
Abstract
Microsatellites or simple sequence repeats (SSRs) constitute a significant portion of genomes and play an important role in gene function and genome organization. The availability of a complete genome sequence for yak (Bos grunniens) has made it possible to carry out genome-wide analysis of microsatellites in this species. We analyzed the abundance and density of perfect SSRs in the yak genome. We found a total of 723,172 SSRs with 1–6 bp nucleotide motifs, indicating that about 0.47 % of the yak whole genome sequence (2.66 Gb) comprises perfect SSRs, the average length of which was 17.34 bp/Mb. The average frequency and density of perfect SSRs was 272.18 loci/Mb and 4719.25 bp/Mb, respectively. The proportion of the six classes of perfect SSRs was not evenly distributed in the yak genome. Mononucleotide repeats (44.04 %) with a total number of 318,435 and a average length of 14.71 bp appeared to be the most abundant SSRs class, while the percentages of dinucleotide, trinucleotide, pentanucleotide, tetranucleotide and hexanucleotide repeats was 24.11 %, 15.80 %, 9.50 %, 6.40 % and 0.15 %, respectively. Different repeat classes of SSRs varied in their repeat number with the highest being 1206. Our results suggest that 15 motifs comprised the predominant categories with a frequency above 1 loci/Mb: A, AC, AT, AG, AGC, AAC, AAT, ACC, ATTT, GTTT, AATG, CTTT, ATGG, AACTG and ATCTG.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Microsatellites, also known as simple sequence repeats (SSRs), are tandemly repeated DNA sequences that are generally 1–6 bp in length per unit (Tautz and Renz 1984). As one of the most popular source of genetic markers, SSRs are widely employed in population genetics, biogeography and microevolutionary studies (Guichoux et al. 2011). The yak (Bos grunniens) is endemic to central Asia, being adapted to the cold and high altitude environment. As one of the local important domestic animals, yaks play an indispensable role in the region. More than 14 million domestic yaks provide meat, milk, transportation, dung for fuel and hides for Tibetans and other nomadic pastoralists living at high altitudes (Wiener et al. 2003). In the past few years, using cattle-specific SSR markers, some researchers have studied the population and evolution genetics of yak (Ritz et al. 2000; Wang et al. 2003; Nguyen et al. 2005; Zhang et al. 2008; Qi et al. 2005, 2010; Ramesha et al. 2012). Recently, Cai et al. (2014) reported 19 novel yak-specific polymorphic microsatellites including nine perfect microsatellites and ten imperfect or compound repeats. These studies contributed a great deal of valuable information for the assessment, protection and management of yak as a genetic resource.
The availability of complete genome sequence for the yak has made it possible to carry out genome-wide analysis (Qiu et al. 2012). However, to date there are no reports on the abundance and density of microsatellites (1–6 bp) repeats in the yak genome. We screened the entire yak genome sequence to study the distribution and density of perfect SSRs, in order to facilitate the understanding of structure of the yak genome, and to build up a foundation for the isolation and identification of more yak-specific SSRs.
Materials and methods
The complete yak genome sequence with a total length of 2.66 Gb was downloaded (Hu et al. 2012) in FASTA file format to generate SSRs data. MSDB 2.4.2 (Microsatellite Search and Database) (http://msdb.biosv.com/) (Du et al. 2013) was used to scan the entire yak genome for abundance and density of perfect SSRs, using the “perfect” search mode. We identified six classes of microsatellites: mono-, di-, tri-, tetra-, penta- and hexa-nucleotide SSR motifs at a minimum repeat number of 12, 7, 5, 4, 4 and 4, respectively. The length of flanking sequence was constrained to 200 bp. Microsatellite statistics were selected using the “whole” mode, which means the program will generate one statistical Excel file for all sequence files as a whole. Repeats with unit patterns being circular permutations and/or complements were considered as one type for statistical analysis. For example, AGC denotes AGC, GCA, CAG, GCT, TGC and CTG in different reading frames or on the complementary strand. The software SPSS 19.0 was used to perform the data analysis and mapping. To facilitate the comparison among different repeat types or categories, the relative frequency, (SSR number per Mb of the sequence analyzed), and the relative density, [SSR length (in bp) per Mb of the sequence analyzed] were evaluated.
Results
Frequency and density of six classes of microsatellites
After scanning the genome sequence for six classes of SSRs, a total of 723,172 SSRs were identified in the yak genome assembly (Table 1). The total and mean lengths were 12,539,047 bp and 17.34 bp, respectively. The relative frequency and density were 272.18 loci/Mb and 4719.25 bp/Mb, respectively. About 0.47 % of the yak whole genome (2.66 Gb) was occupied by the perfect SSRs.
The counts, length, frequency, density and percentages of the six classes of perfect SSRs are summarized in Table 1. Mono-nucleotides were the most abundant type, with the highest relative frequency (119.85 loci/Mb) and density (1762.75 bp/Mb), accounting for 44.04 % of all SSRs, followed by dinucleotides (24.11 %), tri-nucleotides (15.80 %), penta-nucleotides (9.50 %) and tetra-nucleotides (6.40 %). Hexa-nucleotides were much less abundant, accounting for only 0.15 % of all SSRs (Table 1).
Abundance and repeat numbers for different microsatellite categories
Mononucleotide repeats
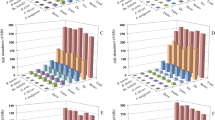
Poly (A) [or poly (T)] was the predominant mononucleotide repeat category, with 312,471 loci accounting for 98.13 % of the mononucleotide SSRs. The total length, frequency and density of poly (A) was 4.59 Mb, 117.60 loci/Mb and 1727.86 bp/Mb, respectively, and the average length was 14.69 bp (Table 2). However, poly (C) [or poly (G)] was far less abundant than poly (A) [or poly (T)], accounting for only 1.87 % of the total. The abundance of poly (C) was also lower (namely 2.25 loci/Mb and 34.90 bp/Mb, respectively). The repeat numbers of mononucleotide repeats ranged from 12 to 967 times. But the repeat times ranged from 12 to 29 were predominant, which numbered 317,538 accounting for 99.72 % of the total count of mononucleotide SSRs (Fig. 1A).

Repeat times of different types of SSRs in yak genome
Dinucleotide repeats
Dinucleotide repeats include AC, AT, AG and CG categories of SSRs. Results showed that the frequencies of AC and AT were highest (40.22 loci/Mb and 19.00 loci/Mb, respectively). AG had the middle frequency of 6.29 loci/Mb (Table 2). These three categories of SSRs numbered 174,048 and accounted for 99.82 % of the total number of dinucleotide repeats. The CG repeat had the lowest frequency of 0.12 loci/Mb and numbered 315. The repeat times of dinucleotide repeats ranged from 7 to 1206 times. However, the predominate repeat times ranged from 7 to 25 which numbered 173,346 and accounted for 99.42 % of the total count of dinucleotide SSRs (Fig. 1B).
Trinucleotide repeats
Statistical analysis of all trimer repeats including AAC, AAG, AAT, ACC, ACG, ACT, AGC, AGG, ATC and CCG showed that AGC had the highest frequency of 32.61 loci/Mb. Three categories of AAC, AAT and ACC had the middle frequencies that were 3.48 loci/Mb, 2.57 loci/Mb and 1.68 loci/Mb (Table 2), respectively. The others had the lower frequencies which ranged from 0.01 to 0.82 loci/Mb. The repeat times of trinucleotide SSRs ranged between 5 and 1033 times. But 5–11 repeat times were predominant and numbered 113,808 and accounted for 99.59 % of the total count of trinucleotide SSRs (Fig. 1C).
Tetranucleotide repeats
A total of 33 categories of tetranucleotide repeats were obtained in this study. Analysis of frequencies and densities of each tetrameric repeat categories revealed that ATTT, GTTT, AATG, CTTT and ATGG were predominant across the genome, and had frequencies of 4.58 loci/Mb, 2.82 loci/Mb, 1.48 loci/Mb, 1.44 loci/Mb and 1.15 loci/Mb (Table 2), respectively. The overall frequencies of 24 tetrameric repeats namely ATGC, ACAT, CCTT, AGAT, AGTG, CCCT, ACTG, AATT, CTGT, AACC, AGGC, AAGT, GGGT, AATC, ACGC, AGCC, CTTG, ACCT, AGTT, AGCT, AGCG, CCCG, ACGG and CCGG were at the middle level, which ranged between 0.01 loci/Mb and 0.71 loci/Mb. There were four categories of tetrameric repeats namely CGAA, GGTC, GTAC and TCGA which had low densities (namely 0.01 bp/Mb). The repeat times of tetranucleotide SSRs ranged between 4 and 248 times but 4–8 repeat times were predominant, and numbered 46,201 and accounted for 99.78 % of the total count of trinucleotide SSRs (Fig. 1D).
Pentanucleotide repeats
In pentanucleotide repeats categories, AACTG and ATCTG had a higher frequency of 18.79 loci/Mb and 4.04 loci/Mb, and density of 401.32 bp/Mb and 86.46 bp/Mb (Table 2), respectively. The frequencies of remainder categories were <0.76 loci/Mb. The repeat times of pentanucleotide SSRs ranged between 4 and 339 times. However, repeats ranging between 4 and 6 times were predominant, and numbered 68,439 and accounted for 99.62 % of the total count of pentanucleotide SSRs (Fig. 1E).
Hexanucleotide repeats
The frequencies of all hexanucleotide repeat categories were lower than that of above five types of repeats, and ranged between 0.03 loci/Mb and 0.00 loci/Mb. The repeat times of hexanucleotide SSRs ranged between 4 and 89 times. However, the predominate repeat times ranged between 4 and 7, and numbered 1072 and accounted for 97.90 % of the total count of hexanucleotide SSRs (Fig. 1F).
Discussion
Currently, a SSR scan for the entire yak genome sequence using bioinformatics methodology has not been reported. Our research firstly examined the abundance of perfect SSRs composed of 1–6 bp motifs in yak genomic sequence. In our study, approximately 0.47 % of the yak genome comprised perfect SSRs from mono- to hexa-nucleotide repeats. This percentage is similar to the results that reported before on the cattle (0.48 %) (Qi et al. 2013), sheep (0.48 %) (Qi et al. 2013) and chicken (0.49 %) (Huang et al. 2012), but smaller than that of other species genomes such as human (3 %) (Subramanian et al. 2003), mosquitoes (2.14 %) (Yu et al. 2005) and mouse (2.85 %) (Tong et al. 2006). These differences also could be due to the variation in search criteria, size of the database and bioinformatics software tools used in different studies for identification of SSRs.
Unsurprisingly, the proportion of the six classes of perfect SSRs was not evenly distributed in the yak genome. Mononucleotide repeats, accounting for the largest proportion (44.04 %) in six types of SSRs, had the highest frequency (119.85 loci/Mb) and maximum density (1762.75 bp/Mb), followed by dinucleotide, trinucleotide, pentanucleotide and tetranucleotide repeats. Hexanucleotide repeats had the lowest frequency (0.41 loci/Mb) and minimum density (12.04 bp/Mb) (Table 1). This trend is similar to what has been found in human, cattle, sheep and chicken genomes (Subramanian et al. 2003; Huang et al. 2012; Qi et al. 2013), but is different from that of mouse, silkworm, drosophila, mosquito and zebra fish (Katti et al. 2001; Li et al. 2004; Yu et al. 2005; Tong et al. 2006). This difference in abundance might be due selection for or against mono-, di- and trimers to tetra-, penta- and hexamers repeats.
In the present investigation, the number and density of certain repeat categories are greater than others within each type of repeats. In the case of mononucleotide repeats, Poly (A) [or Poly (T)] exhibited a strong over-representation, accounting for 98.13 % of total number of mononucleotide SSR categories. Similarly, in the other five classes of SSRs, fourteen categories including AC, AT, AG, AGC, AAC, AAT, ACC, ATTT, GTTT, AATG, CTTT, ATGG, AACTG and ATCTG in yak genome were the predominant repeats, which all had a normal frequency above 1.00 loci/Mb (Table 2). It is possible that during SSR evolution the poly (A) stretches present in the genome might have mutated to produce the A-rich repeats. It is also possible that the abundance of repeats is influenced by their secondary structures and the effect on DNA replication. In addition, the repeat times of different categories of SSRs was also different. For example, the repeat times for mononucleotide SSRs mainly ranged between 12 and 29, for dinucleotide SSRs ranged between 7 and 25 times, for trinucleotide SSRs ranged between 5 and 11 times, and for tetranucleotide, pentanucleotide and hexanucleotide SSRs the repeats ranged between 4–8, 4–6 and 4–7 (Fig. 1), respectively. (Schlotterer 1998) showed that nucleotide sequences with higher GC content possessed fewer SSRs than those of higher AT content. Our results are consistent with this research, indicating that SSRs in yak genome are also AT-rich.
It should be noted that although the complete genome sequence of yak was obtained in 2012, it has not yet been physically mapped. So, in the future studies, after assembling the yak genome sequence to each chromosome, the following areas need to be further explored. Firstly, comparative analysis of abundance of SSRs on different chromosomes, and the association between the length of chromosomes and the distribution of SSRs on each chromosome needs to be investigated. Moreover, the difference in abundance of different classes of SSRs in coding and non-coding regions of yak genome (i.e. exon, intron and intergenic regions) should be studied. Some studies showed that SSRs plays an important role in the structure and function of the genome and may be associated with some diseases (Hefferon et al. 2004; Campregher et al. 2010). Therefore, another research focus should be to reveal genetic mechanisms, the function of SSRs in the yak genome and correlative analysis between some diseases and SSRs. Lastly, at present, the genome sequence of yak Y chromosome has not yet been obtained as the present complete genome sequence came from a female yak (Qiu et al. 2012). Pian Niu or Cattle-yak (Bos taurus × Bos grunniens), the first filial generation of yak and ordinary cattle, showed obvious hybrid vigor. However, an issue with crossbreeding and improvement of yak is that the males are sterile, thus it is not possible to reliably utilize the heterosis. Until now, although many studies have been done on the sterility of the male Pian Niu both at home and abroad, there has been no solution to the problem of male sterility (Luo et al. 2014). Therefore, it is necessary to obtain the genome sequence of yak Y chromosome and mine more Y-chromosome-specific SSR markers. Then, combining to the Y chromosome information from cattle, Pianniu (Bos taurus × Bos grunniens), zebu and others, the problem of male sterility of Pianniu can be explored.
References
Cai X, Mipam T, Zhao FF, Sun L (2014) Isolation and characterization of polymorphic microsatellites in the genome of Yak (Bos grunniens). Mol Biol Rep 41:3829–3837
Campregher C, Scharl T, Nemeth M, Honeder C, Jascur T, Boland CR, Gasche C (2010) The nucleotide composition of microsatellites impacts both replication fidelity and mismatch repair in human colorectal cells. Hum Mol Genet 19:2648–2657
Du L, Li Y, Zhang X, Yue B (2013) MSDB: a user-friendly program for reporting Distribution and building databases of microsatellites from genome sequences. J Hered 104:154–157
Guichoux E, Lagache L, Wagner S, Chaumeil P, Léger P, Lepais O, Lepoittevin C, Malausa T, Revardel E, Salin F, Petit RJ (2011) Current trends in microsatellite genotyping. Mol Ecol Resour 11:591–611
Hefferon TW, Groman JD, Yurk CE, Cutting GR (2004) A variable dinucleotide repeat in the CFTR gene contributes to phenotype diversity by forming RNA secondary structures that alter splicing. Proc Natl Acad Sci USA 101:3504–3509
Hu QJ, Ma T, Wang K, Xu T, Liu JQ, Qiu Q (2012) The yak genome database: an integrative database for studying yak biology and high-altitude adaption. BMC Genomics 13:600
Huang J, Du LM, Li YZ, Li WJ, Zhang XY, Yue BS (2012) Distribution regularities of microsatellites in the Gallus gallus genome. Sichuan J Zool 31:358–363 (in chinese with english abstract)
Katti MV, Ranjekar PK, Gupta VS (2001) Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol Biol Evol 18:1161–1167
Li B, Xia QY, Lu C, Zhou ZY, Xiang ZH (2004) Analysis on frequency and density of microsatellites in coding sequence of several eukaryotic genomes. Genomics, Proteomics Bioinform 2:24–31
Luo XL, Song HF, Guan JQ (2014) Investigation on mechanism of sterility of male hybrids between yak and cattle. J Appl Anim Res 42:395–399
Nguyen TT, Genini S, Ménétrey F, Malek M, Vögeli P, Goe MR, Stranzinger G (2005) Application of bovine microsatellite markers for genetic diversity analysis of Swiss yak (Poephagus grunniens). Anim Genet 36:484–489
Qi XB, Han JL, Lkhagva B, Chekarova I, Badamdorj D, Rege JE, Hanotte O (2005) Genetic diversity and differentiation of Mongolian and Russian yak populations. J Anim Breed Genet 122:117–126
Qi XB, Han JL, Wang G, Rege JEO, Hanotte O (2010) Assessment of cattle genetic introgression into domestic yak populations using mitochondrial and microsatellite DNA markers. Anim Genet 41:242–252
Qi WH, Jiang XM, Xiao GS, Huang XY, Du LM (2013) Seeking and bioinformatics analysis of microsatellite sequence in the genomes of cow and sheep. Acta Veterinaria et Zootechnica Sinica 44:1724–1733 (in chinese with english abstract)
Qiu Q, Zhang GJ, Ma T, Qian W, Wang J, Ye Z, Cao C, Hu Q, Kim J, Larkin DM, Auvil L, Capitanu B, Ma J, Lewin HA, Qian X, Lang Y, Zhou R, Wang L, Wang K, Xia J, Liao S, Pan S, Lu X, Hou H, Wang Y, Zang X, Yin Y, Ma H, Zhang J, Wang Z, Zhang Y, Zhang D, Yonezawa T, Hasegawa M, Zhong Y, Liu W, Zhang Y, Huang Z, Zhang S, Long R, Yang H, Wang J, Lenstra JA, Cooper DN, Wu Y, Wang J, Shi P, Wang J, Liu JQ (2012) The yak genome and adaptation to life at high altitude. Nat Genet 44:946–949
Ramesha KP, Biswas TK, Jayakumar S, Das S, Gupta N, Kataktalware MA, Gupta SC (2012) Application of cattle microsatellite markers to assess genetic diversity of Indian yaks. Indian J Anim Sci 82:770–772
Ritz LR, Glowatzki-Mullis ML, MacHugh DE, Gaillard C (2000) Phylogenetic analysis of the tribe Bovini using microsatellites. Anim Genet 31:178–185
Schlotterer C (1998) Genome evolution: are microsatellites really simple sequences? Curr Biol 8:132–134
Subramanian S, Mishra RK, Singh L (2003) Genome-wide analysis of microsatellite repeats in humans: their abundance and density in specific genomic regions. Genome Biol 4:R13
Tautz D, Renz M (1984) Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Res 12:4127–4138
Tong XL, Dai FY, Li B, Yu QY, Xia QY, Lu C (2006) Microsatellite repeats in mouse: abundance, distribution and density. Curr Zool 52:138–152
Wang MQ, Weigend S, Barre-Dirie A, Carnwath JW, Lu ZL, Niemann H (2003) Analysis of two Chinese yak (Bos grunniens) populations using bovine microsatellite primers. J Anim Breed Genet 120:237–244
Wiener G, Han JL, Long RJ (2003) The yak. Regional Office for Asia and the Pacific of the Food and Agriculture Organization of the United Nations, Bangkok, Thailand
Yu QY, Li B, Li GR, Fang SM, Yan H, Tong XL, Qian JF, Xia QY, Lu C (2005) Abundance and distribution of microsatellite in the entire mosquito. Prog Biochem Biophys 32:435–441
Zhang GX, Chen WS, Xue M, Wang ZG, Chang H, Han X, Liao XJ, Wang DL (2008) Analysis of genetic diversity and population structure of Chinese yak breeds (Bos grunniens) using microsatellite markers. J Genet Genomics 35:233–238
Acknowledgments
The author thank Prof. Michael W. Bruford, Dr. Penny C. Gardner and Dr. David Stanton (Cardiff university, UK) for their assistance in improving English on the manuscript. This study was supported by the National Natural Science Foundation of China (No. 31360267), the 123 high-level personnel training project of Qinghai university, the scientific and technological innovation platform of bovine (milk, meat, wool) industry of Qinghai Province and the Technology Foundation for Selected Overseas Chinese Scholar, Department of Human Resources and Social Security of Qinghai Province.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ma, Z. Genome-wide characterization of perfect microsatellites in yak (Bos grunniens). Genetica 143, 515–520 (2015). https://doi.org/10.1007/s10709-015-9849-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-015-9849-y