
Abstract
The genus Aegilops L. has been intensively investigated due to its close relationship with wheat (Triticum L.) as contributor of B and D subgenomes. Because of their vast genetic diversity, Aegilops species represent a rich source of alleles of agronomic interest, which could be used to widen the wheat gene pool and improve tolerance to diseases, pests, drought, cold and other environmental stresses. We report the isolation and characterization of the Dehydration Responsive Factor 1 (DRF1) gene in three accessions of Ae. speltoides coming from different regions of the Fertile Crescent. The DRF1 gene belongs to the DREB gene family and encodes transcription factors which play a key role in plant response to water stress. As in other cereals, the DRF1 gene in Aegilops speltoides consists of four exons and three introns and undergoes alternative splicing. A processed pseudogene was also identified and compared with the sequence of an actual mRNA transcript, breaking new ground in the understanding of the complex regulation mechanism of this gene. The genetic diversity was evaluated by comparison of inter- and intra-species variation among some Aegilops and Triticeae, by considering both the whole gene and exon 4 sequences. The phylogenetic analyses were able to cluster the sequences in well-supported clades attributable to the genomes analysed. The overall results suggest that there is a high similarity between the B and S genome copies of the DRF1 gene but also features indicating that the two genomes have evolved independently.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genus Aegilops L. has been intensively investigated due to the close relationship with wheat (Triticum L.), of which it forms the largest part of the so-called secondary gene pool (GP-2) (Harlan and de Wet 1971). Following the commonly accepted system suggested by Van Slageren (1994), the genus comprises 22 species arranged in five sections (Aegilops, Comopyrum, Cylindropyrum, Sitopsis and Vertebrata) and includes diploid, tetraploid and hexaploid genomes. The Sitopsis section is particularly interesting because its five species (Ae.bicornis-SbSb, Ae. longissima-SlSl, Ae. searsii-SsSs, Ae. sharonensis-SshSsh and Ae. speltoides-SS) share the S genome found to be closely similar to the B genome in T. durum (Kerby and Kuspira 1987; Haider 2013; Ruban and Badaeva 2018), whereas Ae. tauschii (Vertebrata section) has been involved in the evolution of the wheat D genome (Marcussen et al. 2014; Pont et al. 2019). The Aegilops genus thus represents a natural laboratory for the study of the effects of the evolution and domestication of cultivated wheat.
Aegilops species are naturally distributed in an area ranging from the Mediterranean to Western and Central Asia. From the Transcaucasus, thought to be their centre of origin, diploid species migrated west and east, the centre of diversity being in the Fertile Crescent (modern day Iraq and parts of Turkey, Syria and Iran), which contains the majority of Aegilops species (Van Slageren 1994). Such a wide spread implies a great adaptability to different environments and a strong resistance to biotic and abiotic stresses. Indeed, many Aegilops species have a strong resistance to disease and insect pests, high drought and salt tolerance and/or good grain quality, thus representing a significant reservoir of new alleles with valuable traits for broadening the genetic diversity of wheat (Schneider et al. 2008; Brozynska et al. 2016; Zhang et al. 2017; Kumar et al. 2019). A number of studies have analysed and compared the nucleotide diversity of a single gene inside the Triticeae tribe; for instance Adderley and Sun (2013) analysed the nuclear phosphoglycerate kinase (pgk1) gene, Liu et al. (2016) studied the Wcor15 gene and Salse et al. (2008) investigated the storage protein activator (SPA) genomic region. In this work, we have analysed the Dehydration Responsive Factor 1 (DRF1) gene in some Aegilops species, with a focus on Aegilops speltoides. DRF1 belongs to the Dehydration Responsive Element Binding (DREB2) gene family and encodes important transcription factors (TFs) involved in plant response to drought. It is characterized by the presence of the APETALA2 (AP2)/Ethylene-Responsive element binding Factor (ERF) motif, through which the TFs bind to the drought-responsive element (DRE)/C repeat (CRT) element in the promoters of several stress responsive genes, thus modulating their expression (Agarwal et al. 2006). DREB2 genes were first identified in Arabidopsis thaliana by Yamaguchi-Shinozaki and Shinozaki (1994), and later on in various cereals, such as Hordeum vulgare (HvDRF1) (Xue and Loveridge 2004), Triticum aestivum (Wdreb2) (Egawa et al. 2006) and Triticum durum (TdDRF1) (Latini et al. 2007). In these cereals, the gene is structurally organized in four exons and three introns, and produces three transcripts through the alternative splicing mechanism (AS). Two of the three transcripts encode putative TF proteins endowed with the AP2 domain, while the other, the DRF1.2 transcript (exon 1, exon 2 and exon 4), encodes an abortive protein, due to the presence of a premature termination codon (PTC), caused by a frameshift mutation.
Here, the whole DRF1 gene was amplified, cloned and sequenced from three accessions of Aegilops speltoides, its nucleotide diversity was analysed and compared with the B subgenome copy of the DRF1 gene in T. durum (Cantale et al. 2018). Furthermore, the nucleotide diversity of the DRF1 exon 4, containing the AP2 domain, was analysed in several accessions of Aegilops speltoides and other diploid and polyploid Aegilops species and compared with available corresponding Triticeae sequences to investigate the reciprocal relationships.
Materials and methods
Plant material
The Aegilops accessions used in the present study are listed in Table 1. Seeds were provided by ICARDA (International Center for Agricultural Research in the Dry Areas), except one accession provided by the Leibniz Institute of Plant Genetics and Crop Plant Research (IPK, DE).
Ten seeds from each accession were germinated in Petri dishes; the plantlets were then transferred into small pots and grown in a greenhouse under controlled standard light and temperature conditions. After 25–30 days, there were sufficient plant leaves (about 100 mg) for harvesting for the molecular analyses.
DNA/RNA extraction and PCR amplification
Genomic DNA was extracted from the leaves using the CTAB method (Murray and Thompson 1980), including a treatment with RNase I to avoid RNA contamination. Total RNA was extracted from the leaves of plants (Ae. speltoides ssp. speltoides, accession IG 47280) grown under normal conditions, using the RNAfast-II Isolation System (Molecular Systems, San Diego, CA, USA). The quality and integrity of the DNA/RNA extracts were checked using agarose gel electrophoresis and their final concentration was measured using a spectrophotometer (OD 260, 280, 260/280). Primer design was based on TdDRF1 gene sequence (NCBI ID: EU089819). The whole gene sequence was isolated by amplifying two gene fragments in optimized specific PCR reactions. The first amplicon was a ~ 2.5 Kb gene fragment obtained using the pair of primers drebfor1 (5′-CATGACGGTAGATCGGAA-3′) and drebrev1 (5′-TGCGCAGGGAAGTTGGTA-3′). This ~ 2.5 Kb gene fragment mapped exon 1, including the ATG start codon, up to part of exon 4 (experimental conditions: annealing temperature 55 °C for 1 m, extension temperature 72 °C for 5 m). The second amplicon was a ~ 1 Kb gene fragment, obtained using the pair of primers drebfor2 (5’-CATGATCCACAGGGTGCAA-3’) and E4rev-down (5′-GGTCCACCATTTGATCTTCATT-3′). This ~ 1 Kb gene fragment overlapped the previous one and completed exon 4, including the 3’-terminal of the gene (experimental conditions: annealing temperature 58 °C for 1 m, extension temperature 72 °C for 2 m). This latter pair of primers was also used to amplify the exon 4 fragment from 42 Aegilops accessions (see Table 1). The PCR reactions were performed in an overall volume of 25 μl, containing 12.5 μl REDTaq® ReadyMix™ PCR reaction mix (20 mM Tris-HCl, pH 8.3, 100 mM KCl, 3 mM MgCl2, 0.002% gelatin, 0.4 mM dNTP mix, stabilizers and 0.06 unit/ml of Taq DNA polymerase), 20 pmol of each primer and 200 ng of gDNA. The PCR thermal cycle conditions included an initial denaturation at 95 °C for 5 m, then 35 cycles, with melting at 94 °C for 1 m, followed by the annealing and extension steps as reported above for each pair of primers, ending at + 4 °C.
Furthermore, the pair of primers drebfor1 and E4rev-down was used to amplify transcripts from the RNA extract, following the Latini et al. procedure (2007).
In Online Resource 1, the annealing regions of the forward and reverse primers used are represented.
PCR products purification and bacterial transformation
The PCR products were purified using the Gel Band Purification kit (Amersham, USA), followed by Sepharose CL-6B affinity chromatography (Pharmacia Corp., NYC, NY, USA), and then cloned into a pCR®II-TOPO® vector (Invitrogen, USA). After transformation of E. coli cells, positive clones were screened by using agar plates containing ampicillin (50 µl/ml), X-Gal (40 mg/ml) and IPTG (40 µl of 100 mM IPTG - Isopropyl β-D-1-thiogalactopyranoside solution). Plasmids were extracted using the SDS plasmid isolation protocol (Sambrook et al. 1990). In order to confirm the presence of the expected cloned fragment, plasmid DNA clones were digested before sequencing by the EcoRI restriction enzyme and visualized on agarose gel.
Sequences from public databases
Various relevant DRF1 sequences, belonging to the Aegilops and Triticum genera, were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/) and other data sources. A list of these sequences is provided in Table 2.
Data analyses
Electropherograms were analyzed using FinchTV (Geospiza Inc., USA) and sequences were aligned using Clustal Omega at EMBL-EBI (Sievers et al. 2011) and assembled using CAP3 (Huang and Madan 1999). FGENESH (by Softberry) at http://www.softberry.com/berry.phtml?topic=fgenesh&group=programs&subgroup=gfind (Soloviev et al. 2006) was used to identify the gene structure (parameter settings: organism = triticum; suboptimal exon cutoff = 0.1).
DnaSP v6 (Rozas et al. 2017) was used to assess the diversity and genetic relationships among sequences. Analyses were carried out using alignments manually edited to improve their accuracy and excluded gaps and missing data.
Phylogenetic analyses were carried out using MEGA 7 (Kumar et al. 2016). The Neighbor-Joining (NJ) method was applied and the evolutionary distances were computed using the Maximum Composite Likelihood model. A bootstrap analysis based on 1000 re-samplings was used to determine the confidence values of the tree branches. All positions containing gaps and missing data were eliminated. For completeness, a separate analysis was also conducted by applying the Maximum Likelihood method and PHYML algorithm at http://www.atgc-montpellier.fr/phyml/ (Guindon and Gascuel 2003; Lefort et al. 2017).
Results
The DRF1 gene in Aegilops speltoides and comparison with the B subgenome copy of T. durum
The whole DRF1 gene was isolated and sequenced from three Ae. speltoides accessions. Representative PCR amplification and restriction results after cloning of the gene fragments are shown in Fig. 1. The three gene sequences were submitted to GenBank: two accessions of Ae. speltoides ssp. ligustica (ICARDA accessions IG 48994 with NCBI ID FJ843102, 3214 bp long and IG 47948 with NCBI ID FJ858187, 3229 bp long) and one accession of Ae. speltoides ssp. speltoides (ICARDA accession IG 110757 with NCBI ID FJ858188, 3214 bp long).

Electrophoresis gel of products obtained by PCR amplifications of genomic DNA. a 2.5 Kb and 500 bp fragments amplified using the pair of primers drebfor1 and drebrev1. Lane 1: Ae. speltoides ssp. speltoides IG 110757; lane 2: Ae. speltoides ssp. ligustica IG 47948; lane 3: Ae. speltoides ssp. ligustica IG 248994. b 1 Kb fragment amplified using the pair of primers drebfor2 and E4rev-down. Lane 1: Ae. speltoides ssp. ligustica IG 47948. c EcoRI restriction analysis of clones containing the ~ 500 bp fragments. The samples in lanes 2 and 4 (Ae. speltoides ssp. speltoides IG 126225 and IG 116071 accessions, respectively) represented two positive clones, whose sequences were deposited (lateral arms of vector were included in the bands, thus justifying 600 bp size), while lanes 1 and 3 were found to be empty plasmids. d 1 Kb fragment of exon 4 amplified using the pair of primers drebfor2 E4rev-down primers, from various accessions of Ae. speltoides. Lane 1: IG 117905, seed 1; lane 2: IG 117905, seed 2; lane 3: IG 116079; lane 4: IG 135337. MM stands for 1 kb molecular marker size
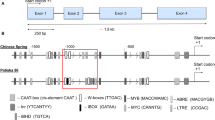
The FGENESH gene finder tool, used for the prediction of the gene structure of the Ae. speltoides sequences, recognized the presence of four exons and three introns, with a high confidence, showing higher scores for exons 1 and 4 than for exons 2 and 3. The same structure had been already observed in wheat and barley. As an example, the prediction obtained using the AeslDRF1-FJ843102 sequence as the template is shown in Fig. 2. An analogous result was obtained when using the other two sequences as templates.

The FGENESH prediction of gene structure using AeslDRF1-FJ843102 as template. CDSf stands for first exon including start codon, CDSi stands for internal exon, CDSl stands for last coding segment, ending with stop codon, CDSo stands for genes with only one CDS (starting with start codon and ending with stop codon) TSS stands for the position of transcription start, PolA stands for polyA segment
The three Ae. speltoides sequences were aligned and the genetic diversity quantification is summarized in Table 3. 3210 sites were analysed and 20 out of 30 identified polymorphisms were located in exon 4, with an overall nucleotide diversity of Pi = 0.006. A total of 4 indels (mutations due to the insertion or deletion of nucleotides) were observed and the lack of a triplet in the simple sequence repeat (SSR) region of exon 1 in AeslDRF1-FJ858187 was particularly interesting, as it caused a shorter alanine stretch in the putative protein, whereas the other three indels were located in non-coding regions. Worth noting is that the AessDRF1-FJ858188 sequence showed a nonsynonymous mutation at nucleotide position 2431, within the AP2 domain coding region, resulting in the replacement of a Thr (ACG) in an Ala (GCG). All the above-reported polymorphisms are highlighted in the alignment shown in Online Resource 1.
Afterwards, the Ae. speltoides sequences were aligned with two different B subgenome copies of T. durum, namely KM520370 and KM504245, recently published (Cantale et al. 2018). A very high similarity (more than 93% identity) was observed among the sequences. The analysis involved 2989 sites and identified 5 haplotypes and 135 polymorphic sites, the overall nucleotide diversity being Pi = 0.025 (Table 3). Focusing on the coding regions, the SSR region in exon 1 of the Aegilops group is shorter and characterized by the GCG triplet, while the B genome copies of T. durum showed the distinctive GCC triplet (both triplets coding for alanine). Furthermore, two in-frame deletions, each 15 bp long, were present in exon 4 of the B genome copies of T. durum. As regards the non-coding regions, two different insertions at alignment position 325 bp (intron 1) distinguished the Aegilops and Triticum groups, the former with a length of 65 bp and the latter of 25 bp, characterized by completely different nucleotide sequences. There were also few other differences among the indels of the two groups (SSR and indels are highlighted in light grey in Online Resource 1).
Transcription of the DRF1 gene in Aegilops speltoides
The activity of the DRF1 gene in Aegilops speltoides was ascertained with RNA extraction and reverse transcription PCR (RT-PCR) which revealed the presence of the DRF1.2 transcript (Online Resource 2). Its exonic combination (exon 1, exon 2 and exon 4) generated a frameshift causing a premature termination codon (PTC). The sequence of this AesDRF1.2 transcript, isolated from accession IG 47280, was deposited at GenBank (NCBI ID: EU197054) and used as the template in a BLAST search. High identity values were observed between this and DRF1 transcripts of different varieties of T. durum, such as Atil (NCBI ID EU089824, 95.43%), Ciccio (NCBI ID FJ560492, 93.78%) and Karalis (NCBI ID FJ560496, 93.77%), T. aestivum, such as TaDREB3C (NCBI ID AY781351, 95.34%), TaDREB4C (NCBI ID AY781356, 95.16%) and TaDREB5C (NCBI ID AY781357, 95.52%), and also the T. dicoccoides DRE-binding transcription factor (NCBI ID HM015904, 95.43%). The DRF1.2 transcript was the only one detectable since plants were grown in normal conditions, as reported in our previous work (Latini et al. 2013).
Identification of a DRF1 pseudogene in Aegilops speltoides
The genomic DNA amplification using the pair of primers drebfor1 and drebrev1 always amplified shorter fragments, ~ 500 bp, in addition to the expected ~ 2.5 Kb gene fragment (Fig. 1a). These amplicons were cloned (Fig. 1c) and sequenced. For each sample, some sequences turned out to be unspecific genomic fragments, but one corresponded to a DRF1 gene copy lacking introns. The analysis showed that this genomic sequence joined exons 1, 2 and 4, thus looking like the mature DRF1.2 transcript. This structure pointed to the presence in the Ae. speltoides genome of a pseudogene linked to its cognate DRF1 gene. Two sequences, obtained from the accessions IG126225 and IG116071, were deposited at GenBank (NCBI IDs MN639698 and MN639699, respectively). The sequences alignment of these two pseudogenes, the above described mRNA of DRF1.2 of Ae. speltoides and a DRF1.2 transcript of the B genome of T. durum, is shown in Fig. 3.

Alignment of two about 500 bp long fragments with two DRF1.2 transcripts. MN639698 and MN639699 are the NCBI ID of two sequences, isolated from Ae. speltoides accessions IG126225 and IG116071, respectively; EU197054 is the sequence of the AesDRF1.2 transcript (accession IG 47280); KM520370 is the sequence of a TdDRF1.2 transcript from the B subgenome copy of T. durum. Exon 1 is shown in bold, exon2 using white letters and black background and exon 4 using white letters and grey background. The SSR region, consisting of 15 bp, GCG pattern, plus further 6 bp (all coding for alanine) and the premature stop codon are highlighted in boxes
Nucleotide diversity analysis of exon 4 in Aegilops
A genomic DNA fragment of about 1 Kb, mapping in exon 4 (Fig. 1d), was isolated from 42 Aegilops accessions, obtaining a total of 95 cleaned sequences; then, after removing identical sequences obtained from different seeds of the same accession, 81 unique sequences were submitted to GenBank (see Table 1 for Aegilops accessions and the GenBank accession numbers). Three other sequences of Aegilops mapping DRF1 exon 4 were downloaded from public databases (see Table 2) and added to the set. The nucleotide diversity of these 84 sequences was analysed and the results are reported in Table 4. Sequences were analysed as a sole group (bulked group) and also split into subgroups, in accordance with their subspecies (Ae. speltoides ssp. speltoides and Ae. speltoides ssp. ligustica, Ae. speltoides, Aegilops). A further analysis was carried out by merging all Ae. speltoides sequences, irrespective of subspecies (Ae speltoides merged group). Interestingly, the nucleotide diversity of diploid Aegilops speltoides sequences was Pi = 0.018, when considering both the merged and individual subgroups, and it was 4.5 times higher than the nucleotide variation observed when analysing the same region in various Triticum durum varieties (nucleotide diversity Pi = 0.0040, data not shown). Most sequences represented a single, non-shared haplotype (71 haplotypes out of 84 sequences) (Table 4), except a few cases shared inside and between taxa, with Haplo 4 being the most common one (Table 5). Furthermore, a comparison between the Ae. speltoides ssp. ligustica and Ae. speltoides ssp. speltoides subspecies highlighted the presence of several SNPs, even if most of them were synonymous and did not cause replacements (data not shown). A phylogenetic analysis was carried out based on these 84 sequences using both the Neighbor-Joining (NJ) and Maximum Likelihood (ML) methods and the inferred NJ tree is shown in Fig. 4. The inferred ML tree showed the same topology (data not shown). Aegilops speltoides and the other Aegilops sequences formed two well-supported clusters and no noticeable separation could be observed between Ae. speltoides ssp. speltoides and Ae. speltoides ssp. ligustica subspecies sequences.

The optimal Neighbor Joining tree based on 84 sequences of DRF1 exon 4 from Aegilops species (sum of branch length = 0.4504). There was a total of 812 positions in the final dataset. The bootstrap support values (> 50%) are shown close to the node. The species, the NCBI IDs and the simplified genome codes are reported on each branch. Genome clusters are indicated in bold to right of tree. Asterisks denote branches shared with the tree inferred by the Maximum-Likelihood method
To investigate the relationships among DRF1 exon 4 sequences in Aegilops and Triticum, 16 DRF1 exon 4 sequences from the Triticeae tribe (7 out of 16 were isolated, sequenced and submitted to GenBank by our laboratory) were added to the dataset (see Table 2) and the total 102 sequences, including 2 outgroups, were aligned and analysed. Two further sequences of Psathyrostachys juncea and Brachypodium distachyon mapping DRF1 exon 4 were also added as outgroups. The NJ phylogenetic tree inferred is represented in Fig. 5. The 102 sequences formed distinct clades: all Ae. speltoides sequences clustered in a single clade (S genome), which was graphically collapsed to better highlight the relationships with the other taxa; four sequences grouped together with three B copies of the gene in T. durum, forming a well-supported clade (96%, B genome), sister to the S one; four other sequences grouped together with two A copies of the gene in T. durum, forming a well-supported clade (99%, A genome); two sequences grouped together with three Ae. tauschii sequences, forming the D genome clade (99%) and finally all other Aegilops sequences, sharing the U genome copy, formed a single clade (92%, U genome) distinct from the Ae. cylindrica sequence.

The optimal Neighbor Joining tree based on 102 DRF1 exon 4 sequences from Aegilops and Triticum species (sum of branch length = 0.9089). For clarity, the Aegilops speltoides group of 68 sequences was graphically collapsed. There was a total of 730 positions in the final dataset. The bootstrap support values (> 50%) are shown close to the node. The species, the NCBI IDs and a single capital letter indicating the genome type are reported on each branch. When the genome type was unknown, the simplified genome code was indicated: AB for T. durum and T. dicoccoides or ABD for T. aestivum. Genome clusters are indicated in bold on the right of the tree. Asterisks denote branches shared with the tree inferred by Maximum-Likelihood method
Discussion
The study of gene variability in the Aegilops species has recently attracted considerable attention as wild relatives represent a valuable source of gene alleles that can be exploited for wheat improvement. In this context, the DRF1 gene, involved in the drought response in plants, was studied in Aegilops speltoides and in other Aegilops species to investigate its various features. The gene was isolated and sequenced in three accessions of Ae. speltoides and its structure fully corresponded to that previously observed in barley and wheat (Xue and Loveridge 2004; Egawa et al. 2006; Latini et al. 2007), as deduced by sequence similarity and gene structure prediction (Fig. 2).
The detection of the DRF1.2 transcript in Ae. speltoides suggested the occurrence of transcription involving the alternative splicing mechanism as this transcript arises by joining exons 1, 2 and 4. It was the only transcript isolated, as plants were grown in normal conditions and it is reasonable to speculate that the other two transcripts, DRF1.1 and DRF1.3, would also be expressed and detectable in plants when affected by drought stress, as previously demonstrated in Triticeae (Xue and Loveridge 2004; Egawa et al. 2006; Latini et al. 2007). The sequence of this transcript was highly similar to the same transcripts in wheat and barley and does not encode for a transcription factor, but for an abortive short protein whose function is still unknown. Such a kind of transcript variant can often be found in alternatively spliced genes and is reported to play an important role in gene regulation through its modulated expression and degradation by nonsense-mediated mRNA decay (Dubrovina et al. 2013) or other RNA surveillance mechanisms (Reddy et al. 2012; Bedre et al. 2019).
The isolation of a pseudogene suggests an increasingly complex scenario for DRF1 gene regulation in Ae. speltoides. Indeed, during the isolation of the whole gene, an unexpected 500 bp genomic fragment was always amplified and its sequence corresponded to the genomic variant of the mature DRF1.2 transcript, thus fully satisfying the criteria for representing a processed pseudogene (retropseudogene) arising by reverse transcription and reinsertion into the genome (Zou et al. 2009; Tutar 2012). Because they are derived from mature transcripts, retropseudogenes lack upstream promoters, introns and other regulatory elements and were previously considered transcriptionally silent, dead-on-arrival (Vanin 1985). However, more recently, there has been increasing evidence that they are still active and often involved in the regulation of their parental genes, particularly at the post-transcriptional level, through various mechanisms including RNA interference and RNA antisense (Vinckenbosh et al. 2006; Wen et al. 2012; Sen and Ghosh 2013; Xie et al. 2019). To the best of our knowledge, this is the first reported case of a pseudogene belonging to the DREB gene family in Triticeae.
More targeted research is necessary to investigate the potential roles of both the DRF1.2 transcript and pseudogene in DRF1 gene regulation in Ae. speltoides.
The analysis of DNA polymorphisms of the nucleotide sequences of the three Ae. speltoides whole DRF1 gene accessions revealed a relatively low total nucleotide diversity (Pi = 0.006), in line with the observation reported that TFs and particularly those containing specific domains, like the AP2 domain, are more conserved than other gene families (Tatarinova et al. 2016). Interestingly, the nucleotide diversity turned out to be lower in introns than in exons, which was somewhat unexpected, as introns are considered freer to accumulate mutations than exons, which are constrained by their function. This genetic feature (exons more variable than introns) has been observed in animals, in specific gene families involved with the immune system or associated with predator–prey responses, and its role in adaptive evolution has been hypothesised (Nakashima et al. 1995; Samonte and Eichler 2002; Caporale 2003). Accordingly, one can speculate that the molecular evolution of the DRF1 gene, involved in response to abiotic stress, has undergone a similar genetic mechanism, even if more investigation is necessary to confirm this result.
The DNA sequence polymorphism analyses highlighted that there were no differences between the two Ae. speltoides subspecies, ssp. ligustica and ssp. speltoides, when considering either the whole gene or the exon 4 sequences (Table 4). Indeed, the three groups, Ae. speltoides ssp. speltoides, Ae. speltoides ssp. ligustica and Ae. speltoides, showed very similar Pi values and the phylogenetic analysis further confirmed the above observation. In the NJ tree (Fig. 4), clades are well-supported (bootstrap value ≥ 93%) and all Ae. speltoides sequences clustered in a single clade, in which ssp. speltoides and ssp. ligustica were scattered and mixed, but definitively separated from other the Aegilops species. Our results are in line with the conclusions of Belyayev and Raskina (2013) showing that ssp. ligustica and ssp. speltoides were a case of population dimorphism in fruit structure and did not in fact represent two different subspecies, as initially proposed by Zhukovsky (1928).
In the phylogenetic tree (Fig. 4) the sequences of Aegilops species were arranged according to their specific known genome even though the sampling performed of the various diploid and polyploid Aegilops species was somewhat limited and a little unbalanced for experimental reasons. It is worth noting that the two sequences of Ae. sharonensis from the Sitopsis section were found to be sisters of the Ae. speltoides clade and the two sequences of Ae. tauschii clustered together in a separated and well-supported branch. Furthermore, Ae. umbellulata, the only other diploid Aegilops species included in this analysis and donor of the U genome to some of the tetraploid species included, was able to form the highly supported (93%) U genome clade. In this regard, it is reported that the U genome occurring in some polyploid species is very well conserved and similar to the parental diploid one, whereas the second genomes evolved rapidly from their ancestors (Badaeva et al. 2004). Interestingly, the Ae. cylindrica sequence, which has the genomic formula CcCcDcDc (Linc et al. 1999), stood apart and was placed in a single branch, leading us to suppose that this sequence proceeded from the Cc genome, since the D genome of Ae. cylindrica is closely related to the D genomes of both Ae. tauschii and T. aestivum (Caldwell et al. 2004).
Due to the close genetic relationship between the S genome of Aegilops speltoides and the B subgenome of wheat (Haider 2013; Marcussen et al. 2014), the three DRF1 gene sequences isolated from Ae. speltoides (AesDRF1 gene) were aligned with the two DRF1 gene sequences from the B subgenome of T. durum (see the alignment in Online Resource 1) and the comparison appeared very interesting, in particular when considering exon 1, containing a SSR region coding for a variable number of alanines (Di Bianco et al. 2011). We recently found that this region consists of the repetition of two different patterns, both encoding for alanine, namely GCG and GCC, peculiar to the A and B subgenome copies, respectively, thus representing a specific subgenome signature in T. durum (Cantale et al. 2018). The length of the SSR region was also variable and we found it was 21 bp long in the A subgenome and 18 bp in the B subgenome copies, respectively. In Ae. speltoides, the SSR consisted of 15 nucleotides in AeslDRF1-FJ843102 and AessDRF1-FJ858188, and 12 nucleotides in AeslDRF1-FJ858187, always displaying the GCG triplet. It is worth noting that the SSR regions of the two pseudogene sequences, isolated from different accessions, showed the GCG triplet for a stretch of 15 nucleotides, but also two further downstream triplets (GCC GCA), both encoding for alanine, with sequences identical to those observed in the B subgenome copies (see Fig. 3). The presence of a variable repeat number, the GCG triplet and the GCC GCA motif places the Ae. speltoides SSR sequence in an intermediate position between the A and B subgenomes, but the few sequences containing this region currently available do not make it possible to generalize. On the other hand, it is reported that SSRs show an extremely high rate of reversible variation in the repeat number, which can be functionally significant (Gemayel et al. 2010). Recent literature provides evidence of an evolutionary role for SSRs in transcribed regions as important sources of adaptive genetic variation, as they are involved in a number of effects such as the regulation of transcription and translation of their own genes, organization of chromatin and morphological variations (Kashi and King 2006; Viera et al. 2016). All things considered, further research into the SSR region of the DRF1 gene in Ae. speltoides looks particularly interesting, as it may represent another possible player involved in the gene regulation.
To widen the comparison with the Triticum genus, the exon 4 sequences from Aegilops, T. aestivum and T. durum were aligned and a phylogenetic tree was inferred (Fig. 5). Variability of a single gene has been used to investigate the phylogenetic relationship among species (Adderley and Sun 2013; Liu et al. 2016), although this task appears particularly challenging in this tribe, and sometimes large incongruities among the trees built from different genes could be observed, due to the complex nature of allopolyploidy, possible introgressions and the involvement of mechanisms such as gene hybridization, gene flow or horizontal gene transfer (Degnan and Rosenberg 2006; Liberles and Dittmar 2008; Escobar et al. 2011). In spite of the above, and also considering that the number of accessions analysed among the Aegilops and Triticum genera was not balanced, the phylogenetic tree inferred showed well-supported clades attributable to the current genomes (S, A, B, D and U genomes). Furthermore, those sequences of which the subgenome was initially not known placed themselves correctly with respect to a genomic designation consistent with their species, so that the association enriches information about them. The topology observed among the A, B and D clades could be symbolized by B (A, D) as the A clade is closely related to the D clade and both are distant from the B clade. Marcussen et al. (2014) found that the above topology is the most frequent in bread wheat when analysing both genes in the whole genome and a large number of trees from different genes. Furthermore, with coalescent-based genome divergence analyses they estimated that Ae. speltoides is a sister to the B genome, T. urartu is a sister to the A genome and Ae. sharonensis and Ae. tauschii are successive sisters to the D genome (Marcussen et al. 2014).
In conclusion, we report a study focused on the isolation and characterization of the DRF1 gene in Aegilops speltoides and other Aegilops species. The overall results suggest that there is a high similarity between the B and S genome copies of the DRF1 gene, but also some features indicating that both genomes have evolved independently, in line with other observations reporting that B is the most variable among the three subgenomes of current polyploid wheats (Wendel 2000; Petersen et al. 2006). As DRF1 is a key gene in drought response, we intend to continue our studies: the possible involvement of the pseudogene, SSR variability and the DRF1.2 transcript in the gene regulation mechanisms is very interesting and deserves further research. Furthermore, the sequencing of the whole gene in a larger taxon sampling would increase knowledge of DRF1 gene diversity in the Triticeae tribe. The advancement of genomics and the growing availability of wild relative genomes, coupled with the development of new statistical methods for comparative genomics, will make it possible for molecular data to be supplemented by other information that is underutilized in phylogenetic analyses, such as protein structural constraints in specific residues, chromosomal information and population genetics, leading in the end to a better understanding of the evolutionary history of Triticeae. Looking further ahead, the increase in knowledge should provide an important contribution to the widening of crop genetic diversity in support of global food security.
Abbreviations
- AP2:
-
Apetala 2
- AS:
-
Alternative splicing mechanism
- DREB:
-
Dehydration responsive element binding factor
- DRF1:
-
Dehydration responsive factor 1
- ERF:
-
Ethylene-responsive element binding factor
- GP-2:
-
Secondary gene pool
- PTC:
-
Premature termination codon
- SSR:
-
Simple sequence repeat
- TF:
-
Transcription Factor
- Aes :
-
Ae. speltoides
- Aesl :
-
Ae. speltoides ssp. ligustica
- Aess :
-
Ae. speltoides ssp. speltoides
- At :
-
A. thaliana
- Ta :
-
T. aestivum L.
- Td :
-
T. durum (Triticum turgidum ssp. durum L.)
References
Adderley S, Sun G (2013) Molecular evolution and nucleotide diversity of nuclear plastid phosphoglycerate kinase (PGK) gene in Triticeae (Poaceae). Gene 533(1):142–148
Agarwal PK, Agarwal P, Reddy MK, Sopory SK (2006) Role of DREB transcription factors in abiotic and biotic stress tolerance in plant. Plant Cell Rep 25(12):1263–1274
Badaeva ED, Amosova AV, Samatadze TE, Zoshchuk SA, Shostak NG, Chikida NN, Zelenin AV, Raupp WJ, Friebe B, Gill BS (2004) Genome differentiation in Aegilops. 4. Evolution of the U-genome cluster. Plant Syst Evol 246:45–76
Bedre R, Irigoyen S, Petrillo E, Mandadi KK (2019) New era in plant alternative splicing analysis enabled by advances in high-throughput sequencing (hts) technologies. Front Plant Sci 10:740
Belyayev A, Raskina O (2013) Chromosome evolution in marginal populations of Aegilops speltoides: causes and consequences. Ann Bot 111(4):531–538
Brozynska M, Furtado A, Henry RJ (2016) Genomics of crop wild relatives: expanding the gene pool for crop improvement. Plant Biotech J 14(4):1070–1085
Caldwell KS, Dvorak J, Lagudah ES, Akhunov E, Luo MC, Wolters P, Powell W (2004) Sequence polymorphism in polyploid wheat and their d-genome diploid ancestor. Genetics 167(2):941–947
Cantale C, Di Bianco D, Thiyagarajan K, Ammar K, Galeffi P (2018) B genome specific polymorphism in the TdDRF1 gene is in relationship with grain yield. Planta 247(2):459–469
Caporale LK (2003) Darwin in the genome: molecular strategies in biological evolution. McGrow-Hill, New York
Degnan JH, Rosenberg NA (2006) Discordance of species trees with their most likely gene trees. PLoS Genet 2(5):e68
Di Bianco D, Thiyagarajan K, Latini A, Cantale C, Felici F, Galeffi P (2011) Exploring the genetic diversity of the DRF1 gene in durum wheat and its wild relatives. Plant Genet Res 9(2):247–250
Dubrovina AS, Kiseklev KV, Zhuravlev YN (2013) The role of canonical and noncanonical pre-mRNA splicing in plant stress responses. Biomed Res Int. https://doi.org/10.1155/2013/264314
Egawa C, Kobayashi F, Ishibashi M, Nakamura T, Nakamura C, Takumi S (2006) Differential regulation of transcript accumulation and alternative splicing of a DREB2 homolog under abiotic stress conditions in common wheat. Genes Genet Syst 81(2):77–91
Escobar JS, Scornavacca C, Cencil A, Guilhaumon C, Santoni S, Douzery EJP, Ranwez V, Glémin S, David J (2011) Multigenic phylogeny and analysis of tree incongruences in Triticeae (Poaceae). BMC Evol Biol 11:181
Gemayel R, Vinces MD, LegendreM Verstrepen KJ (2010) Variable tandem repeats accelerate evolution of coding and regulatory sequences. Ann Rev Genet 44:445–477
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52(5):696–704
Haider N (2013) The origin of the B genome of bread wheat (Triticum aestivum L.). Russ J Genet 49(3):263–274
Harlan JR, de Wet JMJ (1971) Toward a rational classification of cultivated plants. Taxon 20(4):509–517
Huang X, Madan A (1999) CAP3: a DNA sequence assembly program. Genome Res 9(9):868–877
Kashi Y, King DG (2006) Simple sequence repeats as advantageous mutators in evolution. Trends Genet 22(5):253–259
Kerby K, Kuspira J (1987) The phylogeny of the polyploid wheats Triticum aestivum (bread wheat) and Triticum turgidum (macaroni wheat). Genome 29(5):722–737
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874
Kumar A, Kapoor P, Chunduri V, Sharma S, Garg M (2019) Potential of Aegilops sp. for improvement of grain processing and nutritional quality in wheat (Triticum aestivum). Front Plant Sci 10:308
Latini A, Rasi C, Sperandei M, Cantale C, Iannetta M, Dettori M, Ammar K, Galeffi P (2007) Identification of a DREB-related gene in Triticum durum and its expression under water stress conditions. Ann Appl Biol 150(2):187–195
Latini A, Sperandei M, Cantale C, Arcangeli C, Ammar K, Galeffi P (2013) Variability and expression profile of the DRF1 gene in four cultivars of durum wheat and one triticale under moderate water stress conditions. Planta 237(4):967–978
Lefort V, Longueville J-E, Gascuel O (2017) SMS: smart model selection in PhyML. Mol Biol Evol 34(9):2422–2424
Liberles DA, Dittmar K (2008) Characterizing gene family evolution. Biol Proced Online 10(1):66–73
Linc G, Friebe BR, Kynast RG, Molnar-Lang M, Köszegi B, Sutka J, Gill BS (1999) Molecular cytogenetic analysis of Aegilops cylindrica Host. Genome 42:497–503
Liu F, Si H, Wang C, Sun G, Zhou E, Chen C, Ma C (2016) Molecular evolution of Wcor15 gene enhanced our understanding of the origin of A, B and D genomes in Triticum aestivum. Sci Rep 6:31706
Marcussen T, Sandve SR, Heier L, Spannagl M, Pfeifer M, The International Wheat Genome Sequencing Consortium, Jakobsen KS, Wulff BBH, Steuernagel B, Mayer KFX, Olsen O (2014) Ancient hybridizations among the ancestral genomes of bread wheat. Science 345(6194):1250092
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Nakashima K, Nobuhisa I, Deshimaru M, Nakai M, Ogawa T, Shimohigashi Y, Fukumaki Y, Hattori M, Sakaki Y, Hattori S, Ohno M (1995) Accelerated evolution in the protein-coding regions is universal in crotalinae snake venom gland phospholipase A2 isozyme genes. PNAS 92:5605–5609
Petersen G, Seberg O, Yde M, Berthelsen K (2006) Phylogenetic relationships of Triticum and Aegilops and evidence for the origin of the A, B, and D genomes of common wheat (Triticum aestivum). Mol Phylogenet Evol 39(1):70–82
Pont C, Leroy T, Seidel M, Tondelli A, Duchemin W, Armisen D, Lang D, Bustos-Korts D, Goué N, Balfourier F, Molnár-Láng M, Lage J, Kilian B, Özkan H, Waite D, Dyer S, Lettellier T, Alaux M, Wheat and Barley Legacy for Breeding Improvement (WHEALBI) Consortium, Russel J, Keller B, van Eeuwijk F, Spannagl M, Mayer KFX, Waugh R, Stein N, Cattivelli L, Haberer G, Charmet G, Salse J (2019) Tracing the ancestry of modern bread wheats. Nat Genet 51(5):905–911
Reddy AS, Rogers MF, Richardson DN, Hamilton M, Ben-Hur A (2012) Deciphering the plant splicing code: experimental and computational approaches for predicting alternative splicing and splicing regulatory elements. Front Plant Sci 3:18
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol Biol Evol 34(12):3299–3302
Ruban AS, Badaeva ED (2018) Evolution of the S-genomes in Triticum-Aegilops alliance: evidences from chromosome analysis. Front Plant Sci 9:1756
Salse J, Chagué V, Bolot S, Magdelenat G, Huneau C, Pont C, Belcram H, Couloux A, Gardais S, Evrard A, Segurens B, Charles M, Ravel C, Samain S, Charmet G, Boudet N, Chalhoub B (2008) New insights into the origin of the B genome of hexaploid wheat: evolutionary relationships at the SPA genomic region with the S genome of the diploid relative Aegilops speltoides. BMC Genom 9:555
Sambrook J, Fritsch EF, Maniatis T (1990) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Samonte RV, Eichler EE (2002) Segmental duplications and the evolution of the primate genome. Nat Rev Genet 3(1):65–72
Schneider A, Molnár I, Molnár-Láng M (2008) Utilisation of Aegilops (goatgrass) species to widen the genetic diversity of cultivated wheat. Euphytica 163:1–19
Sen K, Ghosh TC (2013) Pseudogenes and their composers: delving in the ‘debris’ of human genome. Brief Funct Genom 12(6):536–547
Sievers F, Wilm A, Dineen DG, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins D (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol Syst Biol 7:539
Soloviev V, Kosarev P, Seledsov I, Vorobyev D (2006) Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol 7(Suppl 1):S10
Tatarinova TV, Chekalin E, Nikolsky Y, Bruskin S, Chebotarov D, McNally KL, Alexandrov N (2016) Nucleotide diversity analysis highlights functionally important genomic regions. Sci Rep 6:35730
Tutar Y (2012) Pseudogenes. Comp Funct Genomics 424526
van Slageren (1994) Wild wheats: a monograph of Aegilops L. and Amblyophyrum (Jaub & Spach) Eig (Poaceae). ICARDA/Wageningen Agricultural University Papers 94–7
Vanin EF (1985) Processed pseudogenes: characteristics and evolution. Annu Rev Genet 19:253–272
Viera MLC, Santini L, Diniz AL, de Freitas MC (2016) Microsatellite markers: what they mean and why they are so useful. Genet Mol Biol 39(3):312–328
Vinckenbosh N, Dupanloup I, Kaessmann H (2006) Evolutionary fate of retroposed gene copies in the human genome. PNAS 103(9):3220–3225
Wen Y-Z, Zheng L-L, Qu L-H, Ayala FJ, Lun Z-R (2012) Pseudogenes are not pseudo any more. RNA Biol 9(1):27–32
Wendel JF (2000) Genome evolution in polyploids. Plant Mol Biol 42:225–249
Xie J, Li Y, Liu X, Zhao Y, Li B, Ingvarsson PK, Zhanga D (2019) Evolutionary origins of pseudogenes and their association with regulatory sequences in plants. Plant Cell 31:563–578
Xue GP, Loveridge CW (2004) HvDRF1 is involved in abscisic acid-mediated gene regulation in barley and produces two forms of AP2 transcriptional activators, interacting preferably with a CT-rich element. Plant J 37(3):326–339
Yamaguchi-Shinozaki K, Shinozaki K (1994) A novel cis-acting element in an Arabidopsis gene is involved in responsiveness to drought, low-temperature, or high-salt stress. Plant Cell 6(2):251–264
Zhang H, Mittal N, Leamy LJ, Barazani O, Song B-H (2017) Back into the wild—apply untapped genetic diversity of wild relatives for crop improvement. Evol Appl 10:5–24
Zhukovsky M (1928) A critical systematical survey of the species of the genus Aegilops L. Bull Appl Bot Genet Plant Breed 18:417–609
Zou C, Lehti-Shiu MD, Thibaud-Nissen F, Prakash T, Buell CR, Shiu SH (2009) Evolutionary and expression signatures of pseudogenes in Arabidopsis and rice. Plant Physiol 151(1):3–15
Acknowledgements
The authors wish to thank ICARDA (International Center for Agricultural Research in the Dry Areas) and the Leibniz Institute of Plant Genetics and Crop Plant Research (IPK, DE) for providing seeds of the various accessions. The authors thank Prof. Enrico Porceddu (Scuola Superiore Sant’Anna, Pisa, IT) and Dr. Prashant Vikram (CIMMYT, MX) for helpful criticisms during manuscript preparation and Mr. Ian Pace for reviewing the English of the text. KT was supported by an International Doctoral Fellowship in Agrobiodiversity (Plant Genetic Resources) from the Scuola Superiore Sant’Anna, Pisa, Italy and by an ENEA International 2-years postdoc Fellowship at ENEA Casaccia Research Centre, Italy.
Funding
Open access funding provided by Ente per le Nuove Tecnologie, l'Energia e l'Ambiente within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thiyagarajan, K., Latini, A., Cantale, C. et al. Structural characterization of the DRF1 gene of Aegilops speltoides and comparison of its sequence with those of B and other Triticeae genomes. Euphytica 216, 152 (2020). https://doi.org/10.1007/s10681-020-02679-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-020-02679-7