
Abstract
Hereditary endocrine tumor syndromes are rare conditions with overlapping features. It is imperative that healthcare providers differentiate between these syndromes for proper patient care. Advances in genetic testing technologies have increased utilization of genetic counseling and testing in this field; however, few endocrine cancer genetics clinics exist. Two years ago, a genetic counselor (GC) specializing in endocrine cancer genetics was added to the multidisciplinary team of the James Neuroendocrine/Thyroid Clinic at The Ohio State University. Here, we report on this experience. In total, 358 patients were seen. The majority were referred by medical oncology (n = 204; 57%) for a personal history of disease (n = 249; 81%). The most common referral indications were pancreatic neuroendocrine tumors (n = 44; 17%), multiple primary tumors (n = 37; 14%), and pheochromocytoma/paraganglioma (n = 35; 14%). Most patients completed genetic testing after genetic counseling (n = 200; 65%). Targeted gene panel testing was the most common testing ordered (n = 98; 32%). Thirty-one patients (15.5%) had ≥ one likely pathogenic variant (LPV) or pathogenic variant (PV) identified. Approximately 37% (n = 11) did not meet genetic testing guidelines for the gene they tested positive for. The most common genes with LPV/PVs were the SDH genes (n = 8) and MEN1 (n = 7). Referral indications with the highest likelihood of LPV/PVs were paraganglioma, medullary thyroid carcinoma, and multiple primary tumors. We believe this data can provide valuable guidance to healthcare providers who see patients with endocrine neoplasia or who are seeking to establish hereditary endocrine cancer clinics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hereditary endocrine tumor syndromes are rare genetic conditions. While distinct, these syndromes often have similar clinical symptoms and tumor types. Healthcare professionals involved in the care of these patients, including physicians and genetic counselors, must be able to recognize and differentiate between these conditions to ensure appropriate medical care. Early identification of these syndromes is beneficial to the patient and has been shown to decrease morbidity and mortality, avoid unnecessary surgeries, increase surveillance, and help identify at-risk family members [1, 2]. The most common hereditary endocrine tumor syndromes include multiple endocrine neoplasia type 1 (MEN1), multiple endocrine neoplasia type 2 (MEN2), von Hippel-Lindau syndrome (VHL) and hereditary paraganglioma-pheochromocytoma syndrome. Less common hereditary endocrine tumor syndromes include Carney complex, CDC73-related disorders/hyperparathyroidism-jaw tumor syndrome (HPT-JT), multiple endocrine neoplasia type 4 (MEN4), and tuberous sclerosis complex (TSC). Other hereditary tumor/cancer syndromes with endocrine manifestations include familial adenomatous polyposis (FAP), neurofibromatosis type 1 (NF1), and PTEN hamartoma tumor syndrome/Cowden syndrome, among others. A brief review of the endocrine-related tumors and cancers associated with these syndromes can be found in Table 1.
Each of these hereditary endocrine tumor syndromes have known genetic etiologies with high identification rates through genetic testing. With recent advances in genetic testing technologies, such as next-generation sequencing, utilization of genetic counseling and testing for hereditary endocrine tumor syndromes has increased. However, there are only a small number of dedicated endocrine cancer clinics in the U.S. with expertise caring for these patients. In addition, due to the rarity of many of these conditions, few guidelines on referral and genetic testing for endocrine cancer clinics exist. Two years ago, a genetic counselor specializing in endocrine tumor/cancer genetics was added to the multidisciplinary team of the James Neuroendocrine/Thyroid Clinic at The Ohio State University (OSU). In addition to the genetic counselor, the multidisciplinary team includes 5 endocrinologists, 2 endocrine surgeons, 3 medical oncologists, and several nurse practitioners and physician assistants. Genetic counseling appointments are scheduled on the same day as appointments with the physicians. The objective of this study was to report on this unique experience of embedded genetic counseling and evaluation services in a multidisciplinary endocrine cancer clinic.
Materials and methods
After obtaining approval through The Ohio State University (OSU) Institutional Review Board (IRB), the clinical database of the endocrine genetic counselor was reviewed to identify eligible patients. Eligibility criteria included any individual referred and seen for genetic counseling within the OSU James Multidisciplinary Neuroendocrine/Thyroid Cancer Clinic between April 5, 2016 and April 27, 2018. After identification of these patients, their electronic medical records were reviewed and the following information was obtained: referral source, referral indication, tumor pathology, family history/pedigree, genetic testing ordered, genetic counseling note(s), and results from genetic testing. A total of 358 records were reviewed.
Results
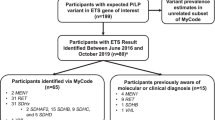
Figure 1 shows the basic workflow of patient data analysis. The embedded genetic counselor was involved with 358 endocrine-specific patients between the dates of 4/5/16 and 4/19/18. Two hundred four patients were referred by medical oncology, 64 patients by endocrinology, 51 patients by a family member for cascade testing, 31 patients by surgery/surgical oncology, and 8 patients by another specialty/entity (self-referral, genetics, otolaryngology, etc.). Those referred by a family member for cascade testing were excluded from the next step of analysis and analyzed separately as their indication for referral/testing was fundamentally different; i.e. a known likely pathogenic variant (LPV) or pathogenic variant (PV) was present in the family. The remaining 307 patients were categorized into one of three groups based on reason for referral: personal history of disease (referred only due to their own personal history), personal and family history of disease (referred due to a combination of personal and family history), or family history of disease (no personal history themselves; referred only due to family history). Two hundred forty-nine patients were referred due to a personal history of disease, 54 patients were referred due to a personal and family history of disease, and 4 patients were referred due to a family history of disease.

Flowchart of patient data analysis. LPV likely pathogenic variant, PV pathogenic variant, VUS variant of uncertain significance, d/t due to
Referral indications for the 249 patients referred for a personal history of disease were analyzed and are depicted in Table 2. Eleven patients had two primary reasons for referral and were counted for each, increasing the total number of indications from 249 to 260. Patients in this clinic were most commonly referred for personal histories of pancreatic neuroendocrine tumors (panNETS; n = 44; ~ 17%), multiple primary tumors in the same individual (n = 37; ~ 14%), and pheochromocytomas (PCC) and/or paragangliomas (PGL; n = 35; ~ 13%). Other reasons for referral included personal histories of lung/bronchial/thymic carcinoid (n = 17; ~ 7%), established molecular diagnosis (positive genetic testing) from another provider (n = 15; ~ 6%), adrenocortical carcinoma (ACC; n = 14; ~ 5%), clinical MEN1 (defined by the presence of 2–3 parathyroid tumors, pituitary tumors, and/or panNETs, hereafter referred to as the “p-triad”; n = 14; ~ 5%), two endocrine tumors (n = 14; ~ 5%), abnormal result on somatic/tumor testing (n = 13; ~ 5%), gastrointestinal (GI) neuroendocrine tumors (NET; n = 12; ~ 5%), parathyroid disease, including hyperparathyroidism, parathyroid adenoma, and parathyroid carcinoma (n = 9; ~ 3%), medullary thyroid cancer (MTC; n = 9; ~ 3%), NET not otherwise specified/site of origin unknown (NOS; n = 8; ~ 3%), and appendix carcinoid (n = 8; ~ 3%). Additionally, 11 patients (~ 4%) were referred for another diagnosis and are included in the “other” category (e.g., clinical diagnosis of VHL, personal history of ganglioneuromas). Individuals in the multiple primary tumors category had 2 or more primary tumors with at least one being an endocrine-related tumor. If the tumors were consistent with clinical MEN1 or if they were 2 endocrine tumors solely, they were included in those respective categories.
In total, 200 patients out of 307 (excluding those patients referred and seen for cascade testing) pursued and completed genetic testing after their visit with the genetic counselor, making the overall uptake rate of genetic testing in this clinic approximately 65%. Of note, 15 patients (~ 5%) had genetic testing performed by another provider prior to coming in for genetic counseling. Of the 200 patients who had genetic testing performed and completed after their visit with the endocrine genetic counselor, 98 (~ 32%) had targeted gene panel testing specific to their symptoms/tumor type/condition, 61 patients (~ 20%) had multi-cancer gene panels (~ 60–80 genes), 16 patients (~ 5%) had common cancer gene panels (~ 30–40 genes), 16 patients (~ 5%) had single gene testing, and 9 patients (~ 3%) had between 2 and 5 genes tested. Examples of targeted gene panel tests that were commonly ordered include, but are not limited to: custom ACC panel (APC, EPCAM, MEN1, MLH1, MSH2, MSH6, PMS2, TP53), Hereditary PCC/PGL panel (MAX, NF1, RET, SDHA, SDHAF2, SDHB, SDHC, SDHD, TMEM127, VHL), and HPT panel (CASR, CDC73, CDKN1B, MEN1, RET). Test selection was based on the patient’s personal and/or family history, as well as patient preference. All decisions were made through a shared decision making model with the patient and genetic counselor. Eighty-six patients (~ 28%) did not have genetic testing performed after genetic counseling. Reasons for not pursuing genetic counseling included: they were determined to be low risk/low yield by the genetic counselor (n = 35), they wanted to think more about testing (n = 14), other family members were determined to be more appropriate for testing (n = 10), testing was not a covered benefit by their insurance company/high out pocket cost for testing (n = 6), they were not interested in testing (n = 6), they wanted to talk with family members before deciding (n = 4), they stated being overwhelmed/had a lot going on (n = 3), and additional records were needed prior to pursuing genetic testing (n = 3). Additionally, 2 patients enrolled in a research study, 2 patients received referrals to other providers, and 1 patient wanted to complete surgery before doing testing.
Out of the 200 patients that completed genetic testing after genetic counseling, 31 patients had at least one likely pathogenic variant (LPV) or pathogenic variant (PV) identified, making the positive rate in this clinic 15.5%. Of those 31 patients, 21 (~ 10%) had one LPV/PV identified, 2 (~ 1%) had more than one LPV/PV identified, and 8 (~ 4%) had one LPV/PV and one variant of uncertain significance (VUS) identified. Forty-three patients (~ 22%) without LPV/PVs had one VUS identified and 17 patients (~ 8%) had more than one VUS identified. The remaining 109 patients (~ 55%) had normal genetic testing results. It should be noted that our positive rate includes MUTYH heterozygotes in which cancer risks are contestable. Our positive rate excluding MUTYH heterozygotes was 13% (26 of 200).
In the 31 patients that tested positive for at least one LPV/PV, 34 total LPV/PVs were identified. There were 7 LPV/PVs in MEN1, 6 in SDHB, 5 in MUTYH (heterozygous state), 2 in RET, 2 in MITF, and one in each of the following genes: ATM, BARD1, CDKN1B, CHEK2, FH, MSH2, MSH6, NBN, SDHC, SDHD, TMEM1, and VHL. The phenotypes of the 31 patients with identified LPV/PVs were reviewed and are displayed in Table 3. The phenotypes of the patients listed are more comprehensive than their referral indication for the purpose of identifying if they met current genetic testing guidelines for the gene they tested positive in.
Of the 15 patients that came in with previously established molecular diagnoses/positive genetic testing from another provider, there were 5 patients with LPV/PVs in MEN1, 5 in RET, 4 in the SDH genes (3 SDHB, 1 SDHA), and 1 in NF1.
To analyze which population of patients were yielding the most LPV/PVs, positive rates for each personal history referral indication were calculated and are displayed in Table 4. To calculate the positive rates, the number of LPV/PVs for each referral indication were divided by the total number of people from each referral indication that pursued genetic testing. Of note, there was one patient who had both a PCC and PGL and was included in both the PCC and PGL categories. Patients with PGLs had the highest positive rate (50%), followed by MTC (40%), multiple tumors (~ 26%), abnormal somatic/tumor testing (25%), clinical diagnosis of MEN1 (presence of 2–3 p-triad tumors; ~ 23%), parathyroid disease (~ 22%), appendix carcinoid (20%), GI NET (~ 18%), PCC (~ 10.5%), ACC (~ 8%), two endocrine tumors (~ 8%), and panNET (~ 4%). One patient in the “other” category had a clinical diagnosis of VHL and tested positive for a LPV/PV in VHL, making the positive rate from the “other” category ~ 13%. No LPV/PVs were identified in any patients with a personal history referral indication of lung/bronchial/thymic carcinoid or a NET NOS.
Lastly, the histories of individuals referred by a family member for cascade testing were analyzed. All 51 patients referred for this indication pursued genetic testing for the LPV/PV known in their family. Twenty-seven patients (~ 53%) tested positive. There were 8 LPV/PVs identified in SDHB, 8 in MEN1, 5 in RET, 2 in VHL, and 1 in SDHA.
Discussion
We found that the majority of patients (~ 65%) in our clinic pursued genetic testing after genetic counseling. Of these, 15.5% tested positive for at least one LPV/PV. The percentage of patients pursuing genetic testing and the positive yield in our clinic is generally in line with other reported cancer specialties which found uptake rates of genetic testing between 60 and 95% and positive yields of genetic testing between 9 and 20% [3,4,5]. Due to the high positive yield found in our clinic and the potential genetic risk many of these genes carry, we believe programs aimed at identifying hereditary risk need to better include endocrine-related tumors in their screening approaches. To our knowledge, this is the first study to examine uptake rates and positive rates of genetic testing in the endocrine cancer genetics field.
Of the 31 patients in our cohort that tested positive for at least one LPV/PV, we found that overall 11, or about 35.5%, did not meet current genetic testing guidelines (such as the National Comprehensive Cancer Network (NCCN) guidelines), for the gene they tested positive in. Previous studies examining efficiency of genetic testing guidelines for other cancer syndromes have found that nearly 50% of patients with an identified LPV/PV do not meet current genetic testing guidelines [6, 7]. This data paired with the data from our clinic further highlights the need for improvements in genetic testing guidelines across cancer genetic specialties, and in particular in endocrine cancer genetics. If our clinic had strictly followed current genetic testing guidelines, over one third of our patients that had an identified LPV/PV would have been missed.
An interesting result from our study was the number of LPV/PVs in unrelated, or incidental, genes. Our rate of incidental findings in this study was 5%. These findings include 5 variants in MUTYH in the heterozygous state, as well as 1 in each of the following genes: ATM, NBN, BARD1, CHEK2, and MITF. While all of these genes are associated with an increased risk for cancer (with some controversy in regards to the cancer risk of MUTYH heterozygotes), none of them were definitively related to the endocrine indication of the patient being tested, although some studies have suggested a possible association with MUTYH heterozygote status and carcinoid risk [8]. This rate of incidental findings is generally in line with previously reported literature on the topic [9]. Additionally, many of the incidental findings in our study are in genes known to have a high frequency of variants in the generation population, such as MUTYH (1–2% population frequency in heterozygous state), ATM (1–2% population frequency in heterozygous state), and CHEK2 (up to 3% population frequency in the heterozygous state) [10,11,12]. Although identification of these LPV/PVs in our cohort will likely not affect endocrine-related risk management, most will affect other management for these patients. As additional research is done on these genes there may be important endocrine implications that are found; however, at this time they are seemingly incidental results.
MEN1 was one of the most common genes with a LPV/PV identified in our study. Of the 7 patients that tested positive for a LPV/PV in MEN1, four of them did not meet clinical criteria for MEN1 at the time of testing. One patient had only a panNET, one had a single NET of unknown origin, one had only HPT, and one had a panNET and a lung carcinoid. Further, if we look at our patients with a clinical diagnosis of MEN1 (i.e. presence of 2–3 p- triad tumors), we found that 10 out of 13 did not have an identifiable LPV/PV in MEN1. Based on this information, the positive predictive value (PPV) for MEN1 testing (i.e. the probability of having a positive gene test given clinical criteria are met) in this cohort was 23%. This PPV suggests that the clinical criteria for MEN1 are ineffective at predicting patients with MEN1 LPV/PVs, at least in our population. These observations highlight both the vagueness of the current MEN1 clinical criteria and also the need for improved genetic testing in this syndrome, whether that be more comprehensive testing of the MEN1 gene and/or identification of other genes that may be causative of this phenotype, among others. Clearly, there is a discrepancy occurring between those with a clinical diagnosis of MEN1 and those that tested positive for a LPV/PV in the MEN1 gene. We believe future research is needed in this area to determine why these discrepancies are occurring.
Based on current recommendations in the field and our clinical experience with embedded endocrine genetic counseling, we propose guidelines for healthcare providers on when to refer patients for endocrine-related genetic counseling and what genetic testing is appropriate for them. We recommend prioritizing patients that are easiest to identify, refer, and have a high likelihood of hereditary risk. In the context of endocrine genetic counseling, this would be any individual diagnosed with a PCC, PGL, and/or MTC. Recently, some endocrine cancer genetics clinics have recommended genetic testing in all patients with parathyroid carcinoma; however we cannot comment on this recommendation as neither of our patients with parathyroid carcinoma (n = 2) had an identified LPV/PV. Additionally, our clinic saw high referral and mutation-positive rates for individuals with multiple primary tumors, indicating that these individuals may benefit from genetic counseling and testing, and should be considered for referral. We recommend a gene panel approach for patients with endocrine-related indications given the many overlapping clinical features/tumor types, variable expressivity, reduced penetrance, and emerging phenotypes of many of the hereditary endocrine tumor syndromes which makes it difficult to identify a single gene that might cause a given tumor. Additionally, we recommend regular follow-up and continued care for patients who test negative through genetic testing but whose histories are suggestive of hereditary risk (i.e. young patients, patients with multiple tumors, patients with concerning endocrine manifestations, etc.) as genetics knowledge continues to grow and improved testing may identify mutations in new genes in patients who previously tested negative.
Limitations and future directions
A limitation of our study is the relatively small sample size. In particular, our number of patients with LPV/PVs was small and thus any conclusions need to be viewed with some caution. However, this study represents findings from the largest clinical cohort of endocrine cancer genetic counseling patients to date that we are aware of. Another limitation of this study is that the referral and practice patterns in this clinic and with this genetic counselor may be different from other clinics and other counselors. For example, the majority of patients diagnosed with MTC at our institution undergo RET testing without the endocrine genetic counselor involved. Therefore, MTC was not a common referral indication seen in our clinic. Additionally, the medical oncologists at our institution readily refer patients with panNETs for endocrine genetic counseling; which may explain why medical oncology was the most common referring specialty in our cohort and why panNETs were the most common referral indication. This study was also conducted at a large, tertiary care center and it is possible that the patients seen at this center are fundamentally different (i.e. more severe or syndromic cases) than those seen at community-based centers. Thus, experiences may differ in other clinical practices.
Future directions for this project are many-fold. Additional studies could determine if/how management changed for those who tested positive in this clinic, and how that impacted clinical outcomes. The VUS data from this population could be examined more in-depth as there were a significant number of patients who had VUS results in our study. Future studies could determine if the VUSs identified in our cohort were suspicious for pathogenicity and/or if they were in line with patient phenotypes, among others. Patient demographics (such as age, gender, ethnicity, education level, and socioeconomic status, etc.) would also be important to look at further to identify any barriers to genetic testing in this field and/or any patterns in genetic testing results. As there are a limited number of endocrine cancer genetics clinics with embedded genetic counselors in the country, it could be beneficial to compare practice patterns as well as uptake and positive rates to identify any potential modifications and/or improvements that could be made in the field. Lastly, there could be benefit in providing resources to non-endocrine genetic counselors that are more representative of overall practice patterns in the endocrine cancer genetics field as opposed to those influenced by individual or institutional factors.
Conclusion
We have reported on our unique experience of embedded genetic counseling and evaluation services in a multidisciplinary endocrine clinic, including referring specialties, referral types, and personal history referral indications. This data may help in the establishment of new hereditary endocrine cancer clinics and in the practice of existing clinics. In our clinic, the majority of referrals (~ 57%) came from medical oncology and the most common reason for referral was for a personal history of disease (~ 81%). The most common personal history referral indications were panNETs (~ 17%), multiple primary tumors in the same individual (~ 14%), and PCC/PGL (~ 13%). The uptake of genetic testing in our clinic was approximately 65% and the positive yield was 15.5%. MEN1 and the SDH genes were the most common positive results. Referral indications that yielded the highest frequency of positive results were PGL, MTC, and multiple primary tumors. Overall, approximately 35% of our patients that tested positive did not meet current genetic testing guidelines for the gene they tested positive in. These data can provide valuable guidance to other healthcare professionals who see endocrine cancer patients or are seeking to establish hereditary endocrine cancer clinics.
References
White HD, Blair J, Pinkney J, Cuthbertson DJ, Day R, Weber A, Macfarlane IA (2010) Improvement in the care of multiple endocrine neoplasia type 1 through a regional multidisciplinary clinic. QJM 103(5):337–345. https://doi.org/10.1093/qjmed/hcq020
Pieterman CR, Schreinemakers JM, Koppeschaar HP, Vriens MR, Rinkes IH, Zonnenberg BA et al (2009) Multiple endocrine neoplasia type 1 (MEN1): its manifestations and effect of genetic screening on clinical outcome. ClinEndocrinol 70(4):575–581. https://doi.org/10.1111/j.1365-2265.2008.03324.x
Susswein L, Marshall M, Nusbaum R, Postula K, Weissman M, Yackowski L et al (2015) Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med 18(8):823–832. https://doi.org/10.1038/gim.2015.166
Walker E, Carnevale J, Pedley C, Blanco A, Chan S, Collisson E et al (2018) Referral frequency, attrition rate, and outcomes of germline testing in patients with pancreatic adenocarcinoma. Fam Cancer. https://doi.org/10.1007/s10689-018-0106-2
Stuckey A, Febbaro T, Laprise J, Wilbur J, Lopes V, Robison K (2016) Adherence patterns to national comprehensive cancer network guidelines for referral of women with breast cancer to genetics professionals. Am J ClinOncol 39(4):363–367. https://doi.org/10.1097/COC.0000000000000073
Brand R, Borazanci E, Speare V, Dudley B, Karloski E, Peters M et al (2018) Prospective study of germline genetic testing in incident cases of pancreatic adenocarcinoma. Cancer 124(17):3520–3527. https://doi.org/10.1002/cncr.31628
Beitsch P, Whitworth P, Hughes K, Patel R, Rosen B, Compagnoni G et al (2019) Underdiagnosis of hereditary breast cancer: are genetic testing guidelines a tool or an obstacle? J ClinOncol 37(6):453–460. https://doi.org/10.1200/JCO.18.01631
Dumanski JP, Rasi C, Björklund P, Davies H, Ali AS, Grönberg M et al (2017) A MUTYHgermline mutation is associated with small intestinal neuroendocrine tumors. EndocrRelat Cancer 24(8):427–443. https://doi.org/10.1530/ERC-17-0196
Roche MI, Berg JS (2015) Incidental findings with genomic testing: implications for genetic counseling practice. Curr Genet Med Rep 3(4):166–176. https://doi.org/10.1007/s40142-015-0075-9
Win AK, Hopper JL, Jenkins MA (2011) Association between monoallelic MUTYH mutation and colorectal cancer risk: a meta-regression analysis. Fam Cancer 10(1):1–9. https://doi.org/10.1007/s10689-010-9399-5
Jerzak KJ, Mancuso T, Eisen A (2018) Ataxia-telangiectasia gene (ATM) mutation heterozygosity in breast cancer: a narrative review. CurrOncol (Toronto, Ont.) 25(2):e176–e180. https://doi.org/10.3747/co.25.3707
Apostolou P, Fostira F (2013) Hereditary breast cancer: the era of new susceptibility genes. Biomed Res Int 2013:747318. https://doi.org/10.1155/2013/747318
Funding
This research did not receive any special grant from any funding agency in the public, commercial, or not-for-profit sector.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Jennifer L. Anderson, Pamela Brock, and Robert Pilarski. The first draft of the manuscript was written by Jennifer L. Anderson and Pamela Brock and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Lawrence Kirschner has a patent on the identification of the PRKAR1A gene (Carney complex; United States Patent 6,759,525 Stratakis, et al. July 6, 2004). There are no additional conflicts of interest to disclose.
Ethical approval
This study was reviewed and approved by The Ohio State University (OSU) Institutional Review Board. This study was performed in line with the principles of the Declaration of Helsinki.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Anderson, J.L., Pilarski, R., Kirschner, L. et al. Genetic evaluation of patients and families with concern for hereditary endocrine tumor syndromes. Familial Cancer 21, 93–100 (2022). https://doi.org/10.1007/s10689-020-00222-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-020-00222-0