
Abstract
In recent years, sorghum (Sorghum bicolor (L.) Moench) has gained attention as a food, feed, and biofuel crop, but yield improvements have lagged behind those in other crops. Here, we investigated quantitative trait loci (QTLs) associated with yield and yield-related traits in sorghum. We measured eight morphological traits related to yield potential in an F2 and F2:3 population derived from a cross between African and Japanese sorghum landraces, and we developed a genetic linkage map of 137 sorghum genome-based simple sequence repeat markers. The total map length was 1,239.2 cM, with an average distance of 9.9 cM between adjacent markers. By using both single- and multiple-QTL approaches to identify the chromosomal regions controlling these traits, we identified a total of 52 QTLs associated with the eight traits (culm length, number of tillers, panicle length, culm diameter, leaf length, leaf width, grain weight/panicle, and 100-grain weight) using F2 phenotypic values while 25 QTLs were confirmed in the F2:3 population. The percentage phenotypic variation explained by individual QTLs ranged from 3.1 to 36.3 % in the F2 and 2.1 to 30.4 % in the F2:3 population. Most of the traits were significantly correlated with one another (P < 0.05). Ten QTLs had a significant association with more than one trait. A QTL for culm length was mapped to the same region of chromosome 7 as the dw3 gene for plant height. The major QTLs identified here are expected to provide useful information toward understanding the genetic mechanisms of important agronomic traits related to yield in sorghum.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sorghum (Sorghum bicolor (L.) Moench) is a very important cereal in semi-arid tropical and subtropical regions that will play an important role in resolving food security problems in resource-poor countries around the world. The adaptability of sorghum to low inputs and adverse conditions makes it an important “failsafe” in the global agro-ecosystem, but yield improvements are needed to ensure its availability. Worldwide, sorghum is the fifth most important grain crop grown based on tonnage, after maize, wheat, rice, and barley. Its relatively small genome size makes it a model for study of complex genetic phenomena in other C4 crops such as maize and sugarcane, which have larger genomes. In developed countries, sorghum has been gaining the attention of researchers, producers, and industry for its potential as a feedstock for biofuels, as both a source of grain-based ethanol and of cellulosic biofuel stock. Here also genetic improvement and increase in yield are the major factors necessary for progress in developing and utilizing sorghum for biofuels. Genetic improvement in yield of sorghum has lagged far behind that of other cereals such as maize, presumably because of the much smaller effort committed to sorghum improvement. There is still much potential for genetic improvement of yields of important products such as grain, sugar, lignocellulose, and total biomass.
Yield is generally believed to be controlled by many genes and is very difficult to manipulate in breeding programs. Yield can be considered to be composed of component parts such as number of tillers, culm length, number of grains, and grain weight. Mapping genes controlling quantitative traits such as yield provides a clearer understanding of the complex genetic nature of the traits. The use of molecular marker technology and recent advances in quantitative trait locus (QTL) detection by new statistical models facilitate the discovery of QTLs controlling yield and yield components.
In sorghum, different kinds of molecular markers [e.g., restriction fragment length polymorphism (RFLP) markers, random amplified polymorphic DNA (RAPD) markers, amplified fragment length polymorphism (AFLP) markers, and simple sequence repeat (SSR) markers] have been used to develop linkage maps. Recently, QTL mapping has been widely used to locate chromosome regions harboring genes for important agronomic traits, including yield and those related to it. For example, Srinivas et al. (2009) identified QTLs for 12 different agronomic traits, including yield and its components, in a recombinant inbred line (RIL) population. Similarly, Rami et al. (1998) investigated two sorghum caudatum × guinea RIL populations in QTL mapping for grain quality, productivity, and other agronomical traits. Hart et al. (2001) used RILs obtained from a cross of two inbred lines, BTx623 and IS3620C, and reported several QTLs for grain quality, productivity, tillering, and other morphological traits. Other traits studied in QTL mapping experiments include those related to sugar (Ritter et al. 2008; Shiringani et al. 2010), stay-green, drought tolerance (Haussmann et al. 2002; Kebede et al. 2001; Tao et al. 2000; Tuinstra et al. 1996, 1997), and several other biotic and abiotic stresses. Feltus et al. (2006) aligned two different genetic maps derived from inter- and intraspecific sorghum populations and reported 61 QTLs detected from 17 traits related to yield.
The present study was aimed at understanding the genetic basis of the most important traits directly or indirectly related to final yield in a sorghum population derived from a cross between two diverse parents. We report here the linkage of genome-wide SSR markers derived from shotgun sequences of the sorghum genome to QTLs for agronomic traits. These findings will be valuable in marker-assisted selection and other sorghum breeding programs for these traits.
Materials and methods
Plant material
We used a population derived from a cross between two sorghum landraces, Red Kafir and Takakibi. Based on yield potential, these genotypes were chosen from among 320 sorghum accessions previously selected from the National Institute of Agrobiological Sciences (NIAS) Genebank (Shehzad et al. 2009a). Red Kafir, which originates from South Africa, has a compact panicle and white grains. The density of grains in the panicle was low and most florets were sterile. Takakibi is a tall, high-yielding accession which originates from Japan. The panicle is an open type and densely covered with red seeds. The cross was made in 2006 at National Institute of Agrobiological Sciences (NIAS), Tsukuba, and the F1 seeds obtained were sown in the consecutive year 2007. The F1 seeds were then interbred to produce F2 seeds at NIAS. During the 2008 growing season of sorghum, all F2 plants were sown in a field at the University of Tsukuba, 30 cm apart from each other. A total of 149 F2 lines were secured and used for linkage mapping. F2 plants were self-pollinated to produce F3 family lines for further evaluation during the 2009 growing season. Some of the F2 plants did not successfully reach the final harvest stage. Similarly, seeds obtained from some F3 lines were of poor quality, and we discarded those lines from the analysis. Finally, we were able to obtain complete data from only 118 F3 lines.
DNA extraction
Young, soft leaves were cut from 40-day-old seedlings and stored at −80 °C. Genomic DNA was isolated from leaf tissues using a modified cetyltrimethylammonium bromide (CTAB) method. DNA extraction buffer was prepared from 2 % (mg/l) CTAB, 50 mM Tris·HCl (pH 8.0), 10 mM EDTA, 0.7 M NaCl, 0.1 % SDS, 0.1 mg/ml proteinase K, 2 % insoluble polyvinylpyrrolidone (PVP), and 2 % 2-mercaptoethanol. Cellular debris and proteins were removed by extraction in chloroform: isoamyl alcohol (24:1 v/v). DNA was precipitated by the addition of 2-isopropanol, and the precipitate was washed in 70 % then 90 % ethanol. The final precipitate was dissolved in 50 μl of 1/10 TE solution containing RNase A, incubated at 42 °C overnight, and stored at 4 °C. The DNA concentration was measured by using a V-630Bio (JASCO) spectrophotometer, and the DNA was diluted to a working concentration of 5 ng/μl.
Selection of SSR markers and genotyping
SSR markers were selected from the Yonemaru et al. (2009) study, which reported the development of more than 5,000 SSRs based on sorghum whole-genome shotgun sequences. We randomly selected 672 SSR loci covering the whole genome of sorghum, and after using these to screen the parents, selected the best 159 markers with clear banding patterns. These 159 markers included 14 SSR markers previously mapped by Bhattramakki et al. (2000) (ESM1).
The PCR reaction mixture (10 μl) contained 10 ng DNA template, 10× PCR buffer (Mg2+ concentration: 20 mM), 2 mM dNTPs, 25 ng of each primer, and 0.02 U of Blend Taq Plus polymerase (buffer and polymerase from Toyobo Co., Ltd., Japan). Amplification was performed in an Eppendorf Mastercycler (ep 384 thermal cycler). The annealing temperature was determined for all primers by using an Eppendorf Mastercycler (model ep Gradient S). The thermal cycler protocol was set as follows: denaturation at 94 °C for 5 min; 40 cycles of 94 °C (10 s), 55 °C (30 s), and 72 °C (30 s); followed by final extension of 7 min at 72 °C and then cooling at 4 °C. PCR products were run in either 10 % non-denaturing polyacrylamide gel or 3 % superfine resolution (SFR) agarose gel. In the case of polyacrylamide, a small uniform gel (10 cm in size) and a high-efficiency genome scanning (HEGS) gel composed of stacking and running gel (24 cm) containing 10× TBE buffer were used in making the gel. The tank was filled with 1× Tris·glycine buffer. The gel was stained in ethidium bromide solution and photographed with a Kodak Digital Science EDAS 290 camera with Kodak ID Image analysis v. 3.5 software. Different bands produced by the same SSR primer were classified by size against a 100-bp DNA size marker ladder. Genotyping was done visually, and genotypes were identified as “A” for the female parent (Red Kafir) allele, “B” for the male parent (Takakibi) allele, and “H” for heterozygote.
Evaluation of yield and other related traits
The 149 F2 lines and their parental lines were planted in a field at the University of Tsukuba during the 2008 growing season. Phenotypic measurements were performed for eight important yield and yield-contributing traits: (1) culm length (CL), the length from the ground to the neck node of the panicle of the main stem; (2) number of tillers per plant (NT), specifically tillers longer than 1/3 the height of the main stem; (3) panicle length (PL), the length from the neck node to the tip of the panicle of the main stem; (4) culm diameter (CD), the long diameter of the middle of an internode of the main stem at 10–15 cm above the ground; (5) leaf length (LL), the length of the longest leaf blade; (6) leaf width (LW), the width of the widest part of the longest leaf blade; (7) grain weight per panicle (GWP), the total weight of cleaned grains per panicle on the main stem; (8) weight of 100 grains (100GW), the average weight of two 100-grain samples collected after threshing and removal of the glumes.
The two parental lines (Red Kafir and Takakibi) and 118 F3 family lines were grown in the field in 2009 growing season at the University of Tsukuba. The seeds were dusted with fungicidal/insecticidal dust before sowing, and the soil was fertilized with 750 kg per hectare base fertilizer (15 %N + 15 %P + 15 %K). F3 lines were sown in a randomized complete block design (RCBD) with two replications. Each replication had five plants, and individual plants were spaced 25 cm apart in rows spaced 100 cm apart. Data were recorded for seven yield and yield-related traits (CL, PL, CD, LL, LW, GWP and 100GW) for individual plants in both replications.
Statistical analysis
Phenotypic data for all traits were analyzed by using JMP v. 5 statistical software (SAS Institute Inc. 2002). One-way analysis of variance (ANOVA) and non-parametric correlation were performed for the traits studied. A linkage map was constructed using Mapmaker Exp/3.0b software (Lander et al. 1987). Recombination frequencies were converted into map distances in cM by the Kosambi mapping function.
Broad sense heritability (h 2) of the phenotypic traits investigated in F2:3 lines was calculated as the ratio of genetic variance \(\left( {\sigma_{\text{g}}^{ 2} } \right)\) to phenotypic variance \(\left( {\sigma_{\text{p}}^{ 2} } \right)\) (Hanson et al. 1956). Moreover, the expected genetic advance (R) was also calculated for each trait using the formula [R = i h 2 σ p] as described in Falconer and MacKay (1996), where i = 1.40 at 20 % selection intensity.
Two different approaches to QTL analysis were used to identify chromosomal regions associated with yield and yield-contributing traits. The first approach was based on a single-QTL method, composite interval mapping (CIM), and the second was a multiple-QTL method based on a Markov chain Monte Carlo (MCMC) algorithm.
For the single-QTL approach, QTL analysis was performed with WinQTLCart 2.0 software (Zeng 1994) by CIM. Empirical experiment-wise threshold values for significance (P = 0.05) were estimated from 1,000 permutations of the data for each trait. A stringent limit-of-detection threshold (LOD ≥ 2.5) was set to identify the putative presence of QTLs related to the traits. After the genetic map had been scanned and the likelihood of each QTL and its corresponding effects every 1 cM estimated, CIM was performed using Model 6 of WinQTLCart 2.0. The significant marker cofactors were used to adjust the phenotypic effects associated with other positions in the map with forward-backward stepwise regression and a window size of 10 cM. Adjacent QTLs on the same chromosome for the same trait but with non-overlapping intervals were considered different QTLs. The contribution rate (R 2) was estimated as the percentage of variance explained by each QTL. The results were reconfirmed with the R/qtl package (Broman et al. 2003) by interval mapping.
A multiple-QTL MCMC approach based on Bayesian interval mapping (BIM) was also used to identify QTLs and compared with the single-QTL approach. These QTL analyses were performed using the R software package R/qtlbim (Yandell et al. 2007). Bayes factor (BF) profiles were used to estimate the number of QTLs and the QTL effects together. Bayes factors were rescaled as 2logeBF, and negative values were truncated as zero. The threshold was set as 2logeBF ≥ 3.0. QTLs were designated with italicized symbols consisting of “qtl,” one or two digits corresponding to the chromosome number, a hyphen followed by an extra digit if more than one QTL was found on the same chromosome, and a trait designator (composed of two capital Roman letters).
Marker localization and homology with known genes
SSR markers associated with traits were physically localized by BLAST searches of sequences in http://www.phytozome.net/sorghum, http://www.plantgdb.org/SbGDB/, and http://www.gramene.org/. Markers previously identified as linked to known genes were localized to the genome-based sequence information provided in the Map Viewer on the NCBI website (http://www.ncbi.nlm.nih.gov/mapview/). The sequences with maximum matching were then used to search in Map Viewer; if the position was not returned in Map Viewer, the primer sequences were then used to search the sorghum genome database at http://www.phytozome.net/sorghum. Protein sequences predicted from genes were also used to search by BlastP, and the homologous sorghum genes were identified in http://www.plantgdb.org/SbGDB/.
Results
Phenotypic data analysis
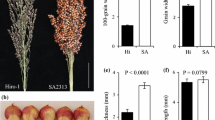
The F2 population showed a significantly greater range of variation than the parents (i.e., transgressive segregation) for all eight traits studied (Fig. 1a; Table 1). In all but four cases, the morphological traits were significantly and positively correlated with one another (P < 0.05), and many correlations were significant at P < 0.001 (Table 2). CL showed a highly significant correlation (P < 0.001) with all traits except for NT, which was non-significant. NT also had non-significant correlations with two other traits, GWP and 100GW, significance at P < 0.01 with CD, and significance at P < 0.05 with traits PL, LL, and LW. Similarly, CD was highly significantly (P < 0.001) correlated with LL, LW, and GWP while significantly (P < 0.05) correlated with 100GW. LL also had highly significant correlations with LW and GWP (P < 0.001) and significant (P < 0.01) correlation with 100GW. The same pattern was followed by LW, i.e., it was correlated with GWP at a high significance level (P < 0.001) and significantly (P < 0.01) correlated with 100GW. Similarly, GWP showed a highly significant correlation with 100GW (P < 0.001).

a Distribution of eight yield and yield-contributing traits studied in 149 F2 populations. b Distribution of seven yield and yield-contributing traits studied in 118 F2-derived F3 populations
The phenotypic measurements for seven traits investigated in the F2:3 family lines also showed a wider range of variations than the parental lines (Fig. 1b). According to the correlation analysis, all the traits had significant correlations except CL, which showed non-significant correlations with PL and LW. Similarly, 100GW showed no correlation with PL measured in F2:3 lines. Univariate analyses of variance (ANOVA) on the yield components revealed significant differences among genotypes for seven traits studied, as shown in Table 3. All of the seven phenotypic traits showed high values for the estimate of heritability (h 2) ranging from 0.72 for 100GW to 0.96 for PL. These traits also exhibited high genetic advance (R) at 20 % selection intensity (Table 3).
Construction of linkage map and QTL mapping
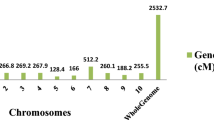
We selected 159 SSR markers for the construction of a linkage map. Of these, 22 could not be linked with other markers, so the genetic map was composed of 137 SSR loci (ESM1) mapped onto the ten chromosomes of sorghum (Fig. 2). The average distance between markers was 9.9 cM; the longest distance was 28.4 cM, and the shortest was 0.6 cM. Out of the 14 SSR markers previously mapped by Bhattramakki et al. (2000), 12 were mapped onto the same chromosomes, but two were mapped to different locations: Xtxp56 and Xtxp287 were mapped to chromosome (Chr) 1 and Chr5, respectively, whereas they were previously mapped to Chr2 and Chr9, respectively (Bhattramakki et al. 2000). This consistency supports the accuracy of the linkage map reported here.

Linkage map generated from 149 F2 plants by 137 SSR makers. Markers with bold face represent the interval where QTLs for CL, NT, PL, CD, LL, LW, GWP, and GW are located. Asterisk represents QTLs identified by both the CIM and BIM methods, plus represents QTLs detected with the CIM method, and open circle represents QTLs detected with the BIM method of QTL analysis. QTLs with regular face were identified using phenotypic values of 137 F2 line; QTLs in the lined box were identified only using 118 F3 lines; phenotypic data and QTLs with bold face were identified in both F2 and F3 generations
QTLs for culm length
In the F2 population, CIM identified two QTLs for CL, on Chr7 (qtl7CL) and Chr8 (qtl8CL), with LOD scores of 16.1 and 17.1 and R 2 values of 18.4 and 19.5 %, respectively (Fig. 2; Table 4). BIM also detected the major QTL, qtl7CL, with a 2logeBF value of 10.0. In contrast, BIM did not identify qtl8CL as significantly associated with this trait. Using phenotypic trait values of F2:3 family lines, qtl7CL was the only QTL identified for this trait with a LOD score of 11.2 and R 2 value of 13.1 % in the CIM method of analysis as shown in Fig. 2; Table 4. BIM also detected the same QTL in this population with a 2logeBF value of 8.3.
QTLs for number of tillers
Four QTLs (qtl4-1NT, qtl4-2NT, qtl6NT, and qtl8NT) were detected for NT in the studied F2 plants, two on Chr4 and one each on Chr6 and Chr8. LOD values for these QTLs ranged from 4.4 to 14.7 and explained 6.0 to 17.2 % of the variance. BIM did not identify any significant QTLs for this trait. This might be due to the non-normal pattern of phenotypic distribution for this trait (Fig. 1). This phenotypic parameter has not been evaluated in the F2:3 population because of the low range of variation among genotypes.
QTLs for panicle length
CIM identified seven QTLs for PL studied in F2s, including qtl1PL, qtl2bPL, and qtl6PL, on Chr1, Chr2b, and Chr6, respectively. Two closely linked QTLs, qtl7-1PL and qtl7-2PL, were identified on Chr7. These first five QTLs had LOD scores ranging from 7.5 to 22.2. Two minor QTLs, qtl8PL and qtl10PL, were located on Chr8 and Chr10 and had LOD scores of 4.4 and 2.8, respectively. R 2 values ranged from 3.4 to 24.1 %. BIM detected all but qtl8PL and qtl10PL (the two with the lowest LOD scores). One QTL (qtl9PL) detected by BIM was not found by CIM (Fig. 2).
Four QTLs out of the previously mentioned seven were also identified for the traits studied in F2:3 lines. These include qtl1PL, qtl2bPL, qtl6PL, and qtl7-2PL with LOD scores ranging from 7.2 to 13.0 and R 2 from 7.0 to 14.4 %. These four QTLs were also detected as significant for PL using BIM with 2logeBF ranging from 3.0 to 9.5.
QTLs for culm diameter
CIM and BIM identified five of the same QTLs for CD. Two of these (qtl1-1CD and qtl1-2CD) were located in different regions of Chr1 (57.2–65.9 and 209.0–214.9 cM, respectively). Single QTLs were identified on Chr1b (qtl1bCD), Chr3 (qtl3CD), and Chr4 (qtl4-2CD). Two QTLs were uniquely identified by CIM: one on Chr4, with flanking markers SB3664 and SB2596 at 0.0–12.1 cM (LOD = 9.5), and one on Chr5, with flanking markers SB3163 and SB3280 at position 77.5–95.6 cM (LOD = 14.5). A QTL on Chr8 (qtl8CD; Fig. 2) was only detected by BIM (2logeBF = 3.5).
While using F2:3 lines, CM and BIM identified the same five QTLs out of seven QTLs detected in the F2 population, including qtl1-1CD, qtl1bCD, qtl3CD, qtl4-2CD, and qtl5CD. The LOD scores ranged from 4.1 (qtl3CD) to 16.1 (qtl4-2CD) with phenotypic variance ranging from 5.5 to 3.4 %. In case of BIM, a minimum 2logeBF value, i.e., 3.4, was recorded for qtl5CD (Chr5), whereas the maximum value of 14.2 was for qtl1bCD located on Chr1b.
QTLs for leaf length
In the CIM analysis, five QTLs had a significant association with LL investigated in F2 lines. Two of them were closely linked on Chr1: qtl1-1LL and qtl1-2LL (Fig. 2). CIM identified three other QTLs: qtl1bLL on Chr1b, qtl3LL on Chr3, and qtl8LL on Chr8. All of these QTLs except for qtl8LL were also detected by BIM. BIM also found two more QTLs for LL: qtl6LL (Chr6) and qtl9LL (Chr9).
In case of F2:3 family lines, three QTLs were significantly related to LL by both CIM and BIM methods of analysis. These QTLs (qtl1-1LL, qtl3LL and qtl8LL) are among the five QTLs identified as significant in the F2 population. The LOD scores ranged from 3.8 to 12.4 in CIM, whereas 2logeBF varied from 2.6 to 6.8 as calculated by BIM.
QTLs for leaf width
Using phenotypic values of F2 lines, CIM identified two QTLs for LW: qtl1bLW (Chr1b) and qtl3LW (Chr3). The total phenotypic variation for LW explained by these two QTLs was 16.4 %. BIM also identified these QTLs, along with four others not detected by CIM: qtl2bLW (Chr2b), qtl5LW (Chr5), qtl8LW (Chr8), and qtl10LW (Chr10), with 2logeBF values of 3.0–5.9.
QTL analyses with CIM and BIM in F2:3 lines revealed the same QTLs (qtl1bLW and qtl3LW) controlling the trait LW. However, one QTL named qtl5LW (Chr5) was only identified by CIM in this population with a LOD value of 5.6 and R 2 value of 9.5 % (Table 4; Fig. 2).
QTLs for grain weight per panicle
For the trait GWP investigated in the F2 population, CIM mapped five QTLs, with LOD scores ranging from 3.1 to 11.2: qtl2GWP (Chr2), qtl2bGWP (Chr2b), qtl3GWP (Chr3), qtl7-1GWP (Chr7), and qtl7-2GWP (Chr7). The cumulative phenotypic variance controlled by these QTLs was 41.7 %, and individual R 2 values ranged from 4.5 % to 13.1 %. BIM mapped the same QTLs for this trait, with 2logeBF values of 2.5–3.9. In addition, BIM identified one unique QTL (qtl6GWP) on Chr6, with a 2logeBF value of 4.4.
CIM and BIM detected four QTLs: qtl2GWP (Chr2), qtl2bGWP (Chr2b), qtl3GWP (Chr3), and qtl7-2GWP (Chr7) for the trait GWP investigated in F2:3 family lines. These QTLs were the same as found significant in the F2 population. The LOD values ranged from 3.1 to 11.2 with phenotypic variance ranging from 2.5 to 22.1 %. BIM gave 2logeBF values for these QTLs ranging from 3.4 to 7.7.
QTLs for 100-grain weight
CIM identified nine QTLs for 100GW using phenotypic data of the F2 population. This was the largest number of QTLs for any trait identified by CIM. These QTLs had large LOD scores, ranging from 14.4 to 33.8. Single QTLs were located on Chr1b (qtl1bGW), Chr2 (qtl2GW), Chr3 (qtl3GW), and Chr5 (qtl5GW). Two QTLs were found on Chr4 (qtl4-1GW, 0.0–12.1 cM; qtl4-2GW, 17.9–30.9 cM), and three QTLs were found on Chr7 (qtl7-1GW, 0.0–12.3 cM; qtl7-2GW, 76.3–93.6 cM; and qtl7-3GW, 93.6–109.6 cM) (Table 4; Fig. 2). All of the R 2 values for these QTLs were around 7 % except for qtl2GW, for which R 2 equaled 36.3 %, the highest value obtained in this study. In contrast, BIM did not detect the three QTLs detected on Chr7 by CIM. In addition, BIM uniquely identified another QTL, qtl6GW, on Chr6 (2logeBF = 3.7). The 2loge(BF) scores for the other markers ranged from 2.1 (qtl1bGW) to 5.4 (qtl4-2GW).
Both CIM and BIM could identify five similar chromosomal regions controlling 100GW in F2:3 lines. These five QTLs were included in those previously detected when F2 phenotypic data were used (i.e., qtl2GW, qtl3GW, qtl5GW, qtl7-1GW, and qtl7-2GW). The LOD values for these QTLs ranged from 14.4 to 33.8, whereas 2logeBF scores ranged from 2.8 to 13.7.
Physical colocalization with known genes
To examine the possibility that some of the QTLs identified here are associated with known genes, we physically localized our markers with previously mapped QTLs on sorghum chromosomes. Marker SB0852, which flanks qtl1-1LL, is located on Chr1 at 70,817,033 bp, which is the location of protein-coding gene SB01G047730. This gene is predicted to function in protein and nucleic acid binding processes. Another marker flanking several QTLs, SB0868, on Chr1, was physically located at 71,950,739 bp. Three QTLs (qtl1-2CD, qtl1PL, and qtl1-2LL) were found in this same region, which contains the protein-coding gene SB01G048937. This gene is orthologous to Osl-31480, a gene that controls branch length in the Oryza sativa ssp. indica group. Similarly, two marker loci on Chr1b, Xtxp335 (55,801,722 bp) and Xtxp149 (50,709,235 bp), define a region in which four QTLs were located, one each for CD, LL, LW, and GW. Xtxp335 is located within gene SB01G032850, which is predicted to play a role in the light reactions of photosynthesis. Marker locus SB2596, near QTLs for CD, GW, and NT on Chr4 (14,734,368 bp), contained within a gene (SB04G010640), is predicted to be responsible for catalytic activity during metabolic processes. Marker locus SB3705, on Chr6 (56,121,612 bp), flanking QTLs qtl6NT and qtl6PL, associated with gene SB06G027180, encodes a known protein with inorganic diphosphatase and binding activities. This gene has 57 orthologs in different plants species.
Discussion
Using two approaches to QTL mapping, CIM and BIM, we detected 52 QTLs associated with seven traits related to yield and its components in an F2 population derived from a cross between African and Japanese sorghum accessions with wide origins and morphologies. Among these QTLs, 25 were confirmed using 118 F2:3 phenotypes, and two QTLs were uniquely found in this generation (qtl7CL and qtl5LW).
The linkage map we constructed is composed of 137 mapped SSR markers; an additional 22 markers were tested but could not be mapped. The SSRs reported here were developed from shotgun sequences of the whole sorghum genome (Yonemaru et al. 2009). The 137 markers included 14 previously mapped SSRs reported by Bhattramakki et al. (2000) to show the pattern of recombination with other markers. Only 2 of the 14 loci were mapped to a different location from that previously reported, while the rest were mapped to the same chromosomes as described in earlier studies. Most of the other new loci selected from Yonemaru et al. (2009) were mapped to the same chromosomes as previously reported, with few exceptions (ESM1). SB3664 was mapped on Chr4 and SB3412 on Chr5, but these markers were originally selected from the genome sequence of Chr6. Similarly, we mapped SB4925 and SB4956 on Chr8, but they were localized on Chr9 in the previous report (Yonemaru et al. 2009). Such discrepancies can be explained by sampling variation or the mapping of paralogous loci (i.e., loci arising from gene duplication). The fact that most of the markers were mapped to the same chromosomes as reported earlier supports the accuracy of the linkage map developed here.
We used single- and multiple QTL approaches to validate the results of the analyses. For the single-QTL mapping approach, we selected CIM for its power to reduce more of the residual variance than interval mapping. The multiple-QTL approach used BIM, which is based on the MCMC sampling technique. In this method, samples are drawn from the posterior distribution of genetic architecture, including the number and location of genetic loci, gene action effects at all loci, and G × E interactions (Yi et al. 2007). Most agronomically important traits in plants are quantitatively inherited and hence are controlled by multiple genetic and environmental factors. To elucidate such traits, selection of the proper model plays a vital role in the identification of chromosomal regions with both major and minor effects. The multiple-QTL model provides greater ability to select QTLs associated with particular traits, regardless of whether the effects are strong or weak.
The two QTL mapping approaches identified similar sets of QTLs as significantly associated with the studied traits. This agreement between the methods supports the efficiency and reliability of our findings. In total, 50 QTLs were identified by the two approaches combined, and each chromosome had at least one QTL. CIM detected 41 QTLs, BIM detected 38, and both detected 29. BIM did not detect any QTL for NT, perhaps because of its non-normal pattern of phenotypic distribution and the low level of variation for the trait. Similarly, with only two exceptions, common QTLs were detected in F2 and F2:3 family lines, confirming the stability of the QTLs mentioned in this report.
Some of the QTLs appear to correspond with previously reported QTLs, whereas others appear to be newly identified. One of the four major dwarfing genes in sorghum, dwarf3 (dw3), which is an ortholog of brachytic2 (br2) in maize, has been cloned and sequenced by Multani et al. (2003) and is located on Chr7. We also identified a major QTL for culm length (qtl7CL) on Chr7 between SSR markers SB4096 (93.6 cM) and SB4024 (109.6 cM), which is the same location as dw3. LL and LW together reflect total leaf green area; some of the QTLs identified for these traits were in similar genomic regions as the previously identified stay-green QTL regions StgG, Stg2, and Stg4 (Crasta et al. 1999; Xu et al. 2000).
Several of the QTLs were associated with two or more traits. For example, a QTL in the distal part of Chr1 with flanking markers SB0868 (209.0 cM) and SB1707 (214.9 cM) was associated with LL, PL, and CD. Similarly, the region in Chr1b between 0.0 cM (Xtxp335) and 28.4 cM (Xtxp149) contained a QTL controlling four traits: CD, LL, LW, and 100GW. A region in Chr2b between markers SB0968 (1.9 cM) and SB0999 (25.7 cM) had a significant association with PL, CD, 100GW, and LW. Together, the CIM and BIM methods detected a QTL on Chr7 between SB4096 (93.6 cM) and SB4024 (109.6 cM) controlling CL, PL, GWP, and 100GW. These results might be explained by the pleiotropic effects of genes controlling more than one quantitative trait. Another possibility is that these results reflect the presence of two or more genes in the same region, each controlling a different trait. The relationships among the QTLs and phenotypes studied here need to be tested and quantified further to establish the nature of genetic heterogeneity and pleiotropy underlying the effects of these QTLs.
Several other studies have identified QTLs associated with the traits studied here. Some of our findings are in accordance with previous ones, while others are unique. Srinivas et al. (2009) used genic-microsatellite markers and mapped several QTLs for agronomic traits. They found four QTLs associated with plant height, two of which we found in the same regions: on Chr7 at 10 cM between flanking markers Dsemhsbm7 and Xtxp92 (0–24 cM) and at 39.4 cM between flanking markers Xtxp92 and Xtxp295 with an interval of 24–48.4 cM. Similarly, Srinivas et al. (2009) located major QTLs for panicle length on chromosomes 2, 6, and 7, in accordance with our findings. A QTL identified for panicle weight on Chr6 matched with qtl6GWP in our study. The QTLs for seed weight detected by Srinivas et al. (2009) on chromosomes 1, 4, and 6 are also similar to qtl1bGW, qtl4-1GW, qtl4-2GW, and qtl6GW.
Shiringani et al. (2010) reported four QTLs on Chr1 associated with plant height that explained 4.7–6.3 % of the phenotypic variance, whereas we did not identify any QTL for CL on Chr1. They also found five QTLs for panicle weight, each explaining 3.9–5.8 % of the phenotypic variance. One of these was on Chr2, and two were on Chr6, where we also identified QTLs, but we did not identify the others (on Chr1 and Chr5). Ritter et al. (2008) detected four QTLs for plant height, three on Chr3 (not identified here) and one on Chr7 (as here) at 28.8 cM with flanking markers AGG to CAA2 and Xtxp295 (25.5–28.8 cM). Hart et al. (2001) detected five QTLs associated with culm length; one was on Chr7 (linkage group [LG] E), with estimated map locations of 100 cM and 96 cM (data from two different testing locations), very close to the position of qtl7CL. Similarly, Hart et al. (2001) identified two QTLs for the number of basal tillers per basal-tillered plants on LG-I (Chr6), similar to qtl6NT, which we found in this study.
Feltus et al. (2006) aligned two different genetic maps and identified a large number of loci associated with 17 morphological traits. They located eight QTLs controlling plant height in sorghum; one that had a major effect in two replications was found on Chr7 (LG-E), where we also found a QTL for CL. Feltus et al. (2006) detected three unique leaf-length QTLs that collectively explained 26.3 or 17.8 % of the phenotypic variance (percentages for each of two replicates). Two of the three QTLs, in LGs-F and H, are the positions closer than this as qtl9LL and qtl8LL, respectively. On the other hand, Feltus et al. (2006) reported ten QTLs for leaf width, only one of which, on LG-J, was on the same chromosome as one that we located (qtl5LW) with flanking markers SB3163 (77.5 cM) and SB3280 (95.6). Similarly, eight out of ten QTLs for kernel weight detected in two different populations were on the same chromosomes as QTLs reported here.
In our previous study of association mapping using a sorghum core collection and SSR markers (Shehzad et al. 2009b), 13 QTLs had a strong association with 12 of the 26 morphological traits studied. Some of the QTLs reported here are in accordance with those QTLs. For example, five loci had a strong association with PL—three on Chr1 (LG-A), one on Chr2 (LG-B), and one on Chr9 (LG-F)—and weak associations were found with markers on chromosomes 6 (LG-I), 7 (LG-E), and 10 (LG-G) (Shehzad et al. 2009b). All of these chromosomes also held markers for PL here. Similarly, a QTL in LG-H (Chr8) was strongly associated with LL (Shehzad et al. 2009b); this QTL is in the same position as qtl8LL identified here. A K-model of association analysis (a model taking into account kinship) found the same QTL on Chr8 to be associated with LW, also in correspondence with qtl8LW here. Similarly, the K-model found a QTL in LG-E (Chr7) to be strongly associated with CL (Shehzad et al. 2009b); this QTL falls in the same position as qtl7CL here. Such similarities in the identification of QTLs for important traits in F2-based linkage mapping and population-based association mapping support the results obtained here.
We have also surveyed the physical locations of loci associated with QTLs identified in this study. Some of the locations overlap with known genes functioning in important biological processes. One of the six sorghum maturity genes, Ma 3 , encodes phytochrome B (Childs et al. 1997) and is located at the position of locus Xtxp335 on Chr1b in this study. Xtxp335 is one of the flanking markers for four QTLs (qtl1bCD, qtl1bLL, qtl1bLW, and qtl1bGW). This linkage suggests that Ma 3 might be related to the effects of one or more of the nearby QTLs, but additional data would be necessary to test this possibility.
Breeding for yield and yield-related traits is an important objective for all agronomically important crops. The development of tools in functional genomics such as microarray technology and expressed sequence tag analysis has made it possible to identify candidate genes. The integration of sorghum genetic maps with the physical map will greatly facilitate the map-based cloning and molecular dissection of complex traits, including yield. Our findings can be used in sorghum improvement through breeding programs and in marker-assisted selection strategies. Our study shows the importance of choosing appropriate statistical models to be applied in mapping quantitative traits and the importance of minor QTLs along with major ones.
References
Bhattramakki D, Dong J, Chhabra AK, Hart GE (2000) An integrated SSR and RFLP linkage map of Sorghum bicolor (L.) Moench. Genome 43:988–1002
Broman KW, Wu H, Sen Ś, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890
Childs KL, Miller FR, Cordonnier-Pratt M, Pratt LH, Morgan PW, Mullet JE (1997) The sorghum photoperiod sensitivity gene, Ma 3 , encodes a phytochrome B. Plant Physiol 113:611–619
Crasta OR, Xu WW, Rosenow DT, Mullet J, Nguyen HT (1999) Mapping of post-flowering drought resistance traits in grain sorghum: association between QTLs influencing premature senescence and maturity. Mol Gen Genet 262:579–588
Falconer DS, MacKay TFC (1996) Introduction to qualitative genetics, 4th edn. Longman, London
Feltus FA, Hart GE, Schertz KF, Casa AM, Kresovich S, Abraham S, Klein PE, Brown PJ, Paterson AH (2006) Alignment of genetic maps and QTLs between inter- and intra-specific sorghum populations. Theor Appl Genet 112:1295–1305
Hanson CH, Robinson HF, Comstock RE (1956) Biometrical studies of yield in segregating populations of Korean Lespedeza. Agron J 48:268–272
Hart GE, Schertz KF, Peng Y, Syed N (2001) Genetic mapping of Sorghum bicolor (L.) Moench QTLs that control variation in tillering and other morphological characters. Theor Appl Genet 103:1232–1242
Haussmann BIG, Mahalakshmi V, Reddy BVS, Seetharama N, Hash CT, Geiger HH (2002) QTL mapping of stay-green in two sorghum recombinant inbred populations. Theor Appl Genet 106:133–142
Kebede H, Subudhi PK, Rosenow DT, Nguyen HT (2001) Quantitative trait loci influencing drought tolerance in grain sorghum (Sorghum bicolor L. Moench). Theor Appl Genet 103:266–276
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg I (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:74–81
Multani DS, Briggs SP, Chamberlin MA, Blakeslee JJ, Murphy AS, Johal GS (2003) Loss of an MDR transporter in compact stalks of maize br2 and sorghum dw3 mutants. Science 302:81–84
Rami JF, Dufour P, Trouche G, Fliedel G, Mestres C, Davrieux F, Blanchard P, Hamon P (1998) Quantitative trait loci for grain quality, productivity, morphological and agronomical traits in sorghum (Sorghum bicolor L. Moench). Theor Appl Genet 97:605–616
Ritter KB, David R, Jordan DR, Chapman SC, Godwin ID, Mace ES, McIntyre CL (2008) Identification of QTL for sugar-related traits in a sweet × grain sorghum (Sorghum bicolor L. Moench) recombinant inbred population. Mol Breed 22:367–384. doi:10.1007/s11032-008-9182-6
SAS Institute Inc (2002) JMP statistical and graphics guide, version 5. SAS Institute Inc., Cary
Shehzad T, Iwata H, Okuno K (2009a) Genome-wide association mapping of quantitative traits in sorghum (Sorghum bicolor (L.) Moench) by using multiple models. Breed Sci 59:217–227
Shehzad T, Okuizumi H, Kawase M, Okuno K (2009b) Development of SSR-based sorghum (Sorghum bicolor (L.) Moench) diversity research set of germplasm and its evaluation by morphological traits. Genet Resour Crop Evol 56:809–827. doi:10.1007/s10722-008-9403-1
Shiringani AL, Frisch M, Friedt W (2010) Genetic mapping of QTLs for sugar-related traits in a RIL population of Sorghum bicolor L. Moench. Theor Appl Genet 121:323–336
Srinivas G, Satish K, Madhusudhana R, Reddy RN, Mohan SM, Seetharama N (2009) Identification of quantitative trait loci for agronomically important traits and their association with genic-microsatellite markers in sorghum. Theor Appl Genet 118:1439–1454. doi:10.1007/s00122-009-0993-6
Tao YZ, Henzell RG, Jordan DR, Butler DG, Kelly AM, McIntyre CL (2000) Identification of genomic regions associated with stay green in sorghum by testing RILs in multiple environments. Theor Appl Genet 100:1225–1232
Tuinstra MR, Grote EM, Goldsbrough PB, Ejeta G (1996) Identification of quantitative trait loci associated with pre-flowering drought tolerance in sorghum. Crop Sci 36:1337–1344
Tuinstra MR, Grote EM, Goldsbrough PB, Ejeta G (1997) Genetic analysis of post-flowering drought tolerance and components of grain development in Sorghum bicolor (L.) Moench. Mol Breed 3:439–448
Xu W, Subudhi PK, Crasta OR, Rosenow DT, Mullet JE, Nguyen NT (2000) Molecular mapping of QTLs conferring stay-green in grain sorghum (Sorghum bicolor L. Moench). Genome 43:461–469
Yandell BS, Mehta T, Banerjee S, Shriner D, Venkataraman R, Moon JY, Neely WW, Wu H, von Smith R, Yi N (2007) R/qtlbim: QTL with Bayesian interval mapping in experimental crosses. Bioinformatics 23:641–643
Yi N, Shriner D, Banerjee S, Mehta T, Pomp D, Yandell BS (2007) An efficient Bayesian model selection approach for interacting QTL models with many effects. Genetics 176:1865–1877
Yonemaru J, Ando T, Mizubayashi T, Kasuga S, Matsumoto T, Yano M (2009) Development of genome-wide simple sequence repeat markers using whole-genome shotgun sequences of sorghum (Sorghum bicolor (L.) Moench). DNA Res 16:187–193
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Acknowledgments
We cordially thank Drs. Masahiro Yano, Junichi Yonemaru, and Takashi Matsumoto (NIAS, Japan) for providing information on the SSR markers used in this study ahead of its publication. We also acknowledge the efforts of Mss. Mannai Yousra and Janaki Khaple Bhandari, graduate students at the University of Tsukuba, for their help in collecting morphological data in the field.
Conflict of interest
The authors have no conflict of interest, and the plant material used is freely available at the National Institute of Agrobiological Sciences (NIAS) Genebank.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shehzad, T., Okuno, K. QTL mapping for yield and yield-contributing traits in sorghum (Sorghum bicolor (L.) Moench) with genome-based SSR markers. Euphytica 203, 17–31 (2015). https://doi.org/10.1007/s10681-014-1243-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-014-1243-9