds of their morphology data, suggested that fin whale resembles the common minke whale (Balaenoptera acutorostrata) more strongly than other baleen whale species.
Next, we also tried to identify which gene has stronger phylogenetic signal by comparing 16 individual region trees with the mitochondrial genome trees. Although control region (Arnason et al. 1993; Hatch et al. 2006) and cytochrome b (Arnason and Gullberg 1994; May-Collado and Agnarsson 2006) sequences have been used as a molecular marker for elucidating the Mysticeti phylogeny, our results indicated that topologies of NADH5 tree were the most closest to those of the mitochondrial genome tree, rather than control region or Cytochrome b. Accordingly, we propose that NADH5 is the most significant phylogenetic marker to reconstruct the Mysticeti phylogeny.
On the time of the most recent common female ancestor of fin whale, our findings indicated that this species is about 9.88 My old. This is much younger than the morphological character-based ancestor of 16 My old (Gatesy et al. 2013), while it is much older than other previous reports: 1.94 My old on the ground of mitochondrial genome (Archer et al. 2013), 7 My old based on mitochondrial genome (Dornburg et al. 2011) and morphological and molecular (Marx and Fordyce 2015a) evidences. Our estimate for fin whale was also much younger than 29.86 Mya of modern baleen whales, which is calculated in this study. Subsequently, we found that North Pacific fin whales occurred first (7.48 Mya), and then moved sequentially to Southern Hemisphere (6.63 Mya) and North Atlantic (4.42 Mya). Especially, 1.76 Mya, some Southern Hemisphere females returned to the North Pacific, and independent lineage of North Pacific fin whales seem to appear 1.42 Mya.
The mean evolutionary rate estimated in this study is 1.002 × 10−3 substitutions/site/My; this value is lower than the results from previous fin whale studies, 1.1 × 10−2 substitutions/site/My (Dornburg et al. 2011) and 2.94 × 10−3 substitutions/site/My (Archer et al. 2013). This value is also lower than our estimate of 2.252 × 10−3 substitutions/site/My for modern baleen whales.
In terms of the effective population size changes of fin whale, our BSP illustrates that the fin whale population has experienced rapid increase from ~ 2.5 Mya to the present day. This period coincides with the Quaternary glaciation stage, the “Ice Age”(Gradstein et al. 2004), which is characterized by repeated glacial cycles. Especially, cooling water derived from upwelling regime is responsible for the changes in the ocean ecosystem. Phytoplanktone, the base of mysticete food change, were fertilized and this high level of nutrient resulted in population size expansion and gigantism of fin whale (Marx and Fordyce 2015b; Slater et al. 2017).
Molecular genetic studies are used to infer patterns (of distribution and migration), population structure change, and the influences of climate change as well as taxonomic classification. Especially, to develop conservation and management strategies, the first important step is to clarify characteristics of the unique populations at the phenotypic and genotypic levels (Prystupa et al. 2012). Fin whale (B. physalus) is listed as Endangered (EN A1d), on the IUCN Red List of Threatened Species (IUCN 2003) and therefore understanding the spatial and temporal patterns are essential to improving their conservation. This is the first study on temporal and spatial dynamics of the fin whale within the baleen whale lineages. The present study enhanced our knowledge of the spatiotemporal dynamics of the fin whales and could provide useful information for future fin whale conservation projects.
References
Amaral AR, Jackson JA, Möller LM, Beheregaray LB, Coelho MM (2012) Species tree of a recent radiation: the subfamily Delphininae (Cetacea, Mammalia). Mol Phylogenet Evol 64:243–253
Archer FI et al (2013) Mitogenomic phylogenetics of fin whales (Balaenoptera physalus spp.): genetic evidence for revision of subspecies. PloS ONE 8:e63396
Arnason U, Gullberg A (1994) Relationship of baleen whales established by cytochrome b gene sequence comparison. Nature 367:726–728. doi:10.1038/367726a0
Arnason U, Gullberg A, Widegren B (1993) Cetacean mitochondrial DNA control region: sequences of all extant baleen whales and two sperm whale species. Mol Biol Evol 10:960–970
Bouetel V, de Muizon C (2006) The anatomy and relationships of Piscobalaena nana (Cetacea, Mysticeti), a Cetotheriidae ss from the early Pliocene of Peru. Geodiversitas 28:319–395
Clarke R (2004) Pygmy fin whales. Mar Mamm Sci 20:329–334
Dornburg A, Brandley MC, McGowen MR, Near TJ (2011) Relaxed clocks and inferences of heterogeneous patterns of nucleotide substitution and divergence time estimates across whales and dolphins (Mammalia: Cetacea.). Mol Biol Evol. doi:10.1093/molbev/msr228
Drummond AJ, Rambaut A (2007) BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Gatesy J et al (2013) A phylogenetic blueprint for a modern whale. Mol Phylogenet Evol 66:479–506
Gradstein F, Ogg J, Smith A (2004) A geologic time scale 2004. Cambridge, Cambridge University Press
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hatch LT, Dopman EB, Harrison RG (2006) Phylogenetic relationships among the baleen whales based on maternally and paternally inherited characters. Mol Phylogenet Evol 41:12–27
Hubbard T et al (2002) The ensembl genome database project. Nucleic Acids Res 30:38–41
IUCN (2003) Red list of threatened species. http://www.redlist.org
Jackson JA et al (2014) Global diversity and oceanic divergence of humpback whales (Megaptera novaeangliae). Proc Biol Sci B 281:20133222
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucl Acids Res 30:3059–3066
LeDuc RG, Dizon AE (2002) Reconstructing the rorquals phylogeny: with comments on the use of molecular and morphological data for systematic study. In: Pfeiffer CJ (ed) Molecular and cell biology of marine mammals. Krieger, Marabar, pp 100–110
Lockyer C, Waters T (1986) Weights and anatomical measurements of northeastern Atlantic fin (Balaenoptera physalus, Linnaeus) and sei (B. borealis, Lesson) whales. Mar Mammal Sci 2:169–185
Lönnberg E (1931) The skeleton of Balaenoptera brydei Ö. Olsen. Almqvist & Wiksell, Stockholm
Marx FG, Fordyce RE (2015a) Baleen boom and bust: a synthesis of mysticete phylogeny, diversity disparity. R Soc Open Sci 2:140434
Marx FG, Fordyce RE (2015b) Baleen boom and bust: a synthesis of mysticete phylogeny, diversity disparity. Open Sci 2:140434
May-Collado L, Agnarsson I (2006) Cytochrome b and Bayesian inference of whale phylogeny. Mol Phylogenet Evol 38:344–354
McGowen MR, Spaulding M, Gatesy J (2009) Divergence date estimation and a comprehensive molecular tree of extant cetaceans. Mol Phylogenet Evol 53:891–906
Mitchell ED (1989) A new cetacean from the late Eocene La Meseta Formation Seymour Island, Antarctic peninsula. Can J Fish Aquat Sci 46:2219–2235
Morin PA et al (2010) Complete mitochondrial genome phylogeographic analysis of killer whales (Orcinus orca) indicates multiple species. Genome Res 20:908–916
Pomilla C et al (2014) The world’s most isolated and distinct whale population? Humpback whales of the Arabian Sea. PloS ONE 9:e114162
Posada D, Crandall KA (1998) Modeltest: testing the model of. DNA substitution. Bioinformatics 14:817–818
Prystupa JM, Hind P, Cothran EG, Plante Y (2012) Maternal lineages in native Canadian equine populations and their relationship to the Nordic and Mountain and Moorland pony breeds. J Hered 103:380–390
Rambaut (2012) Available at http://evolve.zoo.ox.ac.uk/beast
Reilly S et al. (2008) Balaenoptera acutorostrata IUCN red list of threatened species. IUCN, Gland
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Saccone C, De Giorgi C, Gissi C, Pesole G, Reyes A (1999) Evolutionary genomics in metazoa: the mitochondrial DNA as a model system. Gene 238:195–209
Slater GJ, Goldbogen JA, Pyenson ND (2017) Independent evolution of baleen whale gigantism linked to Plio-Pleistocene ocean dynamics. In: Proc R Soc B, vol 1855. The Royal Society, p 20170546
Steeman ME (2007) Cladistic analysis and a revised classification of fossil and recent mysticetes. Zool J Linn Soc 150:875–894
Swofford DL (2003) PAUP: phylogenetic analysis using parsimony, version 4.0b10. Sinauer Associates, Sunderland
Tomilin A (1946) Thermoregulation and the geographical races of cetaceans.(Termoregulyatsiya I geograficheskie racy kitoobraznykh.). Doklady Akademii Nauk CCP 54(5):465–472 (English and Russian)
Acknowledgements
This research was supported by a grant from the Collaborative Genome Program (20140428) funded by the Ministry of Oceans and Fisheries, Korea. This work was also supported by a grant from the National Institute of Fisheries Science (R2017039), Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Jihyun Yu and Bo-Hye Nam have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10709_2017_9988_MOESM1_ESM.pdf
Supplementary Figure 1—The BI tree on the basis of control region sequences of 148 baleen whales. Bayesian posterior probabilities above 0.80 are shown on the nodes. (PDF 141 KB)
10709_2017_9988_MOESM2_ESM.pdf
Supplementary Figure 2—The BI tree on the basis of cytochrome b sequences of 148 baleen whales. Bayesian posterior probabilities above 0.80 are shown on the nodes. (PDF 304 KB)
Rights and permissions
About this article

Cite this article
Yu, J., Nam, BH., Yoon, J. et al. Tracing the spatio-temporal dynamics of endangered fin whales (Balaenoptera physalus) within baleen whale (Mysticeti) lineages: a mitogenomic perspective. Genetica 145, 603–612 (2017). https://doi.org/10.1007/s10709-017-9988-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-017-9988-4