
Abstract
The unique life form of plants promotes the accumulation of somatic mutations that can be passed to offspring in the next generation, because the same meristem cells responsible for vegetative growth also generate gametes for sexual reproduction. However, little is known about the consequences of somatic mutation accumulation for offspring fitness. We evaluate the fitness effects of somatic mutations in Mimulus guttatus by comparing progeny from self-pollinations made within the same flower (autogamy) to progeny from self-pollinations made between stems on the same plant (geitonogamy). The effects of somatic mutations are evident from this comparison, as autogamy leads to homozygosity of a proportion of somatic mutations, but progeny from geitonogamy remain heterozygous for mutations unique to each stem. In two different experiments, we find consistent fitness effects of somatic mutations from individual stems. Surprisingly, several progeny groups from autogamous crosses displayed increases in fitness compared to progeny from geitonogamy crosses, likely indicating that beneficial somatic mutations occurred in some stems. These results support the hypothesis that somatic mutations accumulate during vegetative growth, but they are filtered by different forms of selection that occur throughout development, resulting in the culling of expressed deleterious mutations and the retention of beneficial mutations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mutation is the source of variation for evolution and adaptation, but organisms differ in whether mutations originating during gamete formation (meiosis) or somatic growth (mitosis) contribute to heritable variation. For the vast majority of organisms, including viruses, unicellular microbes, and some multicellular eukaryotes, sexual reproduction is rare or absent. In these organisms, mutations can occur during mitotic cell replication, and the primary mechanism for adaptation and diversification occurs via selection on cell lineages without recombination. By contrast, acquired mutations occurring during somatic growth of animals are not heritable. This is because in most metazoans (but possibly excepting corals; Barfield et al. 2016; Schweinsberg et al. 2014), the germline is determined early in development and relatively few cell divisions occur before the formation of gametes (the Weismann Barrier; Buss 1983). Consequently, heritable mutations typically occur in animals only during the development of gonads and gametes.
Plants differ from animals and microbes, because mutations contributing to heritable variation can arise both during gamete formation and somatic growth (Antolin and Strobeck 1985; Klekowski and Godfrey 1989). This is due to the fact that plants lack a separate germline and grow from the division of a population of undifferentiated meristem cells within the stem tip that is known as the central zone. These germ cell lineages go on to produce future stem, leaf, and reproductive tissues (e.g., flowers). Therefore, as plants grow, individual ramets of the same genet (separate stems or vegetatively propagated plants) can continue to accumulate mitotic mutations, which can make their way into the gametes and thus be passed to the next generation (Bobiwash et al. 2013; Dubrovina and Kiselev 2016; Klekowski 2003; McKnight et al. 2002; Schmid-Siegert et al. 2017; Schultz and Scofield 2009; Watson et al. 2016; Yu et al. 2020). This aspect of plant biology is well known (Ally et al. 2010; Monro and Poore 2009; Monroe et al. 2022; Reusch and Bostrom 2011), and somatic mutation accumulation has been important in agriculture, where the origin of many clonally-derived varieties of fruits, including citrus, apples, and wine grapes, have been cultivated by grafting from genetically differentiated bud tips (Aradhya et al. 2003; Jarni et al. 2015; McKey et al. 2010; Miller and Gross 2011; Pelsy et al. 2015; Vezzulli et al. 2012). Thus, since somatic mutations can be heritable, they may be an important source of genetic variation for evolution. However, there is disagreement over the extent and evolutionary importance of somatic mutation accumulation in plants (Burian et al. 2016; Hanlon et al. 2019; Kuhlemeier 2017; Plomion et al. 2018; Schmid-Siegert et al. 2017; Schultz and Scofield 2009; Watson et al. 2016).
When we consider the potential for meiotic and somatic mutations to contribute to the total mutational load of plant populations—particularly for long-lived plants—it becomes evident that not all of the mutations occurring during a plant’s lifespan are passed to the next generation (Cruzan 2018, pp. 86–98). Indeed, plants have mechanisms of “developmental selection” (Buchholz 1922; Langridge 1958; Williams et al. 1999) that occur during vegetative growth and reproduction to filter the set of mutations that are inherited by progeny (Monroe et al. 2022). New somatic mutations occur as a single copy within the diploid genome, so their fitness effects for the germ cells that carry them will depend on their expression in the heterozygous state. Mathematical models have demonstrated that unexpressed mutations (i.e. neutral and recessive deleterious mutations) are likely to accumulate as germ cells divide, but expressed mutations that reduce cell growth will be eliminated. More rarely, expressed beneficial mutations will arise, such that any mutations that elevate the rate of cell division will tend to increase in frequency until they have replaced the entire germ cell population (clonal selective sweep; Lang et al. 2013; Nowell 1976). As a consequence, the composition of mutations carried by the germ cell population will be altered (Elena and Lenski 2003; Greaves and Maley 2012; Long et al. 2015; Orive 2001; Otto and Hastings 1998; Otto and Orive 1995). This process—referred to as cell lineage selection—will lead to some ramets carrying deleterious somatic mutations, while others could possess beneficial mutations.
In addition to cell lineage selection, some proportion of recessive deleterious mutations may be eliminated during the haploid life stage due to pollen tube attrition and pollen competition (Gemetophytic Selection; Armbruster and Rogers 2004; Arunkumar et al. 2013; Cruzan 1989; Harder et al. 2016; Mable and Otto 1998; Mulcahy 1979). Moreover, a portion of deleterious mutations will be homozygous in zygotes, which can lead to higher rates of seed and fruit abortion (Selective Embryo Abortion; Husband and Schemske 1995; Korbecka et al. 2002), thereby increasing the average fitness of surviving offspring (Cruzan and Thomson 1997; Mena-AlÍ and Rocha 2005). The sum effects of these processes of developmental selection, which include cell lineage selection, gametophytic selection, and selective embryo abortion, may filter the set of mutations that enters the next generation. Therefore, understanding the role of somatic mutations for plant biology is critical to fundamental assumptions concerning the frequencies of deleterious and beneficial mutations in populations, and to the processes of adaptation and diversification.
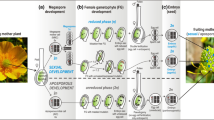
In this study, we make crosses within individual clones of hermaphroditic plants to assess the fitness effects of inherited mutations that accumulated during somatic growth. Although it is often challenging to track the effects of somatic mutations that accumulate within a single generation, plants with separate stems contain distinct germ cell lineages that are derived from the same zygote. As a consequence, each stem can potentially contain different sets of somatic mutations that have originated during growth. By making crosses either within the same flower (autogamy) or between flowers on separate stems of the same plant (inter-ramet geitonogamy—hereafter referred to as geitonogamy), we can produce progeny segregating for somatic mutations unique to each stem (Fig. 1). These crosses are both self-fertilizations, but the offspring of each cross type will differ in the complement of somatic mutations that they inherit. For a diploid plant, we can assume that somatic mutations (a → aʹ) will be in the heterozygous state when they first appear. For progeny generated via autogamy, a somatic mutation will segregate as 25% homozygous (aʹaʹ), 50% heterozygous (aaʹ), and 25% the original (wildtype) homozygote (aa). By contrast, because progeny from geitonogamous crosses will segregate for somatic mutations that are unique to each stem, 50% of offspring will be carrying mutations in the heterozygous state, and none of the progeny will be homozygous for mutations that arose in a single stem. Thus, the average fitness effects of somatic mutations can be evaluated by comparing the difference in fitness of progeny generated by autogamous and geitonogamous crosses (Bobiwash et al. 2013; Schultz and Scofield 2009).

Experimental design to test for the average fitness effects of somatic mutations accumulating in stems of Mimulus guttatus during vegetative growth. A proportion of somatic mutations accumulating during stem growth (dark blue arrows) is made homozygous after within-flower (autogamous) self-pollinations, while all somatic mutations will be heterozygous after between-stem (geitonogamous) self-pollinations. Comparison of the mean fitness of autogamous seedlings (\({\overline{w} }_{k(A)}\)) to geitonogamous seedlings from the same stem (\({\overline{w} }_{k(G)}\)) provides an estimate of the average fitness effects of somatic mutations unique to each stem (δAD(k) = \({\overline{w} }_{k(A)}\) − \({\overline{w} }_{k(G)}\))
The effects of deleterious somatic mutations often are apparent as higher rates of embryo abortion after autogamous compared to geitonogamous pollinations, which is referred to as autogamy depression (Schultz and Scofield 2009; Fig. 1). While autogamy depression for seed and fruit abortion has been observed in several species (reviewed in Bobiwash et al. 2013), no previous study has evaluated the fitness effects of somatic mutations inherited by progeny. In this study, we develop and validate methods that use autogamous and geitonogamous self-pollinations to estimate the fitness effects of somatic mutations segregating in offspring, and then use these methods in two separate experiments to estimate the fitness effects of somatic mutations accumulating during vegetative growth in perennial Mimulus guttatus DC (Erythranthe guttata G.L. Nesom; Phrymaceae).
Methods
Estimating the fitness effects of somatic mutations
As described above, progeny from autogamous self-pollinations will be homozygous for a proportion of somatic mutations, while progeny from geitonogamous crosses will be heterozygous. Consequently, we can estimate the average fitness effects of somatic mutations that are unique to each stem by comparing the average fitness of progeny from autogamous (\({\overline{w} }_{k(A)}\)) and geitonogamous (\({\overline{w} }_{k(G)}\)) crosses to a given stem k as: δAD(k) = \({\overline{w} }_{k(A)}\) − \({\overline{w} }_{k(G)}\). The parameter δAD(k) has been described before in a slightly different format (referred to as Autogamy Depression; Schultz and Scofield 2009; Bobiwash et al. 2013) and summarizes the average magnitude and overall direction of the fitness effects of all expressed somatic mutations that have been transmitted to the next generation. We note that δAD(k) is a quantitative genetic estimate of the sum effect of all somatic mutations that accumulated in a stem (Bobiwash et al. 2013; Schultz and Scofield 2009) and is analogous to estimates of inbreeding depression (Charlesworth and Willis 2009). While standing genetic variation can also lead to fitness effects in offspring, there is no a priori reason to expect these effects to differ in progeny from fruits on different stems of the same plant. Hence, differences in the fitness of progeny from autogamous and geitonogamous pollinations are expected to reflect only the effects of recessive and partially dominant somatic mutations that have accumulated during stem elongation.
Validation of fitness estimates
Given that δAD(k) is based on fitness differences between offspring from autogamous and geitonogamous crosses, it is possible that variation in the dominance of somatic mutations may reduce the reliability of the parameter. In particular, combining somatic mutations from two separate stems in progeny from geitonogamous crosses may reduce fitness and generate estimates of δAD(k) that falsely indicate the presence of beneficial somatic mutations in autogamous progeny, when mutations are actually deleterious (i.e. causing a change in the sign of the estimate). Therefore, to evaluate the reliability of δAD(k), we simulated three different scenarios for variation in the strength of selection and dominance of somatic mutations across multiple stems using randomization functions in Excel (Microsoft Office Professional 2019). First, we assumed that there was only one somatic mutation in each of the two stems, and we calculated expected fitness in the geitonogamous (wG) and autogamous (wA) progeny as;
where s1 and s2 represent the selective effects of mutations, and h1 and h2 represent the dominance coefficients of single somatic mutations in stem 1 and stem 2, respectively. Dominance values were chosen assuming that deleterious somatic mutations with high levels of dominance would be eliminated by cell lineage selection during vegetative growth, and beneficial mutations with high dominance would not display strong fitness differences between autogamous and geitonogamous progeny. For simulations, we randomly chose values of s ranging from -0.2 to 0.2 for 200 pairs of stems. Values of h for s > 0 were allowed to range from 0 to 0.7 for each stem. Because deleterious alleles tend to be recessive (Dudash and Carr 1998; Peters et al. 2003), values of h were constrained to range between 0 and 0.1 for s < 0. To match our experimental design in Mimulus guttatus (see below), we calculated the fitness of autogamous progeny (wA) for one of the stems in each pair. We then calculated wG using information on s and h for both stems in each pair. Then we calculated the difference in fitness between each estimate as a measure of autogamy depression (δAD(k) = wA − wG), compared the estimate of δAD(1) to values of s1, and evaluated the frequency of estimates that were opposite in sign to the actual selective value. This simulation was repeated 20 times (4,000 pairs of stems total) and average values were calculated.
One limitation of the formulation above is that estimates of wG will be out of range when large numbers of mutations are involved (i.e., wG = 1 + 0.5 ∑hi si for a large number of loci segregating for deleterious alleles). To remedy this, we used two sets of fitness calculations for simulations with multiple loci. For the first, we assumed that interactions among somatic mutations would be additive. We estimated the average effect of n loci segregating for deleterious alleles (i.e. ∑hi si /n). If we apply this approach for two loci (one mutation per stem) we obtain,
For a second set of simulations that examined the effects of alleles at multiple loci, we assumed the interactions were multiplicative. In this case, the fitness estimates of geitonogamous and autogamous progeny become,
For both the additive and multiplicative interaction scenarios, we conducted simulations of 200 stems 20 times as described above (a spreadsheet to conduct simulations is available as Appendix 1).
Fitness estimates based on variation among autogamous progeny
To provide additional confirmation of the δAD(k) estimates, we developed an independent approach for estimating the fitness effects of somatic mutations that is based entirely on the variance in mean fitness among autogamous progeny from the same fruit. If somatic mutations affect offspring fitness, variation in fitness should be greater for progeny groups from autogamy than from geitonogamy, as long as mutations do not have complete expression in heterozygotes (i.e. h < 1.0; Appendix 2). This is because somatic mutations will segregate as homozygotes and heterozygotes in autogamous progeny but will remain heterozygous in the progeny of geitonogamous crosses. Therefore, we can estimate the fitness effects of somatic mutations based on the standard deviation in fitness from autogamous crosses (SD), according to the equation: wSD = cSD, where c is the slope of the linear relationship between wSD and SD. For dominance levels of h = 0.0 and 1.0, c = 2.31, and the slope reaches a maximum of 2.83 when h = 0.5 (Appendix 2; Fig. S1). Thus, using only the variation in fitness among progeny from autogamous crosses, we obtain a good approximation of the magnitude of the fitness effects (w) for somatic mutations accumulating in each stem. This approach is independent of δAD(k), which therefore provides an important means of confirming our estimates of the average fitness effects of somatic mutations present in individual stems.
Tests of predictions
From the considerations above, we can identify three predictions for the fitness effects of somatic mutations that occur during stem growth and are passed to offspring. First, on a single stem, the average fitness of progeny deriving from an autogamous cross should be different from the fitness of progeny arising from a geitonogamous cross made using pollen from a different stem. We test for differences in the fitness of progeny from autogamous vs. geitonogamous crosses, and use the mean fitness of each progeny group paired by stem to estimate the sign and magnitude of the average fitness effects of somatic mutations using the formula δAD(k) = \({\overline{w} }_{k(A)}\) − \({\overline{w} }_{k(G)}\) as described above. The second prediction is that somatic mutations unique to individual stems will result in greater variation in mean fitness among autogamous progeny from the same fruit compared to geitonogamous progeny. We test this prediction by comparing the variance among autogamous and geitonogamous progeny groups. Since we expect each stem to possess unique somatic mutations that will be made homozygous in autogamous progeny, there should be greater variation in mean fitness among autogamous progeny from different stems compared to geitonogamous progeny, which will be heterozygous for somatic mutations or homozygous for the non-mutant allele. Consequently, the third prediction is that somatic mutations will result in significant cross type by stem interactions for mean fitness. Below, we test these three hypotheses using two different experiments with Mimulus guttatus.
Study system
Populations of M. guttatus display a wide range of life histories—from annuals to herbaceous perennials that outcross to varying degrees (Wu et al. 2008). We use perennial M. guttatus plants that produce substantial vegetative growth prior to initiation of flowering. These plants are easily propagated from rosettes, and selfing produces substantial numbers of seeds to allow for statistical comparisons. To increase the chances of detecting somatic mutations that impacted fitness, we exposed plants to novel environments. Furthermore, we exposed parental plants and seedlings to the same controlled environments to evaluate the fitness effects of mutations (Baer et al. 2007; Halligan and Keightley 2009; Shaw et al. 2002).
First experiment
We grew plants of M. guttatus in the Research Greenhouse facility at Portland State University from seed collected in July 2013 from three different populations in northern Oregon (Jackson Bottom Wetlands–JB: 45.501794 N, − 122.98776 W; and two from Saddle Mountain–SMB: 45.9861 N, − 123.6859 W, and SMC: 45.9634 N, − 123.6837 W). We assumed that greenhouse conditions were different enough from field environments to provide a novel environment. In August 2013, seeds were cold stratified on moist paper towels at 2 °C for 30 days prior to being sown in soil. Seedlings were transplanted to pots (approximately 10 × 10 × 12 cm) and grown for seven months before the application of pollination treatments. Temperature was maintained between 21 and 26 °C during the day, and 15–21 °C at night. Supplemental HID lights ran for 12 h a day when the seedlings first emerged, and 14 h a day during adult growth.
After plants became established and began producing multiple stems, we conducted autogamous and geitonogamous self-pollinations using flowers on stems 15 to 20 cm in length from two plants from each of four maternal families representing each of the three populations (2 plants × 4 families × 3 populations). Flowers from pairs of stems on individual plants (ramets of the same genet) were reciprocally crossed (geitonogamy), or individual flowers from these same stems were self-pollinated (autogamy). A total of 139 pollinations were conducted across two treatments: limited (pollen was applied to stigmas with one touch from a plastic pipette tip) or excess (where the stigma surface was coated with pollen) in an attempt to manipulate the intensity of gametophytic selection (Cruzan and Barrett 1996). Pollinations were conducted on 12 different days (pollination date) over several weeks in July 2014. Mature fruits were collected and placed individually into paper envelopes, and their contents were examined under a Leica MZ-16 stereoscope. The first 100 ovules from each fruit were categorized as filled seeds (brown, almond-shaped), unfertilized ovules (small, flattened and light-colored), or aborted (larger than unfertilized, dark-colored, shriveled). Ovules that were flattened and appeared to lack endosperm were assumed to be unfertilized or aborted and were not used in germination tests. Seed set and ovule abortion were analyzed using ANOVA models with the GLM procedure of SAS (SAS 2008), with population, maternal plant nested within population, and pollination date as random effects, and cross type (autogamous or geitonogamous) and pollination treatment as fixed effects. Seed set and ovule abortion data were approximately normal so were not transformed prior to analysis.
We assessed the fitness of progeny arising by autogamy and geitonogamy in the same greenhouse environment that was used to grow the parental plants. Seedlings from a subset of ten maternal plants (at least two plants from each of the three populations) that had fruits from both cross types and at least 20 filled seeds were sown in soil and transplanted to 36-cell trays (blocks) in September in a randomized incomplete block design. After 3 months of growth, the progeny were scored for survival, and above ground biomass was measured after drying at 60 °C for at least 24 h. Since all seedlings germinated within a few days of each other, the final biomass is an estimate of growth rate. The fitness of each progeny was estimated as its final biomass, which was log transformed and weighted by the survival frequency of progeny from the same cross. Growth rate is considered to be an appropriate estimate of fitness for perennials (Younginger et al. 2017). Furthermore, we evaluated fitness of seedlings under novel selection regimes in the greenhouse rather than under field conditions, which allowed us to minimize environmental differences between the growth environments of parental plants and seedlings. These estimates were rescaled relative to the maximum value from all crosses, so that \(\overline{w }\) ranged from 0 to 1 across progeny from all crosses.
Second experiment
To control for variation among genets and to further assess the fitness effects of mutations that accumulated during vegetative growth, a single plant (genet BV; obtained from Willamette Gardens native plant nursery, Corvallis, OR) was vegetatively propagated to generate 12 plants (ramets) that were exposed to high salinity and control conditions. This genet was originally propagated from wild-collected seed and retains a high level of genetic diversity (J.A. Schwoch, unpub. data). Plants were grown in pea gravel (4–8 mm) in pots placed in four 53 L tubs using a flood and drain hydroponics system (flooding at 15 min intervals). Two tubs had no added salt and were used as controls, and two tubs had high salinity. The initial salt concentration in the high salinity treatment was 5 mM but increased weekly to 25 mM after plants became established. Salt concentrations were monitored using a conductivity meter to ensure stable concentrations. To provide nutrients, 30 ml of hydroponics fertilizer (FloraGrow, Planet Natural, Bozeman, MT) was added per tub. During the course of the experiment, some plants grew substantially. We imposed selection to favor the fastest growing ramets over the next three months by repeatedly removing single rosettes and transplanting them back into the hydroponics system up to four times.
To promote stress recovery, plants were transplanted to soil for six months, which included a two-month vernalization period in a growth chamber (4 °C and 8 h light; Conviron E8, Controlled Environments Ltd., Winnipeg, Manitoba, Canada). After vernalization, plants were returned to the greenhouse to induce flowering. Autogamous and geitonogamous pollinations were made to pairs of flowers at single nodes or consecutive nodes (seven nodes and 14 pollinations total) on the largest ramets in each of the control and high salt treatments. To account for somatic mutation turnover that may occur due to the effects of cell lineage selection during stem growth, we compared progeny from autogamous and geitonogamous pollinations for a subset of four stems that produced fruits from pairs of flowers from the same node. Without a priori knowledge of the expression of somatic mutations in heterozygotes, it is difficult to determine the best pollen donor for geitonogamous crosses. Consequently, we opted to generate progeny from geitonogamy by pollinating flowers with pollen from a potentially more genetically divergent ramet from the other treatment (i.e. salt pollinated with control pollen, and control pollinated with salt pollen; two stems from each treatment). The fruits were collected, and the total number of aborted and mature seeds and unfertilized ovules were counted under a dissecting microscope. Seeds were planted in soil in trays with three seeds per cell. Seeds were cold stratified in moist soil for three weeks before they germinated in the greenhouse.
To determine whether seedlings from autogamous crosses from ramets exposed to salt stress showed improved performance under the same conditions, all progeny were exposed to high salinity. After germination and establishment in soil, seedlings from autogamous and geitonogamous crosses from control and salt stress ramets were transplanted into pots filled with pea gravel and subjected to high salt in the hydroponics system, as described above. A total of 239 seedlings from 11 fruits (five autogamous and six geitonogamous) were randomly and evenly distributed among 12 hydroponic tubs to ensure equal representation across blocks (tubs). Plant size was measured as the product of the length and width of vegetative spread after two months of growth and was used as a proxy for overall plant performance. Since plants germinated within a few days of each other, plant size represents a good estimate of growth rate. Salt concentration increased from 10 to 37.5 mM over the course of the experiment to induce mortality (~ 57% across all progeny groups). Fitness was estimated as plant size (log transformed to improve normality) weighted by the survival frequency for progeny from the same cross.
Data analyses
Data were analyzed to compare the means and variance in autogamous and geitonogamous progeny fitness to test the three predictions described above. We first test the prediction that somatic mutations would generate greater variation among autogamous compared to geitonogamous progeny grouped by fruit separately in each experiment. We test this prediction using two-way ANOVAs and the stem x cross type (i.e. autogamy vs. geitonogamy) interaction for progeny fitness (Prediction 3 above). We then use data pooled from the two experiments (see below) to test the prediction that the fitness of autogamous and geitonogamous progeny paired by stem will differ (Prediction 1), and that, among progeny within fruits, there will be greater variation in fitness among autogamous compared to geitonogamous progeny (Prediction 2).
Results
Validation of fitness estimates
Our simulations revealed that values of δAD(k) were accurate across a range of values and assumptions for the effects of somatic mutations. For the simulations assuming one mutation per stem, estimates of δAD(k) and s1 were strongly correlated, with R2 values ranging from 0.76 to 0.82 (see Fig. S4 for example results). For simulations assuming additive interactions among multiple somatic mutations, the relationship between δAD(k) and s1 was stronger (R2 ranging from 0.86 to 0.90), and was stronger still when we assumed multiplicative interactions (R2 ranging from 0.93 to 0.95).
Overall, these results indicate that our estimate of δAD(k) is a valid estimate of s; however, it does appear that we are underestimating s (i.e., the slope of the line is less than 1; Fig. S4), making δAD(k) a conservative estimate of the fitness effects of somatic mutations. It is also notable that our simulation assumes a wider range of h values than are generally observed for deleterious mutations. Estimates indicate that the product of hs for deleterious alleles is generally around 0.02, and that the relationship between s and h is generally negative (hyperbolic; Lynch et al. 1999). Minor effects of deleterious somatic mutations are expected, because somatic mutations having strong effects would be eliminated by cell lineage selection. Therefore, these simulations confirm that δAD(k) provides a valid estimate of the mean fitness of somatic mutations occurring within a stem.
Experimental results
In the first experiment, there was no effect of cross type on seed set (Table S5 in Appendix 3), but there was higher abortion of developing seeds after autogamous compared to geitonogamous crosses (Table S6). For ramets from both experiments, there was significant variation in mean fitness among seedlings from fruits generated from autogamous crosses, but not among seedling groups derived from geitonogamy. Consequently, tests of the cross-type by stem interaction were significant for both experiments (Tables S1 and S2 in Appendix 3), which is consistent with the third prediction described above. Since the results from these two experiments were qualitatively similar, we combined the data for all of the following analyses (10 stems from Experiment 1 and four from Experiment 2, for a total of 14 comparisons).
To test the first prediction described above, that inheritance of somatic mutations will have larger effects on autogamous progeny fitness (\({\overline{w} }_{k(A)}\)), we compared them with the fitness of geitonogamous progeny from the same stem (\({\overline{w} }_{k(G)}\); Tables S3 in Appendix 3). As validated by our simulations, the difference in mean fitness for the two cross types provides an estimate of the average fitness effects of somatic mutations unique to each stem. The total fitness effects of all mutations (δAD(k)) occurring in each stem (first experiment) or stem/node combination (second experiment) were calculated as the difference in fitness between progeny from autogamous and geitonogamous crosses, as described above (δAD(k) = \({\overline{w} }_{k(A)}\) − \({\overline{w} }_{k(G)}\)). We observed extensive variation in δAD(k) among stems, with nine stems that were significantly different from zero (Fig. 2). Four of the stems had average fitness effects of somatic mutations that were positive, suggesting a net beneficial effect of somatic mutations transmitted to offspring. In addition, the average fitness of progeny from autogamous and geitonogamous crosses on the same stems or nodes was positively correlated (Fig. S2), which indicates that the effects of somatic mutations from the pollen-donor stem were minimal and do not account for the instances of beneficial mutation estimates found in some stems. Note also that the δAD(k) estimates that deviated the most from zero generally had the highest \({\overline{w} }_{k(A)}\) values (Fig. S3). However, this was not always the case, as one stem (stem 9) with a value of δAD(k) close to zero produced progeny with relatively high fitness after both autogamous and geitonogamous pollinations.

Estimates of δAD(k) for fourteen different stems (ramets) of Mimulus guttatus from two separate experiments (Experiment 1–blue bars; Experiment 2–orange bars) based on mean progeny fitness after autogamous \({\overline{w} }_{k(A)}\) and geitonogamous \({\overline{w} }_{k(G)}\) self-pollinations (δAD(k) = \({\overline{w} }_{k(A)}\) − \({\overline{w} }_{k(G)}\)). Horizontal lines represent standard errors. Asterisks indicate values of δAD(k) that are significantly different from zero based on the t value, calculated as \(t={\delta }_{AD(k)}/\sqrt{SE}\) with n-1 df, where n is the mean of sample sizes for progeny from autogamy and geitonogamy. The relationship between \({\overline{w} }_{k(A)}\) and \({\overline{w} }_{k(G)}\) across stems is shown in Fig. S3. Means and sample sizes for progeny groups are available in Table S2 and Fig. S4 in Appendix 3
To make an independent estimate of the average fitness effects for mutations unique to each stem or node, we used the standard deviation in progeny fitness from autogamous crosses based on the relationship\({w}_{SD}=cSD\), where c = 2.83, which assumes dominance close to h = 0.5 (qualitatively similar results are obtained if we choose other values of c between 2.83 and 2.31, and when h = 0 or 1; Appendix 2). Because the sign of wSD could not be inferred directly from this approach, we used the sign estimated from the δAD(k) method (Fig. 3; Appendix 2). For stems with values of δAD(k) greater than zero, there was a strong positive relationship between δAD(k) and estimates of wSD made from the within-family variation among progeny from autogamy (Fig. 3). In contrast, the relationship for negative values of δAD(k) appeared to be driven largely by a single observation. Note that this observation was not supported by a similarly high value of wSD, and the remaining negative fitness effects were more modest based on both estimates. This one highly negative estimate of δAD(k) may be due to a large influence of mutations from the second stem on the fitness of geitonogamous progeny (note that this stem had the highest estimate of\({\overline{w} }_{k(G)}\); Fig. S2). It is also notable that the variation in fitness for progeny from autogamy did not decline to zero for values of δAD(k) close to zero, which could be due to the presence of both beneficial and detrimental mutations, and possibly genetic background effects (i.e. epistasis), but it may also reflect uncontrolled environmental variation. Overall, the results from both approaches reveal consistent estimates of the average fitness consequences associated with the accumulation of somatic mutations in individual stems.

Relationships between estimates of fitness effects of somatic mutations in Mimulus guttatus, based either on the difference in fitness of progeny from autogamy and geitonogamy (δAD(k)), or the standard deviation in fitness within progeny groups from autogamy for each stem (wSD). Estimates of wSD corresponding to negative values of δAD(k) were transformed to negative values. Estimates from Experiment 1 are indicated by blue circles and from Experiment 2 are orange squares. Dashed lines indicate the separate relationships for positive and negative values of fitness estimates
Discussion
In this study, we have provided evidence that somatic mutations accumulating during vegetative growth can affect the fitness of progeny in the next generation. Consistent with previous work (Bobiwash et al. 2013), we observed significant autogamy depression in the form of greater ovule abortion in autogamous relative to geitonogamous crosses. However, we also detected more variation in survival and growth rate for progeny from autogamous compared to geitonogamous self-pollinations, which provides evidence that somatic mutations that accumulated during vegetative growth can have demonstrable effects on the fitness of plants in the next generation. Both the differences in the mean fitness between progeny from autogamy and geitonogamy (δAD(k)) and variation in fitness of autogamous progeny (wSD) provided consistent estimates of the average fitness effects of somatic mutations segregating in progeny. We found evidence for the effects of beneficial mutations in progeny from some stems, with estimates of δAD(k) and wSD exceeding 0.1 in four cases, while estimates for negative fitness effects were more modest (mostly > − 0.15). These results imply that, in these plants, many deleterious mutations can be culled by the various types of developmental selection prior to seed dispersal.
Somatic mutations accumulating during vegetative growth had an overall positive effect for four of the stems tested. This result appears to contrast with widely held views that the appearance of beneficial somatic mutations should be exceedingly rare (Charlesworth and Willis 2009; Crow 1993). However, this finding is consistent with expected outcomes of clonal evolution occurring during vegetative growth. We expect that most somatic mutations in germ cells in the central zone of the apical meristem will be neutral and occur at low frequencies, but some could contribute to higher rates of division for some cell lineages over others (Otto and Hastings 1998; Otto and Orive 1995). This hypothesis is supported by the observation that the majority of somatic mutations tend to occur at low frequencies in stem tissue, and a minority occur at high frequencies (Yu et al. 2020; Schwoch et al. unpublished data), suggesting that clonal selective sweeps due to the appearance of beneficial mutations have occurred during stem elongation. While the stem cell population is small (e.g., ~ 35 in Arabidopsis; Reddy and Meyerowitz 2005), selection on somatic mutations was apparently strong enough to overcome genetic drift, resulting in higher fitness of autogamous progeny from some stems. Thus, it appears that cell lineage selection has the potential to retain beneficial somatic mutations, as individual cell lineages divide at faster rates resulting in clonal evolution during vegetative growth.
Although there may be few opportunities for beneficial changes to alter basic cellular metabolism, it is becoming apparent from experimental evolution studies with microbes that even basic aspects of cellular metabolism can be sensitive to environmental conditions, which can increase the chances that mutations in clonal populations are beneficial (Böndel et al. 2019; e.g., Lang et al. 2013; Lee and Marx 2013; Maharjan et al. 2015). In the experiments described above, we exposed plants to novel environments either in the greenhouse (experiment 1) or in high salinity in hydroponics (experiment 2). Moreover, genomic evidence suggests that clonal selective sweeps, which may be indicative of the spread of beneficial somatic mutations in the meristem tissue, are more common for stems that have recently been exposed to high salinity (J. A. Schwoch, unpublished data). In this regard, cell lineage selection in a plant meristem represents a potentially powerful forum for the removal of deleterious somatic mutations while favoring the retention of beneficial ones. In addition, gametophytic selection and selective embryo abortion can act as prominent additional filters (Armbruster and Rogers 2004; Arunkumar et al. 2013; Cruzan 1989; Harder et al. 2016; Mable and Otto 1998; Mulcahy 1979), but they are most likely to have effects by culling deleterious mutations. Regardless, the combined effects of these different forms of developmental selection appear to have had a considerable effect on filtering of somatic mutations under controlled conditions in the greenhouse, such that the distribution of fitness effects among stems has shifted to include more beneficial mutations than expected. Future studies should test whether similar findings are found under field conditions, which could indicate a prominent role for somatic mutation in local adaptation.
An alternative explanation for the observed fitness differences is that exposure to environmental stress has induced heritable epigenetic modifications (Quadrana and Colot 2016). However, phenotypic responses due to epigenetic modifications are expected to be consistent and predictable, as they are hypothesized to represent an adaptive response to historic exposure to similar stressors (Baulcombe and Dean 2014; Itabashi et al. 2018). The results of the experiments described here are unlikely to support a role for epigenetics, because fitness responses in the next generation were inconsistent in direction and magnitude, and they were not predictable based on environmental exposure of the parent stem. Moreover, there is no a priori reason to expect that epigenetic modifications would differ between fruits from autogamous and geitonogamos crosses developing on the same stem. We observed that the mean fitness of progeny from autogamy displayed both increases and decreases compared to the geitonogamy controls, which is consistent with the hypothesis that individual ramets are accumulating unique complements of somatic mutations. Furthermore, these conclusions are supported by the observation of numerous somatic variants in the transcriptomes of meristem tissue from multiple ramets derived from a single genet (Yu et al. 2020; Schwoch et al. unpublished data). Thus, the results of the current study indicate that somatic mutations accumulating during stem growth are most likely to be responsible for the observed fitness effects.
The potential for the acquisition of mutations during vegetative growth is a well-known aspect of plant biology (Bobiwash et al. 2013; Klekowski 2003; Schultz and Scofield 2009), but no previous study has demonstrated the effects of somatic mutations on the fitness of progeny in the next generation. Moreover, most studies have focused on the detrimental effects of somatic mutations; chloroplast mutants have been observed in a number of species (Klekowski 2003), declines in pollen fertility were found in older clones of quaking aspen (Ally et al. 2010), and higher rates of seed and fruit abortion after autogamous pollinations were found in several studies (reviewed in Bobiwash et al. 2013). In contrast, agriculturalists have taken advantage of beneficial somatic mutations to improve economically important plants (Aradhya et al. 2003; Jarni et al. 2015; McKey et al. 2010; Miller and Gross 2011; Pelsy et al. 2015; Vezzulli et al. 2012), and a handful of studies report phenotypic responses to selection in asexual lineages. For example, Breese et al. (1965) succeeded in selecting for increased tillering ability (production of new grass stems) within genets of perennial ryegrass (Lolium perenne). Similarly, artificial selection on clonal lineages effectively improved branching in the red seaweed, Asparagopsis armata (Monro and Poore 2009). The current study on M. guttatus contributes to this literature by highlighting the potential for plants to exhibit significant levels of clonal variation within a single generation.
The transmission of beneficial mutations in autogamous crosses may explain some heretofore difficult to understand results from mutation accumulation studies. Our results suggest that beneficial somatic mutations are likely to be partially dominant (i.e. not completely recessive), because they would have to be expressed in the heterozygous state to be favored by cell lineage selection. While somatic mutations may become homozygous through mitotic recombination, this appears to be rare in non-cancerous somatic growth (LaRocque et al. 2011). Mutations accumulating during vegetative growth could contribute to standing genetic variation, but autogamy may be more effective for the accumulation of beneficial somatic mutations in populations than geitonogamy or outcrossing, because beneficial mutations could be made homozygous in progeny in a single generation. Depending on the crossing design, outcrossing would take at least two generations for beneficial somatic mutations to be made homozygous, and under geitonogamy they would remain heterozygous. It is striking that high rates of beneficial mutation accumulation have been observed in at least some mutation accumulation studies in the autogamous plant Arabidopsis thaliana (Rutter et al. 2012, 2018, 2010; Shaw et al. 2002), but not in outcrossing and partially-selfing species of Amsinckia (Schoen 2005). With a few exceptions (e.g., Baer et al. 2005; Denver et al. 2010), nearly all mutation accumulation studies on animals consistently show a prevalence of deleterious mutations (Baer et al. 2007; Halligan and Keightley 2009; but see Bao et al. 2022). Our results suggest that the adaptive potential of autogamous plants may be greater than previously thought, which may help explain the wider geographic ranges of selfing compared to closely-related outcrossing species (Grant and Kalisz 2020; Grossenbacher et al. 2015). Although developmental selection has the potential to contribute to adaptation in all plants, its effects may be enhanced in autogamous lineages, because beneficial mutations arising during vegetative growth have a greater chance of becoming homozygous in offspring and being retained across generations.
As stems grow, mutations can be generated during every mitotic cell division, so the potential for somatic mutation accumulation in plants appears substantial. Thus, understanding how long-lived plants, such as trees, avoid mutational meltdown from the accumulation of deleterious somatic mutations remains a longstanding question. Paradoxically, however, the rate of mutation accumulation observed across generations in plant and animal genomes is similar (Gaut et al. 2011). One hypothesis for this pattern is that, similar to animal germlines, a subset of cell lines in the apical meristem undergoes fewer mitotic divisions, which would protect lineages from the negative effects of mutation accumulation during development of the soma (Plant Germline Hypothesis; Burian et al. 2016; Cruzan 2018; page 90). An alternative hypothesis posits that somatic mutations are generated in apical meristems during plant growth, but these mutations are filtered by developmental selection prior to the establishment of offspring (Somatic Mutation Accumulation Hypothesis; Cruzan 2018, p. 86). Plants have retained the capacity to undergo clonal evolution from their algal ancestors, and thus developmental selection during growth and reproduction has the potential to skew the distribution of fitness effects of transmitted mutations to include a larger proportion of beneficial variants than would be expected through random processes. In addition, this provides a reasonable explanation for why longer-lived plants appear to have slower rates of mutation accumulation across generations (Gaut et al. 2011; Yue et al. 2010). This could be due to the fact that longer generation time leads to more time between recombination events, which can lead to more background selection in non-recombining cell lineages during vegetative growth (Cruzan 2018, pp. 94–95). Recent studies in oaks, spruce, and eel grass (Hanlon et al. 2019; Plomion et al. 2018; Yu et al. 2020), as well as our unpublished data from M. guttatus, confirm that multiple somatic variants are likely, even in plants of very different size. Future work that combines information from experiments evaluating the genomic consequences of somatic variation with anatomical estimates of stem cell population dynamics will allow for the development of new models that provide insights into the extent and limitations of somatic evolution in plants.
In conclusion, despite the potential for somatic mutation accumulation to generate novel genetic variation in plant populations, its role in evolution remains almost entirely unexplored. Our estimates of the fitness effects of somatic mutations were consistent across two different methods and indicate that some stems accumulated primarily deleterious mutations while others produced autogamous progeny with high fitness, which likely indicates the presence of beneficial mutations. Even though high levels of mutation accumulation are often believed to be detrimental, the basic biology of plants suggests that the role of somatic mutations in plant evolution should be considered carefully in the future. Future lines of investigation will improve our understanding of these fundamental aspects of plant development and evolution that may have contributed to the remarkable diversification of plants, and may help to account for some of the variation in mutation rates detected among lineages.
Data archiving
Data and all scripts for data analysis will be submitted to DataDryad upon publication.
Data availability
Data and all scripts for data analysis will be submitted to DataDryad upon publication.
Code availability
Included as Appendix 1.
References
Ally D, Ritland K, Otto SP (2010) Aging in a long-lived clonal tree. PLoS Biol. https://doi.org/10.1371/journal.pbio.1000454
Antolin MF, Strobeck C (1985) The population genetics of somatic mutation in plants. Am Nat 126(1):52–62. https://doi.org/10.1086/284395
Aradhya MK, Dangl GS, Prins BH, Boursiquot JM, Walker MA, Meredith CP, Simon CJ (2003) Genetic structure and differentiation in cultivated grape Vitis vinifera L. Genet Res 81(3):179–192. https://doi.org/10.1017/s0016672303006177
Armbruster WS, Rogers DG (2004) Does pollen competition reduce the cost of inbreeding? Am J Bot 91(11):1939–1943. https://doi.org/10.3732/ajb.91.11.1939
Arunkumar R, Josephs EB, Williamson RJ, Wright SI (2013) Pollen-specific, but not sperm-specific, genes show stronger purifying selection and higher rates of positive selection than sporophytic genes in Capsella grandiflora. Mol Biol Evol 30(11):2475–2486. https://doi.org/10.1093/molbev/mst149
Baer CF, Shaw F, Steding C, Baumgartner M, Hawkins A, Houppert A et al (2005) Comparative evolutionary genetics of spontaneous mutations affecting fitness in rhabditid nematodes. Proc Natl Acad Sci USA 102(16):5785–5790. https://doi.org/10.1073/pnas.0406056102
Baer CF, Miyamoto MM, Denver DR (2007) Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat Rev Genet 8(8):619–631
Bao K, Melde RH, Sharp NP (2022) Are mutations usually deleterious? A perspective on the fitness effects of mutation accumulation. Evolut Ecol. https://doi.org/10.1007/s10682-022-10187-4
Barfield S, Aglyamova GV, Matz MV (2016) Evolutionary origins of germline segregation in Metazoa: evidence for a germ stem cell lineage in the coral Orbicella faveolata (Cnidaria, Anthozoa). Proc R Soc B. https://doi.org/10.1098/rspb.2015.2128
Baulcombe DC, Dean C (2014) Epigenetic regulation in plant responses to the environment. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a019471
Bobiwash K, Schultz ST, Schoen DJ (2013) Somatic deleterious mutation rate in a woody plant: estimation from phenotypic data. Heredity 111(4):338–344. https://doi.org/10.1038/hdy.2013.57
Böndel KB, Kraemer SA, Samuels T, McClean D, Lachapelle J, Ness RW et al (2019) Inferring the distribution of fitness effects of spontaneous mutations in Chlamydomonas reinhardtii. PLoS Biol. https://doi.org/10.1371/journal.pbio.3000192
Breese EL, Hayward MD, Thomas AC (1965) Somatic selection in perennial ryegrass. Heredity 20:367. https://doi.org/10.1038/hdy.1965.50
Buchholz JT (1922) Developmental selection in vascular plants. Bot Gaz 73(4):249–286
Burian A, de Reuille PB, Kuhlemeier C (2016) Patterns of stem cell divisions contribute to plant longevity. Curr Biol 26(11):1385–1394. https://doi.org/10.1016/j.cub.2016.03.067
Buss LW (1983) Evolution, development, and the units of selection. Proc Natl Acad Sci USA-Biol Sci 80(5):1387–1391. https://doi.org/10.1073/pnas.80.5.1387
Charlesworth D, Willis JH (2009) The genetics of inbreeding depression. Nat Rev Genet https://doi.org/10.1038/nrg2664
Crow JF (1993) Mutation, mean fitness, and genetic load. In: Oxford surveys in evolutionary biology, vol 9, pp 3–42, Oxford University Press
Cruzan MB (1989) Pollen tube attrition in Erythronium grandiflorum. Am J Bot 76:562–570
Cruzan MB (2018) Evolutionary biology: a plant perspective. Oxford University Press, New York
Cruzan MB, Barrett SCH (1996) Post-pollination mechanisms influencing mating patterns and fecundity: an example from Eichhornia paniculata. Am Nat 147:576–598
Cruzan MB, Thomson J (1997) Effects of pre-dispersal selection on offspring growth and survival in Erythronium grandiflorum. J Evol Biol 10:295–314
Denver DR, Howe DK, Wilhelm LJ, Palmer CA, Anderson JL, Stein KC et al (2010) Selective sweeps and parallel mutation in the adaptive recovery from deleterious mutation in Caenorhabditis elegans. Genome Res 20(12):1663–1671. https://doi.org/10.1101/gr.108191.110
Dubrovina AS, Kiselev KV (2016) Age-associated alterations in the somatic mutation and DNA methylation levels in plants. Plant Biol 18(2):185–196. https://doi.org/10.1111/plb.12375
Dudash MR, Carr DE (1998) Genetics underlying inbreeding depression in Mimulus with contrasting mating systems. Nature 393(6686):682–684. https://doi.org/10.1038/31468
Elena SF, Lenski RE (2003) Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat Rev Genet 4(6):457–469
Gaut B, Yang L, Takuno S, Eguiarte LE (2011) The patterns and causes of variation in plant nucleotide substitution rates. Ann Rev Ecol Evolut Syst 42:245–266
Grant A-G, Kalisz S (2020) Do selfing species have greater niche breadth? Support from ecological niche modeling. Evolution 74(1):73–88. https://doi.org/10.1111/evo.13870
Greaves M, Maley CC (2012) Clonal evolution in cancer. Nature 481(7381):306–313
Grossenbacher D, Briscoe Runquist R, Goldberg EE, Brandvain Y (2015) Geographic range size is predicted by plant mating system. Ecol Lett 18(7):706–713. https://doi.org/10.1111/ele.12449
Halligan DL, Keightley PD (2009) Spontaneous mutation accumulation studies in evolutionary genetics. Ann Rev Ecol Evolut Syst 40:151–172
Hanlon VCT, Otto SP, Aitken SN (2019) Somatic mutations substantially increase the per-generation mutation rate in the conifer Picea sitchensis. Evol Lett 3:348–358. https://doi.org/10.1002/evl3.121
Harder LD, Aizen MA, Richards SA (2016) The population ecology of male gametophytes: the link between pollination and seed production. Ecol Lett 19(5):497–509. https://doi.org/10.1111/ele.12596
Husband B, Schemske DW (1995) Evolution of the magnitude and timing of inbreeding depression in plants. Evolution 50:54–70
Itabashi E, Osabe K, Fujimoto R, Kakizaki T (2018) Epigenetic regulation of agronomical traits in Brassicaceae. Plant Cell Rep 37(1):87–101. https://doi.org/10.1007/s00299-017-2223-z
Jarni K, Jakse J, Brus R (2015) Vegetative propagation: linear barriers and somatic mutation affect the genetic structure of a Prunus avium L. stand. Forestry 88(5):612–621. https://doi.org/10.1093/forestry/cpv029
Klekowski EJ (2003) Plant clonality, mutation, diplontic selection and mutational meltdown. Biol J Lin Soc 79(1):61–67. https://doi.org/10.1046/j.1095-8312.2003.00183.x
Klekowski EJ, Godfrey PJ (1989) Ageing and mutation in plants. Nature 340(6232):389–391. https://doi.org/10.1038/340389a0
Korbecka G, Klinkhamer PGL, Vrieling K (2002) Selective embryo abortion hypothesis revisited: a molecular approach. Plant Biol 4(3):298–310. https://doi.org/10.1055/s-2002-32331
Kuhlemeier C (2017) How to get old without aging. Nat Plants 3(12):916–917. https://doi.org/10.1038/s41477-017-0076-7
Lang GI, Rice DP, Hickman MJ, Sodergren E, Weinstock GM, Botstein D, Desai MM (2013) Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature 500(7464):571–574. https://doi.org/10.1038/nature12344
Langridge J (1958) A hypothesis of developmental selection exemplified by lethal and semi-lethal mutants of Arabidopsis. Aust J Biol Sci 11(1):58–68. https://doi.org/10.1071/BI9580058
LaRocque JR, Stark JM, Oh J, Bojilova E, Yusa K, Horie K et al (2011) Interhomolog recombination and loss of heterozygosity in wild-type and Bloom syndrome helicase (BLM)-deficient mammalian cells. Proc Natl Acad Sci 108(29):11971–11976. https://doi.org/10.1073/pnas.1104421108
Lee M-C, Marx CJ (2013) Synchronous waves of failed soft sweeps in the laboratory: remarkably rampant clonal Interference of alleles at a single locus. Genetics 193(3):943. https://doi.org/10.1534/genetics.112.148502
Long A, Liti G, Luptak A, Tenaillon O (2015) Elucidating the molecular architecture of adaptation via evolve and resequence experiments. Nat Rev Genet 16(10):567–582. https://doi.org/10.1038/nrg3937
Lynch M, Blanchard J, Houle D, Kibota T, Schultz S, Vassilieva L, Willis J (1999) Perspective: spontaneous deleterious mutation. Evolution 53(3):645–663. https://doi.org/10.2307/2640707
Mable BK, Otto SP (1998) The evolution of life cycles with haploid and diploid phases. BioEssays 20(6):453–462. https://doi.org/10.1002/(sici)1521-1878(199806)20:6%3c453::aid-bies3%3e3.0.co;2-n
Maharjan RP, Liu B, Feng L, Ferenci T, Wang L (2015) Simple phenotypic sweeps hide complex genetic changes in populations. Genome Biol Evol 7(2):531–544. https://doi.org/10.1093/gbe/evv004
McKey D, Elias M, Pujol B, Duputie A (2010) The evolutionary ecology of clonally propagated domesticated plants. New Phytol 186(2):318–332. https://doi.org/10.1111/j.1469-8137.2010.03210.x
McKnight TD, Riha K, Shippen DE (2002) Telomeres, telomerase, and stability of the plant genome. Plant Mol Biol 48(4):331–337. https://doi.org/10.1023/a:1014091032750
Mena-AlÍ JI, Rocha OJ (2005) Selective seed abortion affects the performance of the offspring in Bauhinia ungulata. Ann Bot 95(6):1017–1023. https://doi.org/10.1093/aob/mci109
Miller AJ, Gross BL (2011) From forest to field: perennial fruit crop domestication. Am J Bot 98(9):1389–1414. https://doi.org/10.3732/ajb.1000522
Monro K, Poore AGB (2009) The potential for evolutionary responses to cell-lineage selection on growth form and its plasticity in a red seaweed. Am Nat 173(2):151–163. https://doi.org/10.1086/595758
Monroe JG, Srikant T, Carbonell-Bejerano P, Becker C, Lensink M, Exposito-Alonso M et al (2022) Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 602(7895):101–105. https://doi.org/10.1038/s41586-021-04269-6
Mulcahy DL (1979) The rise of the angiosperms: a genecological factor. Science 206:20–23
Nowell PC (1976) Clonal evolution of tumor-cell populations. Science 194(4260):23–28. https://doi.org/10.1126/science.959840
Orive ME (2001) Somatic mutations in organisms with complex life histories. Theor Popul Biol 59(3):235–249. https://doi.org/10.1006/tpbi.2001.1515
Otto SP, Hastings IM (1998) Mutation and selection within the individual. Genetica 102–103:507–524. https://doi.org/10.1023/a:1017074823337
Otto SP, Orive ME (1995) Evolutionary consequences of mutation and selection within an individual. Genetics 141(3):1173–1187
Pelsy F, Dumas V, Bevilacqua L, Hocquigny S, Merdinoglu D (2015) Chromosome replacement and deletion lead to clonal polymorphism of berry color in grapevine. PLoS Genet 11(4):12. https://doi.org/10.1371/journal.pgen.1005081
Peters AD, Halligan DL, Whitlock MC, Keightley PD (2003) Dominance and overdominance of mildly deleterious induced mutations for fitness traits in Caenorhabditis elegans. Genetics 165(2):589–599. https://doi.org/10.1093/genetics/165.2.589
Plomion C, Aury J-M, Amselem J, Leroy T, Murat F, Duplessis S et al (2018) Oak genome reveals facets of long lifespan. Nature Plants 4(7):440–452. https://doi.org/10.1038/s41477-018-0172-3
Quadrana L, Colot V (2016) Plant transgenerational epigenetics. Ann Rev Genet 50:467–491
Reddy GV, Meyerowitz EM (2005) Stem-cell homeostasis and growth dynamics can be uncoupled in the Arabidopsis shoot apex. Science 310(5748):663–667. https://doi.org/10.1126/science.1116261
Reusch TBH, Bostrom C (2011) Widespread genetic mosaicism in the marine angiosperm Zostera marina is correlated with clonal reproduction. Evol Ecol 25(4):899–913. https://doi.org/10.1007/s10682-010-9436-8
Rutter MT, Shaw FH, Fenster CB (2010) Spontaneous mutation parameters for Arabidopsis thaliana measured in the wild. Evolution 64(6):1825–1835. https://doi.org/10.1111/j.1558-5646.2009.00928.x
Rutter MT, Roles A, Conner JK, Shaw RG, Shaw FH, Schneeberger K et al (2012) Fitness of Arabidopsis thaliana mutation accumulation lines whose spontaneous mutations are known. Evolution 66(7):2335–2339. https://doi.org/10.1111/j.1558-5646.2012.01583.x
Rutter MT, Roles AJ, Fenster CB (2018) Quantifying natural seasonal variation in mutation parameters with mutation accumulation lines. Ecol Evol 8(11):5575–5585. https://doi.org/10.1002/ece3.4085
SAS (2008) SAS/STAT 9.2 user's guide. Cary, North Carolina, SAS Institute Inc.
Schmid-Siegert E, Sarkar N, Iseli C, Calderon S, Gouhier-Darimont C, Chrast J et al (2017) Low number of fixed somatic mutations in a long-lived oak tree. Nat Plants 3(12):926–929. https://doi.org/10.1038/s41477-017-0066-9
Schoen DJ (2005) Deleterious mutation in related species of the plant genus Amsinckia with contrasting mating systems. Evolution 59(11):2370–2377
Schultz ST, Scofield DG (2009) Mutation accumulation in real branches: fitness assays for genomic deleterious mutation rate and effect in large-statured plants. Am Nat 174(2):163–175. https://doi.org/10.1086/600100
Schweinsberg M, González Pech RA, Tollrian R, Lampert KP (2014) Transfer of intracolonial genetic variability through gametes in Acropora hyacinthus corals. Coral Reefs 33(1):77–87. https://doi.org/10.1007/s00338-013-1102-5
Shaw FH, Geyer CJ, Shaw RG (2002) A comprehensive model of mutations affecting fitness and inferences for Arabidopsis thaliana. Evolution 56(3):453–463
Vezzulli S, Leonardelli L, Malossini U, Stefanini M, Velasco R, Moser C (2012) Pinot blanc and Pinot gris arose as independent somatic mutations of Pinot noir. J Exp Bot 63(18):6359–6369. https://doi.org/10.1093/jxb/ers290
Watson JM, Platzer A, Kazda A, Akimcheva S, Valuchova S, Nizhynska V et al (2016) Germline replications and somatic mutation accumulation are independent of vegetative life span in Arabidopsis. Proc Natl Acad Sci USA 113(43):12226–12231. https://doi.org/10.1073/pnas.1609686113
Williams JH, Friedman WE, Arnold ML (1999) Developmental selection within the angiosperm style: using gamete DNA to visualize interspecific pollen competition. Proc Natl Acad Sci 96(16):9201–9206. https://doi.org/10.1073/pnas.96.16.9201
Wu CA, Lowry DB, Cooley AM, Wright KM, Lee YW, Willis JH (2008) Mimulus is an emerging model system for the integration of ecological and genomic studies. Heredity 100(2):220–230. https://doi.org/10.1038/sj.hdy.6801018
Younginger BS, Sirová D, Cruzan MB, Ballhorn DJ (2017) Is biomass a reliable estimate of plant fitness? Appl Plant Sci 5(2):10. https://doi.org/10.3732/apps.1600094
Yu L, Boström C, Franzenburg S, Bayer T, Dagan T, Reusch TBH (2020) Somatic genetic drift and multilevel selection in a clonal seagrass. Nat Ecol Evolut. https://doi.org/10.1038/s41559-020-1196-4
Yue J-X, Li J, Wang D, Araki H, Tian D, Yang S (2010) Genome-wide investigation reveals high evolutionary rates in annual model plants. BMC Plant Biol 10(1):1–12. https://doi.org/10.1186/1471-2229-10-242
Acknowledgements
We thank J. Thompson, E. Perez, and our research greenhouse manager Linda Taylor for plant maintenance and assistance with this research in the greenhouse and in the lab. Several people made helpful suggestions on this manuscript including N. Diaz, C.B. Fenster, M. Grasty, J. Persinger, and an anonymous reviewer. This research was supported by an NIH NIGMS BUILD EXITO grant (5TL4GM118965-03, 5UL1GM118964-03, and 5RL5GM118963-03) to Portland State University, and by NSF-DEB awards 2051235 to MBC and 2051242 to MAS.
Funding
This research was supported by an NIH NIGMS BUILD EXITO grant (5TL4GM118965-03, 5UL1GM118964-03, and 5RL5GM118963-03) to Portland State University, and by NSF-DEB awards 2051235 to MBC and 2051242 to MAS.
Author information
Authors and Affiliations
Contributions
JAS conducted the greenhouse experiments, processed samples, and assisted with data analysis and wrote a portion of the manuscript. MAS assisted with data analysis and data presentation, and wrote a portion of the manuscript. MBC conducted data analyses and simulations and wrote the majority of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest.
Consent for publication
All authors have read the manuscript and agree with its contents.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cruzan, M.B., Streisfeld, M.A. & Schwoch, J.A. Fitness effects of somatic mutations accumulating during vegetative growth. Evol Ecol 36, 767–785 (2022). https://doi.org/10.1007/s10682-022-10188-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10682-022-10188-3