
Abstract
Meiotic recombination in female ducks was directly studied by immunolocalization of MLH1 protein, a mismatch repair protein of mature recombination nodules. In total, 6820 crossovers were scored along the autosomal synaptonemal complexes in 122 meiotic nuclei. From this analysis we predict that the female map length of the duck is 2845 cM, with a genome wide recombination rate of 2 cM/Mb. MLH1-focus mapping along the six largest bivalents shows regional variations of recombination frequencies that can be linked to differences in chromosome morphology. From this MLH1 mapping it can be inferred that distally located markers will appear more separated in genetic maps than physically equidistant markers located near the centromeres on bivalents 1 and 2. Instead, markers at interstitial positions on the acrocentric bivalents 3–6 will appear more tightly linked than expected on the basis of their physical distance because recombination is comparatively lower at the mid region of these chromosomes. The present results provide useful information to complement linkage mapping in ducks and extend previous knowledge about the variation of recombination rates among domestic Galloanserae.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Domestic ducks (Anas platyrhynchos) are the most economically important waterfowl as a source of meat, eggs and feathers and, at the same time, their wild conspecifics serve as the principal natural reservoir for influenza A viruses. Because of their significance in agriculture and animal health there are now an increasing number of studies devoted to the structural and functional genomics of the species, especially after the completion of the genome sequencing (Huang et al. 2013). In this sense, two linkage maps were developed by analysis of polymorphic microsatellite markers and AFLP markers (Y. Huang et al. 2006, C. W. Huang et al. 2009). These maps for the duck still have regions with insufficient coverage what may lead to inaccurate estimation of the genetic map lengths. Furthermore, global recombination frequencies and crossover parameters, such as the relationship between genetic and physical distances, have not been described yet in this species.
Genetic linkage maps provide estimates of recombination data linked to genes or DNA sequences but they can be greatly affected by marker density, they are especially sensitive to errors in marker order and require the extrapolation of recombination fractions to genetic distances by the use of mapping functions. As an alternative to the indirect approach of genetic linkage maps, recombination rates in individuals, cells or chromosomes can be analyzed using cytological crossover markers (reviewed by Hultén and Tease 2006). A powerful cytological approach to obtain global recombination frequencies and frequency distribution of crossovers on individual bivalents is the immunofluorescent labeling of pachytene oocytes or spermatocytes with antibodies against the DNA mismatch repair protein MLH1, and the synaptonemal complex (SC). The MLH1 protein forms aggregates (foci) at the positions of crossing over and it is part of the multiprotein complex at late recombination nodules, visible with electron microscopy during pachytene (Anderson et al. 2014). Broad research in different vertebrate species, including humans has shown that MLH1 foci are cytological markers of the crossing over providing an unbiased estimate of the total map length, as well as the frequency and distribution of crossovers in individual chromosomes (Froenicke et al. 2002; Sun et al. 2006; Borodin et al. 2009; Vozdova et al. 2013; Mary et al. 2014; Frohlich et al. 2015; Lisachov et al. 2015). Similar observations were done in birds such as chickens, zebra finches, and Japanese quails (Pigozzi 2001; Calderón and Pigozzi 2006; del Priore and Pigozzi 2015). Among the advantages of this methodology are: (1) the direct observation of crossovers at the sites where they occur along bivalents, (2) the analysis of large numbers of germ cells to yield precise estimates of the global recombination rate for single individuals, and (3) its low costs compared to genotyping many offspring as required by pedigree-based methods. Each crossover marked by an MLH1 focus results in 50 % recombinant progeny; therefore, the total length of the genetic map is equal to the average number of MLH1 foci per genome multiplied by 50 centimorgans (cM).
In the present study we estimated the genetic lengths and crossover rates throughout the duck genome and the six largest linkage groups by scoring MLH1 foci in duck oocytes. These results are compared to available linkage map data for the species and also with MLH1 focus analyses performed in other poultry species.
Materials and methods
Synaptonemal complex (SC) spreads and immunostaining
All procedures involving animals were approved by the Animal Care and Use Committee, School of Medicine, University of Buenos Aires. Duck eggs were provided by a local farm and incubated under standard conditions. Oocyte spreads were prepared from the functional ovary of three females, 2 days before hatching or at hatching. Previous knowledge of duck oogenesis indicates that the peak of pachytene oocytes can be found during these days (Solari and Pigozzi 1993). The method to prepare synaptonemal complex spreads from avian oocytes has been previously described in detail (del Priore and Pigozzi 2012). Briefly, the only functional ovary was dissected, placed in Hanks’ solution at room temperature, minced in one drop of 100 mM sucrose at pH 8.5, and the released cells were suspended in additional sucrose solution. About 30 μl of this cell suspension was dropped onto a layer of 1 % paraformaldehyde fixative and 0.1 % Triton X-100 on clean slides and left in a humid chamber for 1 h. After fixation, slides were washed in Photoflo and air-dried. For immunofluorescence, the primary antibodies were rabbit anti-SMC3 (Chemicon, Millipore) at 1:1000 that labels the cohesin axes underlying the axial elements of the synaptonemal complexes, mouse anti-MLH1 (BD Pharmingen) and CREST human antiserum (Roquel Laboratories, Buenos Aires, Argentina) that binds to kinetochores at 1:100. The secondary antibodies were TRITC-labeled goat anti-rabbit, Cy3-labeled donkey anti-human and FITC-labeled goat anti-mouse (Jackson ImmunoResearch) at 1:100. Immunostained spreads were scanned with 100× magnification objective at a fluorescence microscope equipped with appropriate filter sets for each fluorochrome. Individual images for red and green fluorescence were acquired using an Olympus DP73 CCD camera. Images were corrected for brightness and contrast and merged using Adobe Photoshop 6.0.
Image and data analysis
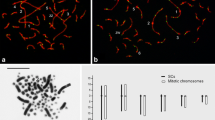
SC lengths, centromere position, and the relative distance of each MLH1 focus from the centromeric signal were scored using the program MicroMeasure 3.3 (available at http://www.colostate.edu/Depts/Biology/MicroMeasure). A total of 122 complete SC sets were used to obtain the number of foci per cell, the total length of the autosomal set, and the relative lengths and number of foci of the six largest autosomal SCs. These measurements and counts were obtained from the composite images merged in Photoshop. After compiling the focus data, the total length of the genetic map results from multiplying the average number of MLH1 foci per genome multiplied by 50 cM. Similarly, the map length of the six largest chromosomes resulted from the average number of foci on the corresponding SCs. The construction of crossover frequency histograms from recombination nodules or MLH1 foci has been explained before in several publications (Sherman and Stack 1995; Pigozzi and Solari 1999; Borodin et al. 2008). MLH1 focus frequencies in each histogram can be converted to cM by multiplying the number of foci in each SC interval by 50, and then dividing by the total number of SCs observed. The cM values for adjacent intervals were added to generate cumulative cM maps along each SC. The six largest SCs of the pachytene shown in Fig. 1a are arranged by size in Fig. 1b after straightening each individual SC in Image J employing a specific plugin (Kocsis et al. 1991).

Immunocytological analysis of crossing over in duck oocytes. a Pachytene oocyte showing the complete set of autosomal synaptonemal complexes and the sex pair (ZW). Centromeres are seen as red dots bulging from the linear SCs and MLH1 foci are labeled in green. The number next to the centromeres indicates the macrobivalents SCs. The arrowhead points to the single MLH1 focus observed near the end of the ZW bivalent. b The first six autosomal SCs of the oocyte in a were digitally straightened to show the differences in lengths and centromere position that allow their identification in measurements. Bar 10 µm
Estimation of genome and chromosome sizes
The haploid DNA content of Anas platyrhynchos is about 1.44 pg. This value is an average calculated from five measurements of the C-value in this species available in the literature (Gregory 2016). We used only the values from references that specified the methodology to measure the DNA content. The DNA content in pg can be converted to millions of base pairs (Mb) multiplying by (0.978 × 109) (Dolezel et al. 2003). From the total genome size of 1408 Mb we subtracted 6 %, corresponding to the Z chromosome since SC lengths and MLH1 foci were computed for the autosomal set. Therefore the haploid genome content of the autosomal set in the duck is about 1323.5 Mb. The size in Mb of the six largest chromosomes was predicted from the relative length of their SCs multiplied by the haploid genome content, as previously done in other organisms (Zhang et al. 2014). This procedure assumes that SC length is proportional to genome size and it is supported by the fact that SC length is a very good predictor of the chromosome size in Mb in the chicken (Pigozzi 2001). Considering the conservation of genomic and cytogenetic features between these two species (Fillon et al. 2007; Skinner et al. 2009), it is reasonable to assume that SC lengths are also good predictors of the DNA content of individual chromosomes in ducks.
Results and discussion
In pachytene oocytes of A. platyrhynchos MLH1 foci were scored in whole cells and also along individual chromosome arms of the six largest bivalents represented by their SCs (Fig. 1a). The SCs of the six largest macrobivalents were identified on the basis of their lengths and centromeric indexes (Fig. 1b). These morphological features are in agreement with those of the mitotic chromosomes of the species: bivalents 1 and 2 are submetacentric, with noticeable length differences between them, while bivalents 3–6 are acrocentric. In a total of 122 immunostained oocytes showing complete SC sets we found that the average number of foci in the autosomal set was 55.9, ranging between 50 and 68 (Fig. 2a). Based on the average number of foci, the map length of the duck female genome is 2845 cM, after the addition of 50 cM to the autosomal map for the obligate crossover in the ZW pairing region. The presence of a single MLH1 focus on the ZW pair (Fig. 1a) is in agreement with the long known presence of a single RN close to the distal end of the bivalent (Solari and Pigozzi 1993). The relative SC lengths of bivalents 1–6 show a direct relationship with the average number of MLH1 foci and the corresponding maps lengths expressed in cM (Fig. 2b; Table 1). These observations are in agreement with data from chicken and quail macrobivalents, where SC lengths are also good predictors of the number of MLH1 foci (Pigozzi 2001; del Priore and Pigozzi 2015). Without additional techniques, the remaining SCs cannot be identified with certainty. The unidentified SCs represent 48 % of the total SC length and most of them showed one MLH1 focus, with only 10 % carrying two foci.

MLH1 focus numbers in whole nuclei and individual SCs. a In the scatter plot each dot represents a single pachytene nucleus with n foci. The horizontal line marks the average number of foci found in 122 oocytes. b The graph shows the linear relationship between number of foci and SC length
Since the estimated haploid DNA content of the duck is 1.44 pg, its genome size would be approximately 1400 Mb. From the cM map length obtained from MLH1 focus data we can estimate a global recombination rate in the duck of 2 cM/Mb. The rate of recombination is about 1.6 cM/Mb along the six largest autosomal SCs and increase to 2.7 cM/Mb for SCs 7–39 (Table 1). The excess of recombination on the shorter bivalents is explained by the fact that they have invariably one crossover, essential for the orderly segregation of the homologs during anaphase I. The global crossover rates calculated here for ducks are lower compared to chickens (2.6 cM/Mb) but closer to quails (1.9 cM/Mb) (Pigozzi 2001; Rodionov and Chechik 2002; Groenen et al. 2009; del Priore and Pigozzi 2015). Altogether the present results add support to the idea that recombination rates observed in domestic chickens are unusually high among birds and that differences between species are largely attributable to macrochromosomes (Calderón and Pigozzi 2006; Stapley et al. 2008).
Besides determining average focus numbers, we also recorded the positions of MLH1 foci in relation to the centromere to build focus frequency distributions and cumulative cM maps of chromosomes 1–6 (Fig. 3). The histograms show higher crossover levels at the distal intervals of the submetacentric bivalents with a rather uniform distribution of MLH1 foci at interstitial arm positions. The acrocentric bivalents show high recombination levels close to the centromere and also near the telomere of the long arm. This crossover distribution is similar to that observed in chickens and quails (Rahn and Solari 1986; Rodionov et al. 2002; del Priore and Pigozzi 2015), two avian species that share similar karyotypic features with ducks, reinforcing the idea that the chromosome architecture constrains crossover location. These observations support the notion that chromosome primary structure plays a role in the distribution of crossing over at coarser level, even when hot spot distribution, sequence content and epigenetic modifications act at a finer scale to modulate the presence of hot and cold regions of recombination (reviewed by Mezard et al. 2015).

Crossover distribution in the macrobivalents of female ducks. Each histogram represents the distribution of MLH1 foci along the six largest autosomal SCs. For each SC, the X-axis indicates the relative positions of the MLH1foci on the SC on the short (left) and long (right) arms. The bin width in each histogram represents a fraction of the total length of each SC, and it is equivalent to 0.5 μm. “C” = centromere. Overlapped to each histogram a line shows the cumulative frequencies of foci as a function of the distance to the telomeric end of the long arm of the SC. The number at the end of each line is the map length expressed in cM
Cumulative cM distributions show that physical and genetic distances are not always linear as a consequence of these regional variations of the crossing over (Fig. 3). The cumulative curves show a steeper slope at regions with higher recombination frequencies such as the distal regions of bivalents 1 and 2. A practical consequence of this uneven crossover distribution is that markers located at proximal regions of the biarmed chromosomes should display much tighter genetic linkage than markers at similar physical distance from each other but located near the telomeres. Along SCs 4–6, recombination frequencies are higher at proximal and distal regions of the long arm, and lower at interstitial positions (Fig. 3). Therefore, more meioses will be necessary in linkage studies to obtain accurate marker positions within interstitial segments of the acrocentric chromosomes. The linkage maps currently available for the domestic duck differ significantly in the genetic lengths estimated for the largest linkage groups probably because of the use of different molecular markers and methodologies (Y. Huang et al. 2006, C. W. Huang et al. 2009). The AFLP map lengths are shorter than those obtained by microsatellite mapping and by MLH1 focus analysis, probably because this map still has chromosomal regions with insufficient coverage. The microsatellite map was made in parallel with a cytogenetic map so linkage groups can be related to chromosomes with certainty. Some linkage groups are longer while others are shorter than expected on the basis of MLH1 focus counts (Table 2), probably reflecting the preliminary state of the linkage data.
In conclusion, we have estimated the global recombination rates for the duck and determined the difference in crossover levels between macro- and microchromosomes. The analysis of crossover distribution along macrochromosomes provides valuable information about the regions with higher/lower recombination levels. Because linkage disequilibrium is expected to be broken down more rapidly in regions of high recombination, the number of markers needed to efficiently cover such regions should thus be larger than for other regions in association mapping studies (Backström et al. 2010).
References
Anderson LK et al (2014) Combined fluorescent and electron microscopic imaging unveils the specific properties of two classes of meiotic crossovers. Proc Natl Acad Sci USA 111:13415–13420. doi:10.1073/pnas.1406846111
Backström N et al (2010) The recombination landscape of the zebra finch Taeniopygia guttata genome. Genome Res. doi:10.1101/gr.101410.109
Borodin PM, Karamysheva TV, Belonogova NM, Torgasheva AA, Rubtsov NB, Searle JB (2008) Recombination map of the common shrew, Sorex araneus (Eulipotyphla, Mammalia). Genetics 178:621–632
Borodin PM, Basheva EA, Zhelezova AI (2009) Immunocytological analysis of meiotic recombination in the American mink (Mustela vison). Anim Genet 40:235–238. doi:10.1111/j.1365-2052.2008.01808.x
Calderón PL, Pigozzi MI (2006) MLH1-focus mapping in birds shows equal recombination between sexes and diversity of crossover patterns. Chromosome Res 14:605–612
del Priore L, Pigozzi MI (2012) Chromosomal axis formation and meiotic progression in chicken oocytes: a quantitative analysis. Cytogenet Genome Res 137:15–21. doi:10.1159/000339133
del Priore L, Pigozzi MI (2015) Sex-specific recombination maps for individual macrochromosomes in the Japanese quail (Coturnix japonica). Chromosome Res 23:199–210. doi:10.1007/s10577-014-9448-2
Dolezel J, Bartos J, Voglmayr H, Greilhuber J (2003) Nuclear DNA content and genome size of trout and human. Cytom A 51:127–128. doi:10.1002/cyto.a.10013 (author reply 129)
Fillon V, Vignoles M, Crooijmans RP, Groenen MA, Zoorob R, Vignal A (2007) FISH mapping of 57 BAC clones reveals strong conservation of synteny between Galliformes and Anseriformes. Anim Genet 38:303–307. doi:10.1111/j.1365-2052.2007.01578.x
Froenicke L, Anderson LK, Wienberg J, Ashley T (2002) Male mouse recombination maps for each autosome identified by chromosome painting. Am J Hum Genet 71:1353–1368
Frohlich J, Vozdova M, Kubickova S, Cernohorska H, Sebestova H, Rubes J (2015) Variation of meiotic recombination rates and MLH1 foci distribution in spermatocytes of cattle, sheep and goats. Cytogenet Genome Res. doi:10.1159/000439452
Gregory TR (2016) Animal Genome Size Database. http://www.genomesize.com
Groenen MA et al (2009) A high-density SNP-based linkage map of the chicken genome reveals sequence features correlated with recombination rate. Genome Res 19:510–519. doi:10.1101/gr.086538.108
Huang Y, Zhao Y, Haley CS, Hu S, Hao J, Wu C, Li N (2006) A genetic and cytogenetic map for the duck (Anas platyrhynchos). Genetics 173:287–296. doi:10.1534/genetics.105.053256
Huang CW, Cheng YS, Rouvier R, Yang KT, Wu CP, Huang HL, Huang MC (2009) Duck (Anas platyrhynchos) linkage mapping by AFLP fingerprinting. Genet Sel Evol 41:28. doi:10.1186/1297-9686-41-28
Huang Y et al (2013) The duck genome and transcriptome provide insight into an avian influenza virus reservoir species. Nat Genet 45:776–783. doi:10.1038/ng.2657
Hultén M, Tease C (2006) Genetic mapping: comparison of direct and indirect approaches. Encycl Life Sci. doi:10.1002/9780470015902.a0006268
Kocsis E, Trus BL, Steer CJ, Bisher ME, Steven AC (1991) Image averaging of flexible fibrous macromolecules: the clathrin triskelion has an elastic proximal segment. J Struct Biol 107:6–14
Lisachov AP, Zadesenets KS, Rubtsov NB, Borodin PM (2015) Sex chromosome synapsis and recombination in male guppies. Zebrafish 12:174–180. doi:10.1089/zeb.2014.1000
Mary N et al (2014) Meiotic recombination analyses of individual chromosomes in male domestic pigs (Sus scrofa domestica). PLoS ONE 9:e99123. doi:10.1371/journal.pone.0099123
Mezard C, Jahns MT, Grelon M (2015) Where to cross? New insights into the location of meiotic crossovers. Trends Genet 31:393–401. doi:10.1016/j.tig.2015.03.008
Pigozzi MI (2001) Distribution of MLH1 foci on the synaptonemal complexes of chicken oocytes. Cytogenet Cell Genet 95:129–133
Pigozzi MI, Solari AJ (1999) Recombination nodule mapping and chiasma distribution in spermatocytes of the pigeon, Columba livia. Genome 42:308–314
Rahn MI, Solari AJ (1986) Recombination nodules in the oocytes of the chicken, Gallus domesticus. Cytogenet Cell Genet 43:187–193
Rodionov AV, Chechik MS (2002) Lampbrush chromosomes in the Japanese quail Coturnix coturnix japonica: cytological maps of macro chromosomes and meiotic crossover frequency in females. Genetika 38:1246–1251
Rodionov AV, Lukina NA, Galkina SA, Solovei I, Saccone S (2002) Crossing over in chicken oogenesis: cytological and chiasma-based genetic maps of the chicken lampbrush chromosome 1. J Hered 93:125–129
Sherman JD, Stack SM (1995) Two-dimensional spreads of synaptonemal complexes from solanaceous plants. VI. High-resolution recombination nodule map for tomato (Lycopersicon esculentum). Genetics 141:683–708
Skinner BM et al (2009) Comparative genomics in chicken and Pekin duck using FISH mapping and microarray analysis. BMC Genom 10:357. doi:10.1186/1471-2164-10-357
Solari AJ, Pigozzi MI (1993) Recombination nodules and axial equalization in the ZW pairs of the Peking duck and the guinea fowl. Cytogenet Cell Genet 64:268–272
Stapley J, Birkhead TR, Burke T, Slate J (2008) A linkage map of the zebra finch Taeniopygia guttata provides new insights into avian genome evolution. Genetics 179:651–667. doi:10.1534/genetics.107.086264
Sun F et al (2006) Variation in MLH1 distribution in recombination maps for individual chromosomes from human males. Hum Mol Genet 15:2376–2391. doi:10.1093/hmg/ddl162
Vozdova M, Sebestova H, Kubícková S, Cernohorska H, Vahala J, Rubes J (2013) A comparative study of meiotic recombination in cattle (Bos taurus) and three wildebeest species (Connochaetes gnou, C. taurinus taurinus and C. t. albojubatus). Cytogenet Genome Res 140:36–45. doi:10.1159/000350444
Zhang L, Liang Z, Hutchinson J, Kleckner N (2014) Crossover patterning by the beam-film model: analysis and implications. PLoS Genet 10:e1004042. doi:10.1371/journal.pgen.1004042
Acknowledgments
Research in our laboratory is funded by grants PIP 003 from Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and PICT 0059 from Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT). Support from the University of Buenos Aires is gratefully acknowledged. M.I.P. is a member of CONICET and L.d.P. is the recipient of a doctoral fellowship from CONICET.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s10709-016-9922-1.
Rights and permissions
About this article

Cite this article
Pigozzi, M.I., del Priore, L. Meiotic recombination analysis in female ducks (Anas platyrhynchos). Genetica 144, 307–312 (2016). https://doi.org/10.1007/s10709-016-9899-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-016-9899-9